
Introduction
Most cases of the breast cancer are unilateral, and bilateral breast cancer (BBC) occurs in approximately 5% of female breast cancer survivors [1, 2]; it is classified as either synchronous or metachronous on the basis of the time interval between the first and second contralateral tumors (generally 6 months) [3]. In clinical practice, approximately 1% of all breast cancers are synchronous and 3%-7% are metachronous [4]. BBC has been considered to possess similar phenotypic features to those of unilateral breast cancer, even though it is more commonly associated with a positive family history of breast cancer, early disease onset, lobular histologic type multicentricity of the first tumor, and BRCA1 and BRCA2 mutations [5-7]. The increasing breast cancer incidence, more favorable prognoses, extended life expectancies, and improvements in detection are expected to lead to an increased survival rate of BBC [8]. Thus, BBC holds intriguing clinical and fundamental aspects in understanding tumor-host relationships, considering the few paired organs in human body with both high cancer incidence and good cancer survival.
Nevertheless, BBC remains less studied than its unilateral counterpart and has posed a great challenge for oncologists due to many unanswered questions. First, the biological relationship between the two breast cancers is not well understood. It can be difficult to discriminate between a second primary tumor and a breast metastasis [9]. Clarifying the issue of BBC clonality has implications for our understanding of breast carcinogenesis and for identifying the optimal breast cancer treatment [5, 6], as the management of primary disease and recurrent or metastatic breast cancer is substantially different [7]. A misdiagnosis of BBC or metastasis may result in different therapeutic regimens and poor disease outcome. BBC clonality has been studied using X chromosome inactivation, p53 mutation detection, and DNA allelotyping [10-15]. The results of these conventional cytogenetic investigations indicate that the vast majority of clinically diagnosed BBCs, if not all, represent clonally independent disease. A few studies have used comparative genomic hybridization (CGH) to determine the clonal association of BBCs [16-18] but reported conflicting results. These were insufficient for drawing a conclusion about the clonal origin of BBC.
Second, although BBCs are subjected to the same environmental and genetic influences, there may be a complex combination of different time spans (synchronous or metachronous), morphologic characteristics, and hormonal receptor statuses [19]. Whether the dominant tumor hypothesis in molecular-genetic profiles stands is debatable [20, 21]. The currently available genetic evidence for breast cancer metastasis suggests that the time interval between the development of the two tumors is important in discriminating between true BBCs and contralateral metastasis [4]. Mostly synchronous metastases present with identical genetic patterns to those of the primary breast tumor. In the case of metachronous metastasis, additional genetic events also drive tumor evolution except for a common load of abnormalities to the primary lesions [4, 14, 22]. Nevertheless, these discordant mutations do not obscure the relationship between the two tumors. Thus, synchronous and metachronous BBC with highly concordant genetic profiles may correspond to contralateral metastasis. However, the results of comparative analyses of the concordance of tumor molecular expression characteristics and genetic patterns in synchronous or metachronous BBC were inconsistent and inconclusive [14, 18, 23, 24]. Further evidence from whole-genome investigations is needed to identify molecular-genetic profiles in synchronous and metachronous BBCs and address whether there is a tendency to concordance of genetic lesions in BBC.
In this study, we analyzed genomic alterations in four BBC tumor pairs and two primary-metastasis tumor pairs from patients with breast cancer and liver metastasis to shed light on the controversial issues of the clonal origins in BBC pathogenesis. With the use of whole-exome high-throughput sequencing and comprehensive array CGH (aCGH), we confirmed that BBC consists of two clonally independent malignancies. Interestingly, neither synchronous nor metachronous BBCs exhibited a tendency towards concordance of genetic routes.
RESULTS
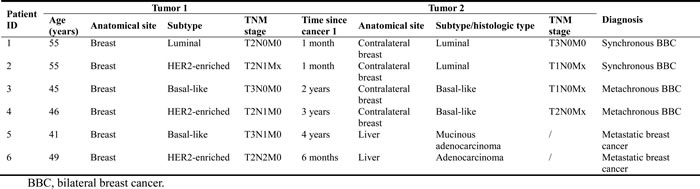
We examined the genomic landscape of BBCs by ultra-deep whole-exome sequencing of the bilateral tumors and matched blood samples. We selected four pairs of BBC samples for sequencing: synchronous 1-1: luminal, 1-2: luminal; synchronous 2-1: Her2-enriched, 2-2: luminal; metachronous 3-1: basal-like, 3-2: basal-like; and metachronous 4-1: basal-like, 4-2: Her2-enriched. We used six months as the cut-point to define synchronous and metachronous cancers [7]. More detailed information on the four pairs of BBC is shown in Table 1. We also sequenced two pairs of primary breast cancer samples and corresponding metastatic liver samples, which served as a control group (Table 1).
