
INTRODUCTION
Prostate cancer (PCa) is the most common noncutaneous cancer and the second-leading cause of cancer-related death in men in the United States [1]. Prior studies have shown that family history, such as a brother or father with PCa or relatives affected at an early age, is a major risk factor [2, 3]. A growing consensus in the field is that inherited factors for PCa are heterogeneous, involving gene mutations of high penetrance that occur in a small number of families, but also low or moderately penetrant alterations that are more common, and which may interact in individuals to promote disease [4–7]. While a few genes such as BRCA2 and HOXB13 are definitively linked to prostate cancer risk in specific patient populations [8–10], a greater proportion of prostate cancer risk may be associated with common alleles of low-to-intermediate penetrance or private alleles in families contributing to cancer risk. While panel testing may be useful in detecting some of these variants, it is difficult to design a panel that adequately captures the rapidly increasingly number of rare variants associated with multiple forms of cancer. With the cost of DNA sequencing rapidly decreasing, analysis of exome and genome data is becoming an alternative approach. However, given the computational complexity of assessing the many rare variants found in every individual [11, 12], particularly if multiple independent variants may be interacting to produce risk [13], it is important to employ a robust analytic pathway grounded in understanding of the physiological basis of the disease.
In addition to genetic heterogeneity of prostate cancer susceptibility, another common scenario in clinical cancer risk evaluation that impacts the assessment of genetic variants is a “case-only” presentation for genetic testing [14]. This may arise when a patient presents for cancer risk evaluation with small family structure, limited family history information, and limited access to specimens from other affected relatives due to death or other causes. In this situation, the ability to clarify cancer susceptibility of genetic variants using family history or by testing a DNA sample from one or more informative blood relatives (affected or unaffected with cancer) is not possible. Such prostate cancer pedigrees will often be characterized by other cancers, raising the possibility that some inherited variants may be risk factors for multiple cancer types. In the clinical cancer risk assessment setting, novel pathway-based approaches to identifying at-risk individuals and families are greatly needed.
In this study, we have tested the hypothesis that germline exome data from individuals with prostate cancer and a family history of one or more cancer types would be enriched in damaging variants falling into two classes. The first class includes variants in genes associated with defects in the DNA damage response (DDR) pathway, reflecting the growing recognition that a number of genes implicated in PCa risk, such as BRIP1, MSH2, MSH3, CHEK2, and PALB2, have general function in contributing to early genomic instability in multiple cancers [15–19]. Of particular relevance to prostate cancer, the androgen receptor has been found to regulate a suite of DDR genes, including some that promote resistance to radiotherapy in prostate cancer in part by promotion of non-homologous end joining (NHEJ) repair [20]. The related second class of variants includes those in genes of sex hormone metabolism previously implicated as relevant to the pathogenesis of non-familial prostate cancer [21], including those that regulate androgen signaling by various mechanisms. Here we present data on specific pathways analysis of exomes of a group of case-only prostate cancer patients who underwent clinical genetic evaluation for inherited cancer risk based on personal and/or family cancer features. We identify likely predisposing variants in every case, with a particular bias towards evidence of DDR-impairing defects in most cases. Based on this work, we perform functional testing that directly demonstrated increased sensitivity to DNA damaging agents in lymphocytes from prostate cancer patients bearing predicted DNA damaging alleles. This work begins to develop an operational framework for exome sequencing in case-only prostate cancer genetic evaluation.
RESULTS
Development of a high value list of candidate genes
To meet the goal of developing an operational framework for assessing case-only patients, we developed a comprehensive, hypothesis-based candidate gene list for particular scrutiny (Supp. Table S1). To this end, we integrated top scoring candidates from a number of existing sources that collated genes based on orthogonal selection criteria (Table 2). The primary hypothesis for this purpose was that rare variants leading to defects in DDR would be important in predisposition for general cancer risk, while the secondary hypothesis was that rare variants damaging genes associated with androgen signaling or prostate function would provide a bias for cancer in the prostate.