Table 1: Clinical characteristics of patients with bilateral breast cancer and metastatic breast cancer
We achieved a median of 427.8X sequence coverage (range: 335.8X~563.6X) of targeted exonic regions, with 97.3% of loci covered at ≥10-fold. On average, 72.8 coding mutations per tumor were identified, 30.1% of which were synonymous (Supplementary Table 1). Of coding point mutations, the observed nonsynonymous:synonymous ratio of 2.33:1 (493:212) was not significantly higher than that expected by chance (right-tail proportional test, P = 0.99) [25], indicating that the majority of coding mutations do not confer a selective advantage to BBC. This is similar to the nonsynonymous:synonymous ratio reported in basal-like breast cancer [26]. The overall BBC mutation rate was comparable to that of other subtypes of breast cancer [27, 28]. TP53 was found to be the most commonly mutated gene in BBC (50% [4 of 8] of the samples). We observed that a nonsynonymous TP53 mutation (p.R43H) occurred in both samples from Patient 6. We also observed that Patient 3 and 4 had two mutations in TP53 at different positions, respectively. Specifically, In Patient 3, we found TP53 p.R81X (stopgain mutation) mutated in tumor1 but not tumor2, whereas TP53 p.R141H was mutated in tumor2 but not tumor1. In Patient 4, TP53 p.Y88C and p.R210X mutations were found in tumor1 and tumor2, respectively. Other known cadre genes that drive breast cancer clusters (i.e., PIK3CA, GATA3, CDH1, and MAP3K1 [27-30]) were not found, perhaps due to small number of samples in this study and/or heterogeneity of breast cancer.
To determine whether BBCs are genetically independent, we first compared the similarities of mutations between paired BBC samples and breast/metastasis samples (Figure 1). The majority of somatic mutations in primary tumors are shared with the corresponding metastatic lesions [31, 32]. In our study, concordant mutations were rare (<10%) in the paired BBC samples, in sharp contrast to around 80% concordant mutations in the paired breast/metastasis samples (median depth: 60.9X, range: 51.8X~81.6X). We have identified 21 mutations shared in the BBC tumor 1 and tumor 2 (Supplementary Table 2). Among these, there are only two non-synonymous somatic mutations identified in Patient 4, i.e. NBPF1 p.D896Y and LILRA6 p. Y297S. The first mutation was predicted to exert functional impact (SIFT score = 0.01) thus considered potential driver mutation, whereas the second did not (SIFT score = 0.11). If BBCs are origin dependent, they will share more SNVs than are expected by chance. Fisher’s exact test was used to determine whether two BBCs shared a significant number of somatic SNVs. The P values for the Fisher’s exact test were not significant for all four pairs of synchronous or metachronous BBCs but were highly significant for the paired breast/metastasis samples (Table 2).
Table 2: Fisher's exact test to determine the clonality between the 1st and the 2nd tumors by evaluating the concordance of their somatic SNVs
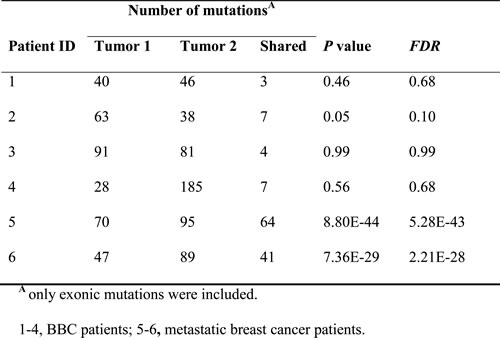
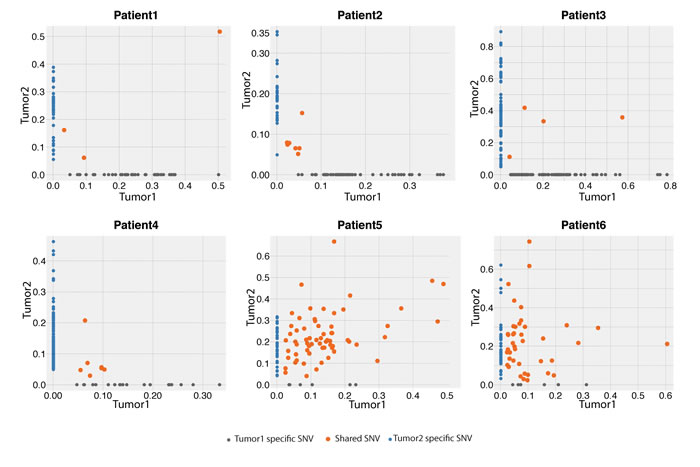
Figure 1: Variant frequency (VAF) distribution of identified SNVs between the first and second tumors from the six pairs of tumors in the exome-sequencing screen.
Different combinations of mutation types, termed mutational signatures, reflect different mutational processes that are operative in cancer [33]. The transition:transversion ratio in BBC was 1.5 in this study, similar to that reported in other types of breast cancer [34]. The mutational signatures of the paired samples are shown in Supplementary Figure 1, with the most common mutations of C>T nucleotide transition (C>T:G>A) in all cases [33]. Consistently, a clustering analysis based on the mutational signature of the 12 samples matched the two pairs of primary/metastasis samples; the BBCs were mismatched except in patient #2 (Figure 2).
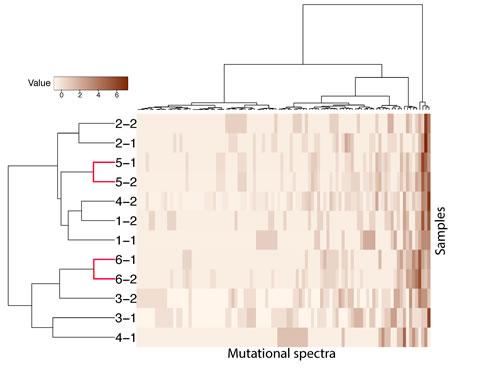
Figure 2: Hierarchical clustering of 12 samples according to their nucleotide context-specific, exonic, and somatic mutation rates in the exome sequencing screen. Mutation spectra in each sample were scaled. Each row represents a sample, and each column represents 1 of 96 strand-collapsed trinucleotide context mutation signatures. Top bar, single-nucleotide context mutational signature; left bar, cluster membership; right gradient, mutation rate scale. “-1”: first tumor, “-2”: second tumor.
To get another line of evidence whether BBCs are origin independent, we used aCGH to study copy number alteration landscape of all these tumors. Although there was inter-patient diversity in the copy number change per chromosome arm among all the six paired cases, the overall pattern of large copy number alterations seen in the first and second samples (Supplementary Figure 2) was consistent with that of these well described alterations. For example, we observed frequent large chromosomal gains in 1q, 3q, 8q, 20q, and 21q and broad losses involving 8p, 11q, 16q, and Xq [16-18, 35, 36]. Thus, these BBC tumor samples, as well as the paired liver samples from metastatic breast cancer, bore many of the hallmark copy number alterations commonly found in breast cancer.
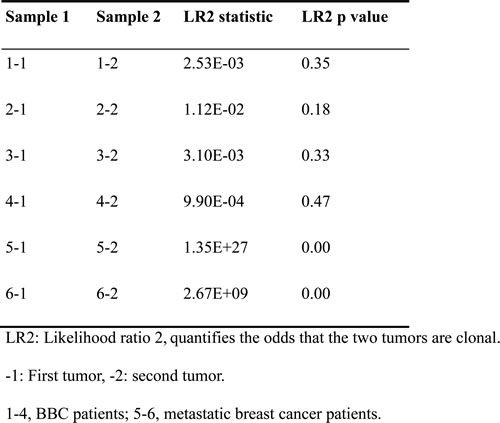
With regard to the inner concordance between the two tumors from each patient, two metastatic breast cancer patients (#5 and #6) had identical CGH ratio profiles in their corresponding tumor pairs. All other BBC tumor pairs had dissimilar CGH ratio profiles (Supplementary Figure 2). More direct evidence resulted from the subsequent clonality analysis using CNV profiles (Table 3). We calculated the LR2 to evaluate the clonal relatedness of the six pairs of tumors and found a significantly higher LR2 value for the two primary-metastasis pairs (P<0.001); however, no obvious relationships were observed for the four synchronous or metachronous BBCs (P value >0.05).