Table 2: Sources for building the candidate gene list
Description |
Source/Reference |
Number of genes |
|---|---|---|
DNA repair genes |
Wood lab |
179 |
AR-regulated DNA repair genes |
(Polkinghorn, et al., 2013) |
144 |
AR interactors |
Human protein reference database |
149 |
BRCA1 interactors |
102 |
|
Genes most frequently mutated in prostate tumors |
(Barbieri, et al., 2012) |
19 |
(Grasso, et al., 2012) |
||
TARGET |
(Van Allen, et al., 2014) |
130 |
Genes linked to androgen and estrogen biosynthesis and metabolism |
(Sun, et al., 2011) |
30 |
Literature mining for genes involved in prostate cancer |
MalaCard prostate cancer, (Rappaport, et al., 2014) |
Top 50 |
Diseases-Jensen lab-University of Copenhagen, (Pletscher-Frankild, et al., 2015) |
Top 150 |
|
Candidate genes list for exome study of LNCaP cell line |
(Spans, et al., 2012) |
Top 50 |
Genes linked to glycosylation disorders |
(Freeze, et al., 2014) |
103 |
In addition to sources noted in the Results section, genes were added from online resources (MalaCards and DISEASES), which use literature mining to identify genes linked to inherited and somatic prostate cancer, a study by Spans et al, describing mutations distinguishing the widely studied LNCaP prostate cancer cell line from normal prostate cells (Spans, et al., 2012), and a group of genes linked to glycosylation disorders (Freeze, et al., 2014), given the growing evidence for defects in proteins involved in glycosylation in the pathogenesis of prostate cancer (Drake, et al., 2015).
To identify a relevant candidate set we queried the Wood group website, which maintains an updated comprehensive list of genes linked to DNA damage response [35]. This list was extended with genes described by Polkinghorn and colleagues, who have noted a subset of DNA repair genes specifically regulated by the androgen receptor (AR) [20] (see Figure 2A). The Human Protein Reference Database (HPRD) [36] provided lists of genes that interacted physically or functionally with the well-validated risk factor BRCA1, as well as the AR. Providing broader context relevant to AR signaling, several recent studies have provided lists of genes mutated at appreciable frequencies in sporadic prostate cancer [37, 38], including castration resistant prostate cancer [39]. The TARGET database (Tumor Alterations Relevant for Genomics-Driven Therapy) [38] provides a broader list of genes of clinical value for cancer treatment, based on their roles as tumor drivers. Sun and colleagues developed a list of single nucleotide polymorphisms (SNPs) affecting sex hormone metabolism, some of which showed significant or near-significant linkage to prostate cancer aggressiveness at diagnosis [21].
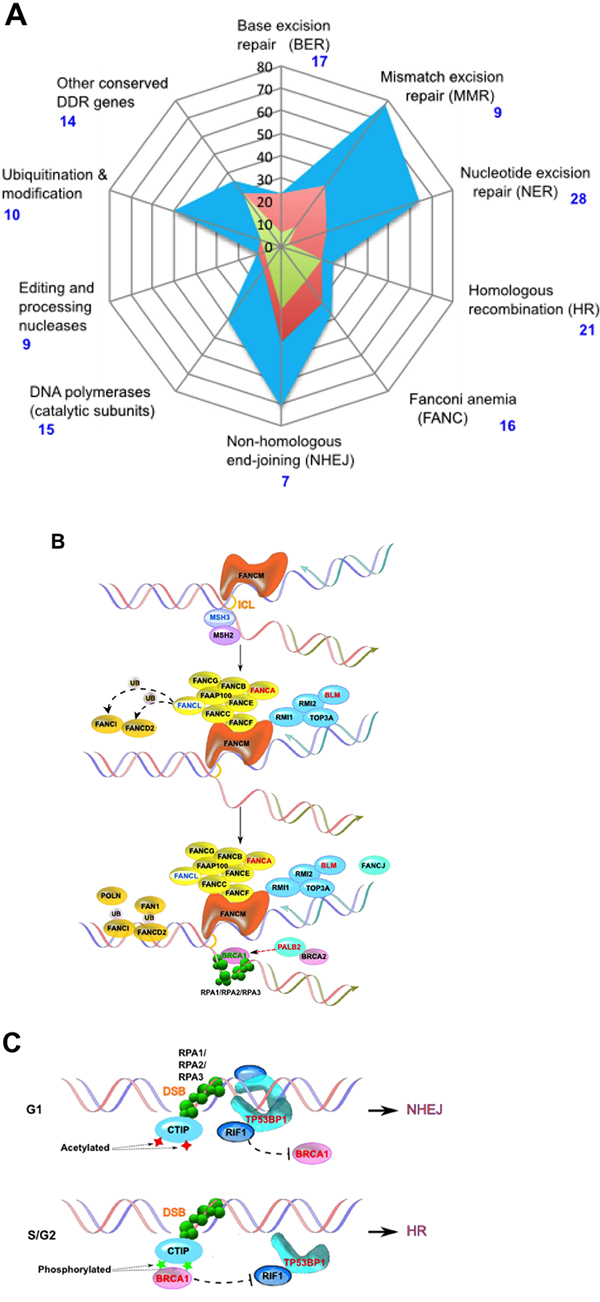
Figure 2: DNA damage response genes in prostate cancer patients. A. Radar plot indicating percent of genes that are AR-associated (blue), induced by androgens (red) or direct AR targets (green) in each class of DNA repair genes, based on [20]. Classes of DDR genes are based on the list posted at the Wood lab Web site (see Table 2), except that the two classes “Base excision repair (BER)” and “Other BER and strand break joining factors” were merged. Vertical black numbering indicates percent of AR-associated genes; blue numbering indicates number of genes in each class. B. Simplified representation of DNA interstrand crosslink damage being repaired by proteins in Fanconi's anemia pathway [101]. Variants found in patient 124604 are indicated in red font; those found in patient 117939, in blue font.C. Alternative binding by TP53BP1 or BRCA1 (shown in red font) specifies NHEJ versus HR DNA repair, with variants in each gene found in patient 129413.
Finally, as a tertiary hypothesis, we also considered that non-rare variants, or variants in genes linked to cancers other than prostate, might contribute to prostate cancer risk in some circumstances. Typically, a threshold of 1% for a minor frequency allele (MAF) is used to filter out non-rare variants as insignificant in the absence of clear clinical indication of phenotypic effect [14]. However, a growing body of evidence suggests that cancer risk reflects the interaction of multiple predisposing factors [13, 40, 41], suggesting such non-rare variants may interact with specific rare variants. We also considered in our analysis non-rare variants in genes predicted as relevant to somatic or hereditary cancers by the Ambry report.
Identification of genetic variants in the prostate cancer patient cohort
Focusing on this candidate list, we analyzed rare single nucleotide variants (SNVs) causing missense mutations, frameshift or nonsense mutations predicted to result in early truncation of protein and short in-frame insertions or deletions (indels) (Supp Tables S2, S3). SNVs were only considered further if they passed an initial test in which multiple predictor algorithms indicated the variant would disrupt protein function (see Methods and Supp Table S2 for comprehensive analysis).
Table 3 summarizes the significant variants found in each of the 12 patients (with extended information in Supp Table S4). All variants listed in Table 3 passed Sanger validation. Each patient had variants affecting 3 to 7 genes on the candidate list. All 12 patients had reported being white and non-Hispanic: Table 3 describes the frequency of each variant in the subset of ~30,000 European non-Finnish individuals of the Exome Aggregation Consortium (ExAC) database; these frequencies were in agreement with those reported in an independent (ITMI) cohort of 634 white non-Hispanic individuals who denied a personal or family history of cancer (Supp Table S4). Thirty alterations identified in the prostate cancer patients in this study were found in fewer than 20 individuals in the ExAC database (< 0.0003%), with 10 alterations never previously reported. In addition, a number of non-rare variants in genes relevant to cancer risk (e.g. in BRCA1, PALB2, BLM, and others) were detected in a significant number of individuals.
Table 3: Selected variants with scores of amino acid damage from 5 predictors and variant frequency in ExAC, by patient
Patient ID |
Variant DNA level |
Gene |
Consequence |
Non-neutral scores |
Representation in ExAC (European non-Finnish) |
|
|---|---|---|---|---|---|---|
Allele count |
Allele number |
|||||
112940 |
9:32989766 G/A |
APTX |
NP_001182178.1 p.R56X |
5† |
0 |
66736 |
17:41246481 T/C |
BRCA1 |
NP_009225.1 p.Q356R |
4 |
4198 |
66734 |
|
4:178274801 T/G |
NEIL3 |
NP_060718.2 p.F460C |
3 |
10 |
66730 |
|
117197 |
22:43933284 CCT/C |
EFCAB6 |
NP_073622.2 p.Q1340Rfs*43 |
5† |
606 |
66684 |
2:38301879 T/A |
CYP1B1 |
NP_000095.2 p.D218V |
5 |
15 |
41314 |
|
10:89503283 C/T |
PAPSS2 |
NP_004661.2 p.P454L |
5 |
0 |
66732 |
|
17:41246481 T/C |
BRCA1 |
NP_009225.1 p.Q356R |
4 |
4198 |
66734 |
|
9:135779052 G/A |
TSC1 |