Table 3: Clonality analysis of 12 samples according to their copy number variations at the probe level
DISCUSSION
Breast cancer patients have a 2- to 6-fold higher risk of developing contralateral breast cancer than women in the general population have of developing a primary breast cancer [1]. BBC is presumed to have a unique genetic background, considering that BBC patients are younger, more commonly have a family history and BRCA germline mutations than unilateral breast cancer cases [8, 9, 37]. Only a few reports have presented the genetic findings of sporadic BBCs involving different aspects and factors in breast carcinogenesis [17].
Another dilemma in BBC is its unclear origin, as it is unknown whether it represents an independent second primary tumor or dependent on the primary tumor. Clarifying this issue will have implications for treating BBC and understanding its carcinogenesis [16, 24, 38]. The clonality of multiple malignancies has long been of interest to cancer biologists. However, the clinical and histopathologic features at the individual patient level described in previous studies, seem to have little diagnostic value in differentiating between an independent primary tumor and a breast-to-breast metastasis [14]. Recent advances in molecular biology have made it possible to define the relationship between the two BBC tumors. A comparative analysis using various cytogenetic techniques presented evidence of the pathogenetic independence of most BBC tumors [10-14, 16, 17]. A few studies have shown concordant genetic alterations in BBC [18, 23, 24], suggesting that comprehensive genome profiling approaches are needed to help us fully understand the discrepancies between contralateral breast metastases and de novo primary tumors. Another reason for these inconsistencies is that different criteria are used to define synchronous and metachronous BBCs [4].
In the present study, we included four paired-samples of BBCs—two synchronous and two metachronous—using the most widely used classification, a 6-month time span from the index tumor to the second tumor. Two paired primary breast tumors and corresponding metastatic liver lesions were used as positive controls for this synchronous and metachronous primary-metastasis model. We achieved a mutational SNV rate in BBCs that was comparable to that in sporadic unilateral breast cancer, as found on high-throughput exome-sequencing [27, 28]. They also shared an identical mutational signature with that of sporadic breast cancers, dominated by the most common mutation of C>T nucleotide transition (C>T:G>A) [33]. It is accepted that different molecular profiles from multiple neoplasms represent distinct clonal origins, while concordant data suggest a monoclonal origin [4]. We observed only a few overlapping variants in each bilateral breast tumor pair, which significantly differed from the obvious common variants shared by the primary and metastatic lesions of two metastatic breast cancers. The bilateral tumors in three of the four BBC patients segregated after hierarchical clustering of mutational signature data, indicating that they were independent. Our breast cancer cases with liver metastases showed proper clustering, allowing us to identify metastatic disease.
One way to reliably determine whether two bilateral breast tumors are two primary carcinomas or metastases of a primary neoplasm is by Fisher’s exact test. It assesses whether two tumors are sharing a significant number of somatic mutations and provides a line of evidence for clonal dependence [39]. Using this guideline, a pathogenetic independence of the bilateral tumors was inferred in the four pairs of synchronous and metachronous BBCs. On the contrary, the likelihood that the shared mutational changes found in the tumors from metastatic breast cancer patients occurred by chance is extremely small. These findings are in keeping with those of previous studies; they exclude the hypothesis of metastatic spread and favor true bilaterality of BBC.
We obtained high-resolution views of all unbalanced chromosomal alterations in this series of six pairs of tumors. The overall pattern of frequently detected genetic alterations in BBC did not differ significantly from that found in previous studies of unilateral, sporadic breast cancers [35, 36]. Compared to the commonly used allelotyping approach (loss of heterozygosity analysis), an elaborate genome-wide copy number arrays (CGH array) may be more valuable for determining the independence or clonality of multiple cancerous lesions. Recently, clonality analysis, a more accurate statistical method with priority over preliminary hierarchical clustering, was developed to compare such genomic profiles [40]. This analysis revealed an overwhelmingly higher degree of clonal relatedness in the cytogenetic profiles of primary and metastatic tumors. By contrast, a nearly negligible association between BBCs was found for all four BBC pairs in our study, leaving little doubt that they are pathogenetically independent. This was in line with the findings that each BBC tumor is the result of a separate carcinogenic event [9].
In summary, with the help of whole-exome sequencing and CGH techniques, we systematically revealed the independent genomes of BBC in Han Chinese women, both synchronous and metachronous. Even the two synchronous tumors that occurred within as short as a month also have very diverse mutation profiles thus representing two independent tumors. This is a revelation at the genomic level that provides new insight into the development of bilateral breast cancer, although we cannot completely rule out the possibility of that they have the same clonal or sub-clonal origin because of our limited sample size. These findings appear to have a practical impact on the therapeutic regimen for BBC, as the clinical management of localized breast cancer is critically different from that of metastatic disease. It is essential to consider BBC as two diseases because similar systemic management for the two may not be applicable. Finally, this proof-of-principle study should be further tested by additional large and comprehensive research to provide important new insights into the biological mechanism of this uncommon disease.
MATERIALS AND METHODS
Patient selection and sample preparation
We searched a retrospective archive of the Tumor Biobank at Tianjin Medical University Cancer Institute and Hospital to identify all patients who had been consecutively histologically diagnosed with BBC from 2004 to 2010.
All patients had been treated by mastectomy or breast conservation therapy according to local protocols and had undergone resection of their bilateral breast tumors. Patients with metastatic cancer in the contralateral breast, as defined using Chaudary’s criteria [41], and who developed distant disease between the development of the first and second primary breast carcinomas were excluded from the study. Synchronous and metachronous breast disease was distinguished by the development of a contralateral cancer within six months or more than six months after the initial tumor diagnosis, respectively [7]. Concomitantly, we also collected samples from 2 metastatic breast cancer patients with liver metastasis for whom both the primary tumor and the sequential hepatic metastasis were available in the Tumor Biobank at Tianjin Medical University Cancer Institute and Hospital (Table 1). Each patient donated 20 mL of blood that was collected into heparinized tubes. The Ethics Committee of Tianjin Cancer Institute approved the study protocol, and written informed consent was obtained from all patients, authorizing the genetic analysis of DNA from their biological samples for research purposes.
Data collected included patients’ characteristics, perioperative age, tumor size, tumor grade, tumor histological type, and clinical stage for both the initial and contralateral tumors, as well as the time interval between the first and second tumors. Patients’ estrogen, progesterone, and HER-2/neu receptor status was also noted by IHC or FISH if the immunohistochemical analysis score was +2 equivocal.
All specimens were snap-frozen in liquid nitrogen during surgery and immediately stored at -80°C for further study. Two pathologists independently confirmed the histopathological diagnosis, tumor grade, and tumor cell content of the hematoxylin-and-eosin-stained tumor sections. For all tumor-tumor pairs, representative fresh-frozen blocks with a tumor cell purity of more than 80% were selected, and genomic DNA was extracted from paired tissues and blood for sequencing using a standard protocol (Qiagen).
Illumina-based whole-exome sequencing and reads alignment
Genomic DNA from the matched tumors and peripheral blood was fragmented and hybridized to commercially available capture arrays for enrichment. The exome capture procedure was performed with Agilent’s SureSelect Human All Exon Kit protocol (Agilent Technologies). The resulting DNA libraries, with an average insert size of 200 bp, were sequenced using the 90-bp paired-end technology on Illumina HiSeq 2000. A real-time image analysis and base calling were performed with HiSeq Control software version 1.1.37 and Real-Time Analysis software version 1.7.45 using standard parameters, respectively. Before aligning reads to the Homo sapiens reference genome, we removed low-quality reads that met the following criteria: (1) reads that included sequencing adaptors; (2) reads with a ratio of ambiguous bases to read length ≥0.1; and (3) reads with more than 5 ambiguous bases. The resultant reads were aligned to reference genome hg19 using Burrows Wheeler Aligner software version 0.5.9 (bwa aln -o 1 -e 50 -m 100000 -t 4 -i 15 -q 10 -I) [42]. SAMtools was used to convert the SAM-formatted alignment results to BAM-formatted alignment files; the Genome Analysis Toolkit (GATK IndelRealigner) was used to calibrate alignment accuracy in local regions, and Picard was used to mark duplicates [43, 44].