NP_000359.1 p.H732Y |
4 |
350 |
66706 |
|
117939 |
1:156212872 T/A |
BGLAP |
NP_954642.1 p.C74X |
5† |
8 |
66696 |
2:58386928 G/GTAAT |
FANCL |
NP_060532.2 p.T367Nfs*13 |
5† |
232 |
65648 |
|
5:80109533 T/C |
MSH3 |
NP_002430.2 p.I929T |
5 |
0 |
66740 |
|
12:124209215 G > T |
ATP6V0A2 |
NP_036595.2 p.K103N |
5 |
15 |
66734 |
|
3:51673972 A/T |
RAD54L2 |
NP_055921.2 p.I730F |
4 |
− |
− |
|
21:16340242 T/C |
NRIP1 |
NP_003480.2 p.E91G |
4 |
− |
− |
|
2:149226489 C/T |
MBD5 |
NP_060798.2 p.A326V |
3 |
− |
− |
|
123136 |
4:1206089 G/A |
CTBP1 |
NP_001319.1 p.421L |
4 |
28 |
14670 |
3:38888684 A/T |
SCN11A |
NP_054858.2 p.F1626Y |
3 |
− |
− |
|
1:63876815 A/G |
ALG6 |
Splice acceptor (-2) |
− |
− |
− |
|
1:120056817 T/TGCA |
HSD3B1 |
NP_000853.1 p.V224_Y225insH |
− |
2 |
66708 |
|
4:153332604 TCTC/T |
FBXW7 |
NP_361014.1 p.E117del |
− |
35 |
66114 |
|
124604 |
16:23634293 C/T |
PALB2 |
NP_078951.2 p.G998E |
5 |
1430 |
66736 |
16:89815152 G/A |
FANCA |
NP_000126.2 p.S1088F |
4 |
4798 |
65430 |
|
15:91326099 C/T |
BLM |
NP_000048.1 p.P868L |
4 |
4239 |
66162 |
|
6:49700908 G/A |
CRISP3 |
NP_006052.1 p.A197V** |
2 |
0 |
66362 |
|
124853 |
18:3452067 G/A |
TGIF1 |
NP_733796.2 p.W30X |
5† |
33 |
66002 |
4:55955969 C/T |
KDR |
NP_002244.1 p.A1065T** |
5 |
52 |
66726 |
|
17:12901781 A/C |
ELAC2 |
NP_060597.4 p.S490A |
5 |
39 |
66734 |
|
19:50766628 C/T |
MYH14 |
NP_001139281.1 p.A882V |
3 |
23 |
27644 |
|
X:110973633 TGAA/T |
ALG13 |
NP_001093392.1 p.E795del |
− |
33 |
41558 |
|
4:103747794 C/T |
UBE2D3 |
Splice acceptor (-1) |
− |
− |
− |
|
125671 |
9:35707745 G/C |
TLN1 |
NP_006280.3 p.L1539V |
4 |
13 |
66734 |
1:145578236 C/T |
PIAS3 |
NP_006090.2 p.R67W |
3 |
13 |
66740 |
|
10:5014483 T/A |
AKR1C1 |
NP_001344.2 p.S221N |
3 |
119 |
66712 |
|
10:5014484 C/A |
AKR1C1 |
119 |
66712 |
|||
11:47237894 CAGA/C |
DDB2 |
NP_000098.1 p.R47del |
− |
− |
− |
|
126002 |
17:35564593 G/A |
ACACA |
NP_942134.1 p.R1182W |
5 |
16 |
66612 |
17:41246481 T/C |
BRCA1 |
NP_009225.1 p.Q356R |
4 |
4198 |
66734 |
|
7:18633593 A/G |
HDAC9 |
NP_001191074.1 p.Y199C |
3 |
0 |
66702 |
|
129413 |
14:50088465 T/G |
MGAT2 |
NP_002399.1 p.I160S |
5 |
610 |
66402 |
17:41246481 T/C |
BRCA1 |
NP_009225.1 p.Q356R |
4 |
4198 |
66734 |
|
15:43762077 TGGGATA/T |
TP53BP1 |
NP_001135451.1 p.I455_P456del |
− |
− |
− |
|
129547 |
2:38298287 T/TGGTGGCATCA |
CYP1B1 |
NP_000095.2 p.T404Sfs*30 |
5† |
*** |
|
10:94297192 C/T |
IDE |
NP_004960.2 p.G72S |
5 |
8 |
66724 |
|
12:124824917 C/T |
NCOR2 |
NP_001070729.2 p.R1794Q |
3 |
2 |
65378 |
|
21:16337279 C/A |
NRIP1 |
NP_003480.2 p.V1079F |
3 |
44 |
66670 |
|
129748 |
16:23632788 TTTTC/T |
PALB2 |
NP_078951.2 p.E1002Tfs*4 |
5† |
− |
− |
6:52657698 C/T |
GSTA1 |
NP_665683.1 p.E168K |
4 |
1 |
66738 |
|
8:48973252 G/A |
UBE2V2 |
NP_003341.1 p.R101Q |
4 |
1 |
65850 |
|
20:31021718 C/T |
ASXL1 |
NP_056153.2 p.R573W** |
4 |
4 |
63434 |
|
12:53776449 G/C |
SP1 |
NP_612482.2 p.G240R |
3 |
26 |
66738 |
|
11:62388048 G/C |
B3GAT3 |
NP_036332.2 p.R60G |
3 |
1 |
60290 |
|
131534 |
9:131709581 A/AT |
DOLK |
NP_055723.1 p.M1? |
5† |
823 |
63350 |
13:28592620 T/C |
FLT3 |
NP_004110.2.Y842C |
5 |
2 |
66710 |
|
10:94274700 A/G |
IDE |
NP_004960.2 p.M254T |
5 |
11 |
66698 |
|
4:70723282 C/G |
SULT1E1 |
NP_005411.1 p.W27C |
4 |
− |
− |
|
1:120478125 A/C |
NOTCH2 |
NP_001186930.1 p.F1209V |
4 |
306 |
66726 |
|
17:33430313 T/C |
RAD51D |
NP_001136043.1 p.E223G |
4 |
874 |
51128 |
|
1:182555767 C/T |
RNASEL |
NP_066956.1 p.G59S |
4 |
379 |
66514 |
|
Nicolas et al., Table 3
5†: score given to variant creating stop gain or frameshift;
**variant causing missense and located in splice site;
***this variant has been described in patients with congenital glaucoma, an autosomal recessive trait usually recognized during the first year of life (Sena, et al., 2004). The mutation is not listed in ExAC as it excludes mutations associated with severe pediatric diseases. In the representation in the ExAC column, (–) denotes that the variant was not found in the database while (0) denotes that the variant was absent in European non-Finnish but detected in other ethnicities, as listed in Supp Table 4. This Table also lists damaging variants in genes shown to be clearly tumor-promoting in some inherited or somatic forms of other cancers, although not currently well validated for prostate, including FLT3, ASXL1, KDR, NOTCH2 (e.g. (Kindler, et al., 2005; Sallmyr, et al., 2008; Antonescu, et al., 2009)); as well as genes which are identified by the candidate criteria noted in Table 2, but for which limited information is available based on functional characterization to date (e.g., the AR-interacting protein IDE interacts directly with AR (Kupfer, et al., 1994)).
High frequency of variants affecting genes involved in DNA repair and androgen signaling
19 variants affecting the function of genes involved in DDR (with some known to be androgen regulated), and AR-dependent transcription were identified in the patient cohort, with 11/12 patients having at least 1 affected gene in this category, and 5/12 having 2 or more. These genes included PALB2 (also known as FANCN), APTX, BLM, BRCA1, CTBP1, DDB2, FANCA, FANCL, MBD5, MSH3, NEIL3, RAD51D, RAD54L2 (also known as ARIP4), SP1, TP53BP1, UBE2D3, UBE2V2 (also known as MMS2). Many of the proteins encoded by these genes interact to mediate DNA repair functions (Figure 2B, 2C)
For some patients, a single variant seemed likely to pose substantial risk, such as the frameshift variant E1002Tfs*4 in PALB2 found patient 129748. This patient was diagnosed at age 41, with a father who was also diagnosed with prostate cancer at age 67. PALB2 truncating mutations have been detected in patients with Fanconi's anemia and various cancers [18], including prostate [15]. Importantly, PALB2 p.E1002Tfs*4 lacks part of the WD40 repeat domain (amino acids 853–1186) known to mediate protein interactions with key proteins involved in homologous recombination (HR) such as BRCA2 and RAD51 [42, 43]. Potentially magnifying the effect of the PALB2 truncation, this patient also has a mutation (p.R101Q) predicted to damage in the ubiquitin conjugating enzyme UBE2V2, identified as a factor required for avoidance of UV damage [44], with expression of UBE2V2 linked to prognosis in breast cancers following treatment with DNA damaging therapies [45].