Detection of somatic SNVs and indels
MuTect was used to detect somatic SNVs from the germline SNVs in blood for both the discovery and validation cohorts. This sensitive tool detects somatic point mutations and addresses tumor impurity and heterogeneity [45]. The minimum coverage was set at 10X for both the tumor and germline genomes; the mutation allele fraction was ≥5%, and ≥5 reads supported this mutation. The somatic mutations were annotated with ANNOVAR [46]. VarScan2 [47] was used to detect somatic indels by comparing the tumor bam file with its matched blood bam file, using the following parameters: min-coverage, 10; min-coverage-blood, 10; min-coverage-tumor, 10; min-var-freq, 0.05; min-freq-for-hom, 0.75; somatic-p-value, 0.05; and min-avg-qual, 0. False-positive indels were removed through a filtering pipeline and manual inspection.
Identification of somatic SNVs for multiple tumors in a patient
To detect sequential somatic SNVs in patients with more than 1 tumor and a shared normal control, the MuTect algorithm [45] was used. An SNV superset was compiled by concatenating all the detected somatic SNVs. We examined this SNV superset in the tumors with a FDR corrected binomial probability of 0.05, assuming a background sequencing error rate of 1%; SNVs with a FDR<10% were called. In this way, a few SNVs that were less sampled during sequencing were recovered.
Copy number variation analysis by aCGH
The chromosome copy number of all tumor samples and their paired blood samples was assayed using an Agilent SurePrint G3 Human CGH microarray kit, 1x1 M, with an average probe spacing of 2.1 Kb (Agilent Technologies). Genomic DNA labeling and chip processing were performed according to Agilent’s recommended protocols. After the hybridization step, microarray slides were washed and scanned using an Agilent Technologies DNA Microarray Scanner with Surescan High-Resolution Technology. Raw expression data, along with tif images, were extracted using Feature Extraction software.
Array CGH data were analyzed using Agilent CytoGenomics Edition version 2.7.22.0 software (Agilent Technologies, CA, USA), using the paired peripheral blood from each patient as germline reference. The QC metrics table was used to check the signal intensities and background noise. A derivative log ratio score above 0.20 was set as the cutoff criterion to exclude the poor quality of array data and the possibility of false copy number variation (CNV) calling [48]. A CNV analysis was performed using the Aberration Detection Method 2 algorithm with a sensitivity threshold of 6.0 and a minimum of three probes. The copy number profiles were visualized using the Integrative Genomics Viewer (IGV).
Clonality analysis
For SNVs, we determined whether two tumors from the same patient shared a significantly higher than expected number of somatic mutations. Specifically, we created a 2-by-2 contingency table that represented the number of somatic mutations that were specific or shared between tumors from the same patients. Fisher’s exact test was used to determine whether they shared a significantly higher number of somatic mutations than would be expected by chance; this was followed by the Benjamini Hochberg method of FDR control.
With respect to CNV, we used the R software package Clonality [49], which uses tumor copy number profiles at the probe level, to determine whether two tumors from the same patient were clonally or origin independent using a likelihood ratio 2 (LR2) statistic [49] (quantifying the odds that the two tumors are clonal). To run Clonality, we used DNAcopy [40] to create a copy number array object. The copy number array object was used as input for Clonality.
Acknowledgments
We thank Ms. Ann M. Sutton of the Department of Scientific Publications at MD Anderson for editing this manuscript. The tissue bank is jointly supported by Tianjin Cancer Institute and Hospital and the National Foundation for Cancer Research (USA).
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81172762 to K.C., no. 81302293, 81202275 and 81302487 to F.S., and no. 81473039 to F.S.), the Chinese 863 Program (no. 2012AA02A207 to K.C. and J.W.), and the National Key Scientific and Technological Project (2015BAI12B15 to K.C. and 2014BAI09B09 to F.S., ). This work was partially supported by the Program for Changjiang Scholars and Innovative Research Team in University in China (IRT_14R40 to K.C.).
ConflictS of interest
None of the authors has any conflict of interest to disclose regarding this manuscript.
References
1. Hartman M, Czene K, Reilly M, Adolfsson J, Bergh J, Adami HO, Dickman PW, Hall P. Incidence and prognosis of synchronous and metachronous bilateral breast cancer. J Clin Oncol. 2007; 25:4210-4216.
2. Chen W, Zheng R, Zhang S, Zhao P, Li G, Wu L, He J. The incidences and mortalities of major cancers in China, 2009. Chin J Cancer. 2013; 32:106-112.
3. Schwentner L, Wolters R, Wischnewsky M, Kreienberg R, Wockel A. Survival of patients with bilateral versus unilateral breast cancer and impact of guideline adherent adjuvant treatment: a multi-centre cohort study of 5292 patients. Breast. 2012; 21:171-177.
4. Imyanitov EN, Suspitsin EN, Grigoriev MY, Togo AV, Kuligina E, Belogubova EV, Pozharisski KM, Turkevich EA, Rodriquez C, Cornelisse CJ, Hanson KP, Theillet C. Concordance of allelic imbalance profiles in synchronous and metachronous bilateral breast carcinomas. Int J Cancer. 2002; 100:557-564.
5. Hartman M, Hall P, Edgren G, Reilly M, Lindstrom L, Lichtenstein P, Kaprio J, Skytthe A, Peto J, Czene K. Breast cancer onset in twins and women with bilateral disease. J Clin Oncol. 2008; 26:4086-4091.
6. Phillips KA, Milne RL, Rookus MA, Daly MB, Antoniou AC, Peock S, Frost D, Easton DF, Ellis S, Friedlander ML, Buys SS, Andrieu N, Nogues C, et al. Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2013; 31:3091-3099.
7. Liang X, Li D, Geng W, Cao X, Xiao C. The prognosis of synchronous and metachronous bilateral breast cancer in Chinese patients. Tumour Biol. 2013; 34:995-1004.
8. Heron DE, Komarnicky LT, Hyslop T, Schwartz GF, Mansfield CM. Bilateral breast carcinoma: risk factors and outcomes for patients with synchronous and metachronous disease. Cancer. 2000; 88:2739-2750.
9. Baker B, Morcos B, Daoud F, Sughayer M, Shabani H, Salameh H, Almasri M. Histo-biological comparative analysis of bilateral breast cancer. Med Oncol. 2013; 30:711.
10. Noguchi S, Motomura K, Inaji H, Imaoka S, Koyama H. Differentiation of primary and secondary breast cancer with clonal analysis. Surgery. 1994; 115:458-462.
11. Shibata A, Tsai YC, Press MF, Henderson BE, Jones PA, Ross RK. Clonal analysis of bilateral breast cancer. Clin Cancer Res. 1996; 2:743-748.
12. Stenmark-Askmalm M, Gentile M, Wingren S, Stahl O. Protein accumulation and gene mutation of p53 in bilateral breast cancer. South-East Sweden Breast Cancer Group. Acta Oncol. 2001; 40:56-62.
13. Scholes AG, Woolgar JA, Boyle MA, Brown JS, Vaughan ED, Hart CA, Jones AS, Field JK. Synchronous oral carcinomas: independent or common clonal origin? Cancer Res. 1998; 58:2003-2006.
14. Saad RS, Denning KL, Finkelstein SD, Liu Y, Pereira TC, Lin X, Silverman JF. Diagnostic and prognostic utility of molecular markers in synchronous bilateral breast carcinoma. Mod Pathol. 2008; 21:1200-1207.
15. Banelli B, Casciano I, Di Vinci A, Gatteschi B, Levaggi A, Carli F, Bighin C, Salvi S, Allemanni G, Ghiorzo P, Pronzato P, Venturini M, Romani M, et al. Pathological and molecular characteristics distinguishing contralateral metastatic from new primary breast cancer. Ann Oncol. 2010; 21:1237-1242.
16. Teixeira MR, Ribeiro FR, Torres L, Pandis N, Andersen JA, Lothe RA, Heim S. Assessment of clonal relationships in ipsilateral and bilateral multiple breast carcinomas by comparative genomic hybridisation and hierarchical clustering analysis. Br J Cancer. 2004; 91:775-782.
17. Agelopoulos K, Tidow N, Korsching E, Voss R, Hinrichs B, Brandt B, Boecker W, Buerger H. Molecular cytogenetic investigations of synchronous bilateral breast cancer. J Clin Pathol. 2003; 56:660-665.
18. Park SC, Hwang UK, Ahn SH, Gong GY, Yoon HS. Genetic changes in bilateral breast cancer by comparative genomic hybridisation. Clin Exp Med. 2007; 7:1-5.
19. Kurian AW, McClure LA, John EM, Horn-Ross PL, Ford JM, Clarke CA. Second primary breast cancer occurrence according to hormone receptor status. J Natl Cancer Inst. 2009; 101:1058-1065.
20. Nichol AM, Yerushalmi R, Tyldesley S, Lesperance M, Bajdik CD, Speers C, Gelmon KA, Olivotto IA. A case-match study comparing unilateral with synchronous bilateral breast cancer outcomes. J Clin Oncol. 2011; 29:4763-4768.