Some patients had suggestive combinations of rare variants. For example, patient 117939 has three independent mutations likely to directly impact DNA damage response: FANCL p.T367Nfs*13, MSH3 p.I929T, and RAD54L2 p.I730F (Figure 2B). FANCL is an E3 ubiquitin ligase of the Fanconi Anemia (FA) core complex. The mutation T367Nfs*13 produces a protein with premature truncation and three novel amino acids at the C-terminus, and has been described in an FA patient [46]. This mutation produces a hypomorphic mutation with only partial correction of mitomycin C sensitivity and chromosomal defects [46]. MSH3 heterodimerizes with MSH2 to mediate mismatch repair; variants in MSH3 have been associated with risk of some forms of cancer, including prostate [47, 48]. Importantly, crosstalk between FA and MSH2/MSH3 in the mismatch repair pathway has been well documented, with the two operating as redundant DNA damage sensors [49–51]. RAD54L2/ARIP4 is a DNA helicase that binds the AR, and modulates AR-dependent transactivation in a promoter-dependent manner, and has been linked to a role in DNA repair [48, 52, 53]. I730 is just downstream of one of the three LXXLL motifs, also known as nuclear boxes, that mediate protein-protein interactions.
In some cases, combinations involving non-rare variants of DDR genes were suggestive. Patient 124604, diagnosed with prostate cancer at age 43, has a pedigree that shows cases of prostate cancer over 3 generations. This patient carries 3 non-rare missense variants in genes involved in DDR: BLM p.P868L, PALB2 p.G998E, and FANCA p.S1088F. BLM encodes a 3′-5′ DNA helicase which functions in maintenance of genomic stability, with inactivating mutations associated with a progeria, Bloom Syndrome (BS). BLM p.P868L has been characterized as an allele that is unlikely to cause BS, but causes partial loss of function manifested by an intermediate sensitivity to hydroxyurea [54], and has been associated with increased rectal cancer risk [55]. Interestingly, an uncle of patient 124604 had colorectal cancer. The PALB2 p.G998E variant in this patient was reported at a similar frequency of ~ 10% in a population of BRCA1- and BRCA2-negative male breast cancer patients in Northern Italy as was observed in healthy individuals [56]. A similar high rate of occurrence in normal populations was observed for FANCA p.S1088F (9/97 in breast cancer cases vs 11/94 in controls) [57]. However, the assortment of three independent alleles affecting DNA repair in the pedigree of this patient may have an additive effect, given the involvement of all three proteins in related DNA repair pathways (Figure 2B).
In a similar case, Patient 112940 had a rare variant causing a stop at amino acid 56, eliminating function of APTX (aprataxin), involved in the repair of multiple forms of DNA breaks and implicated in therapeutic response in cancer [58–60], and a second rare variant (p.F460C) damaging NEIL3, a DNA glycosylase involved in the base excision repair pathways that protects cells from genotoxic stress and has been associated with prostate cancer risk [61, 62]. Interestingly, this patient as well as 3 other unrelated individuals (Patients 117197, 126002, and 129413) all had the same non-rare variant in BRCA1, p.Q356R, which some prior studies have linked to prostate cancer risk [63]; an observed incidence of 33%, versus the expected incidence of this variant should be 9%, based on ExAC. Like patient 112940, the other patients also possessed multiple additional candidate rare variants affecting DNA damage response and/or genes related to androgen function. Patient 129413 had a mutation disrupting TP53BP1 (p.I455_P456del, predicted to be deleterious by PROVEAN), TP53BP1 competes with BRCA1 for directing proteins down the non-homologous end joining (NHEJ) versus homologous recombination (HR) repair pathway (Figure 2C) [64–67]; in the context of impaired TP53BP1, or other DDR defects, and as discussed further below, the BRCA1 variant may have a more deleterious effect.
In some cases, patients have mutational profiles in which disruption of DDR and AR signaling is closely linked. For example, patient 125671 has an in-frame deletion (p.R47del) in the AR-interacting protein DDB2: the R47 residue mediates high affinity binding of DDB2 to damaged DNA [68, 69]. This patient also has an S221N variant predicted to be highly deleterious (Figure 3A) in AKR1C1, a member of an enzyme family that controls concentrations of active androgens and other steroidal hormones, regulates trans-activation of AR in the prostate, and has been shown to regulate resistance to the anti-androgen enzalutamide, recently approved for treatment of castration resistant prostate cancer [70, 71].