21. Irvine T, Allen DS, Gillett C, Hamed H, Fentiman IS. Prognosis of synchronous bilateral breast cancer. Br J Surg. 2009; 96:376-380.
22. Lichy JH, Dalbegue F, Zavar M, Washington C, Tsai MM, Sheng ZM, Taubenberger JK. Genetic heterogeneity in ductal carcinoma of the breast. Lab Invest. 2000; 80:291-301.
23. Suspitsin EN, Sokolenko AP, Togo AV, Lazareva YR, Turkevich EA, Matsko DE, Henrich KO, Borresen-Dale AL, Schwab M, Cornelisse CJ, Imyanitov EN. Nonrandom distribution of oncogene amplifications in bilateral breast carcinomas: Possible role of host factors and survival bias. Int J Cancer. 2007; 120:297-302.
24. Chunder N, Roy A, Roychoudhury S, Panda CK. Molecular study of clonality in multifocal and bilateral breast tumors. Pathol Res Pract. 2004; 200:735-741.
25. Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003; 300:949.
26. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010; 464:999-1005.
27. Network. CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
28. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405-409.
29. Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015; 6:7597-7607.
30. Wheler JJ, Parker BA, Lee JJ, Atkins JT, Janku F, Tsimberidou AM, Zinner R, Subbiah V, Fu S, Schwab R, Moulder S, Valero V, Schwaederle M, et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: implications for current treatment paradigms. Oncotarget. 2014; 5:2349-2354.
31. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114-1117.
32. Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, Fulton R, Magrini V, Schmidt H, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012; 366:1090-1098.
33. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415-421.
34. Newburger DE, Kashef-Haghighi D, Weng Z, Salari R, Sweeney RT, Brunner AL, Zhu SX, Guo X, Varma S, Troxell ML, West RB, Batzoglou S, Sidow A. Genome evolution during progression to breast cancer. Genome Res. 2013; 23:1097-1108.
35. Salhia B, Kiefer J, Ross JT, Metapally R, Martinez RA, Johnson KN, DiPerna DM, Paquette KM, Jung S, Nasser S, Wallstrom G, Tembe W, Baker A, et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One. 2014; 9:e85448.
36. Ueno T, Emi M, Sato H, Ito N, Muta M, Kuroi K, Toi M. Genome-wide copy number analysis in primary breast cancer. Expert Opin Ther Targets. 2012; 16 Suppl 1:S31-S35.
37. Newman LA, Sahin AA, Cunningham JE, Bondy ML, Mirza NQ, Vlastos GS, Whitman GJ, Brown H, Buchholz TA, Lee MH, Singletary SE. A case-control study of unilateral and bilateral breast carcinoma patients. Cancer. 2001; 91:1845-1853.
38. Tse GM, Kung FY, Chan AB, Law BK, Chang AR, Lo KW. Clonal analysis of bilateral mammary carcinomas by clinical evaluation and partial allelotyping. Am J Clin Pathol. 2003; 120:168-174.
39. Begg CB, Eng KH, Hummer AJ. Statistical tests for clonality. Biometrics. 2007; 63:522-530.
40. Ostrovnaya I, Seshan VE, Olshen AB, Begg CB. Clonality: an R package for testing clonal relatedness of two tumors from the same patient based on their genomic profiles. Bioinformatics. 2011; 27:1698-1699.
41. Chaudary MA, Millis RR, Hoskins EO, Halder M, Bulbrook RD, Cuzick J, Hayward JL. Bilateral primary breast cancer: a prospective study of disease incidence. Br J Surg. 1984; 71:711-714.
42. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754-1760.
43. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078-2079.
44. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297-1303.
45. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213-219.
46. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164.
47. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568-576.
48. Du X, An Y, Yu L, Liu R, Qin Y, Guo X, Sun D, Zhou S, Wu B, Jiang YH, Wang Y. A genomic copy number variant analysis implicates the MBD5 and HNRNPU genes in Chinese children with infantile spasms and expands the clinical spectrum of 2q23.1 deletion. BMC Med Genet. 2014; 15:62.
49. Ostrovnaya I, Olshen AB, Seshan VE, Orlow I, Albertson DG, Begg CB. A metastasis or a second independent cancer? Evaluating the clonal origin of tumors using array copy number data. Stat Med. 2010; 29:1608-1621.