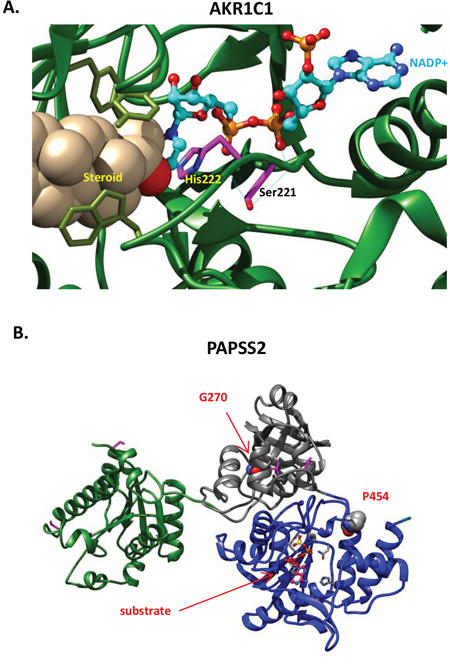
Figure 3: Functional defects associated with novel missense variants. A. AKR1C1, S221N. AKR1C1 catalyzes the inactivation of progesterone to the less potent 20α-hydroxyl-pregn-4-ene-3-one. The reaction is NADPH dependent with an obligatory requirement for the cofactor to bind before the steroid substrate can bind to form the central complex. The progesterone is maintained in a steroid binding site at H222; an H222I mutation decreases the Km value for NADPH 95-fold [102]. Here, AKR1C1 (PDB code: 1MRQ) is shown with bound steroid 20alpha-hydroxy-progesterone, and the cofactor, NADP+ in ball-and-stick representation with cyan carbons, and orange phosphorus atoms. S221 and adjacent catalytic residue H222 of AKR1C1 are shown with magenta sticks. S221 is involved in 2 hydrogen bonds (shown with cyan thin lines) with adjacent residues and one with the NADP+ cofactor. Though predicted to be benign by several conservation based servers, the S221N substitution disrupts the hydrogen-bonding network required to maintain the catalytic active site configuration. B. Shown are the PAPSS2 kinase domain in green, PUA (PseudoUridine synthase and Archaeosine transglycosylase) domain in gray and sulfate adenylyltransferase domain in blue. The position of the P454L and G270D missense variants are indicated.
Patient 123136 comes from a family with a high burden of cancer, with a sister affected with melanoma and breast cancer, father with lung cancer, and mother with colon cancer. This patient has a rare variant (p.P421L) of CtBP1, a coregulator of BRCA1, which has been linked to risk of prostate cancer [72, 73]. Activation of the AR regulator HIPK2 (Homeodomain-Interacting Protein Kinase 2) [74] by genotoxic stress triggers apoptosis in part through phosphorylation of CtBP1, which causes CtBP1 degradation [75]; loss of this signaling could plausibly cause predisposition to multiple forms of cancer. P421L destroys the HIPK2 phosphorylation site on CtBP1.
Other classes of variants
In addition to the selected examples described above, rare variants were identified in all of the categories of interest from the candidate list. Due to space limitations, all profiles cannot be fully detailed. However, some observed allelic combinations are of interest, particularly in regard to androgen availability and AR-dependent transcription. Consideration of these variant combinations may be particularly important in suggesting a possible explanation for why mutations in the DDR machinery, which theoretically could increase risk of any type of cancer, could result in a familial predilection for prostate cancer.
As selected examples, two patients, 129547 and 117939, had disruptive mutations in NRIP1 (also known as RIP140), a co-receptor for estrogen, androgen, and other classes of nuclear hormone receptor. Disruption of NRIP1 function has been reported to lead to hyperactivation of AR signaling [76], and variants in NRIP1 have been linked to risk in breast, endometrial, and other cancers [77, 78]. Interestingly, patient 129547 also has a rare variant affecting a second AR co-repressor, NCOR2 (also known as SMRT) that limits AR signaling [79], while patient 117939 has a rare variant in an alternative AR cofactor, RAD54L2 (also known as androgen receptor interacting protein 4, ARIP4) [80]. These variant pairs may interact to de-repress AR signaling in these two patients. Another patient, 117197, has a frameshift variant (Q1340Rfs*43) in EFCAB6 (also known as DJBP), which encodes a protein that recruits histone-deacetylase (HDAC) complexes to repress AR-dependent transcription [81]; the variant eliminates the HDAC-interaction domain. Patient 124604, noted above as having 3 variants in DDR-related proteins (BLM, PALB2, and FANCA), has a family with prostate cancer over 3 generations. This patient also has a splice site-disrupting variant in CRISP3 (cysteine-rich secretary protein 3). Expression of CRISP3 is prostate-specific, and CRISP3 is up-regulated in a subset of prostate cancers [82], especially prostate cancer with the TMPRSS2-ERG fusion gene [83]. Mis-splicing due to the G/A mutation in 6:49700908 would destroy the CRISP domain (pfam: 08562), which allows CRISP3 to regulate ryanodine receptor Ca2+ signaling. Interestingly, ExAC data indicates the position is multi-allelic with another allele (T) mainly represented in African populations.
A major function of the prostate is in production of glycoproteins, including PSA, that support sperm production, and changes in glycosylation are associated with prostatic neoplasms [84] and response to androgen treatment [85]. Among a number of variants in genes linked to glycosylation defects (PAPSS2, ATP6V0A2, ALG13, MGAT2, B3GAT3, DOLK), the variant in PAPSS2 seems the most interesting. This variant (p.P454L; Figure 3B) is strongly predicted to destroy the catalytic function of PAPSS2, a kinase and ATP sulfurylase that catalyzes two sequential reactions to synthesize PAPS, the sulfate source for sulfation of the androgen precursor didehydroepiandrosterone (DHEA). Of particular relevance to prostate cancer, two mutations in PAPSS2 have been reported as causing androgen excess via complete (W362Cfs*3) or partial (G270D) disruption of DHEA sulfation [86].
Functional defects in response to DNA damaging agents in the lymphocytes of patients of prostate cancer patients with familial risk
Given the strong implication of defects in genes involved in DNA repair as causative factors for numerous classes of hereditary cancer, these genes were of particular interest. PBLs were available for 9/12 patients, allowing us to assess whether the response of these cells to low doses of DNA damaging agents differed from those of 10 age and gender-matched individuals without a cancer diagnosis or a family history of cancer. Immunofluorescence was used to assay the formation of DSB-associated γ (phospho)-H2AX foci in cells with and without treatment with the DNA polymerase inhibitor aphidicolin or the topoisomerase II inhibitor etoposide. Under baseline conditions without drug treatment, the patient and control groups were statistically indistinguishable (p = 0.746) (Figure 4A). However, treatment of cells with low doses of aphidicolin (p = 0.0337) or etoposide (p = 0.007) revealed significant differences in γH2AX induction between cases and controls, with a higher magnitude of induction seen in prostate cancer patients (Figure 4B). Receiver operating characteristic (ROC) curve analysis for the combination of both treatments indicated specificity and selectivity of observed differences were 87% (Figure 4C).
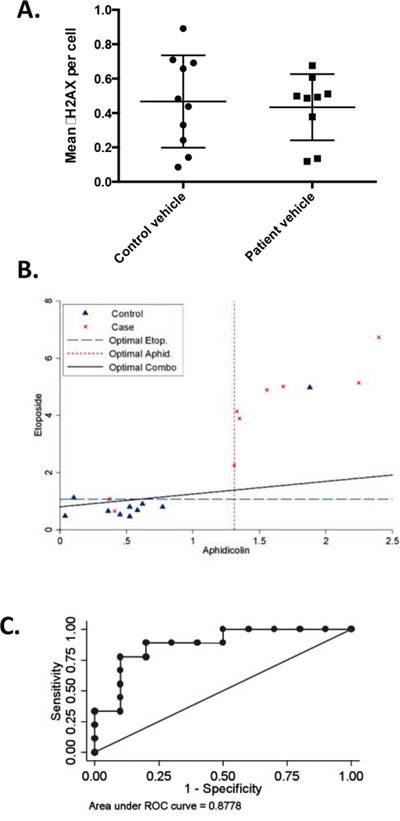
Figure 4: Elevated γH2AX in T-cells from patients with prostate cancer following treatment with DNA damaging agents. Primary T-cells from 9 patients and 10 age- and sex-matched controls were stimulated by PHA and IL-2, then treated with vehicle, aphidicolin or etoposide, and stained for nuclear γH2AX foci. A. Mean number of γH2AX foci in vehicle treated patients and matched controls (p = 0.746, not significant). B. Mean number γH2AX foci per cell are depicted for cases and controls following drug treatment. Cases: red ‘x's, controls: blue triangles. Dashed lines: statistically optimal cutoff points to discriminate between samples with high and low γH2AX levels for each treatment or the two tests combined, as indicated. Using the combination (solid black line), 7/9 patients exhibited high γH2AX levels versus 1/10 controls. C. Area under the receiver operating characteristic (ROC) curve (AUC) for the combined γH2AX scores for aphidicolin and etoposide demonstrates assays discriminate between patients and controls, AUC = 0.8778.
DISCUSSION
With the growing availability of powerful technologies for genomic analysis, cancer risk assessment is at a key point of potentially benefitting from advances in sequencing. Some large families with detailed family cancer history are in the unambiguous situation of having a well-defined mutation in a highly penetrant gene. However, a significant proportion of families have limitations in family cancer history information, making exploratory studies such as this one useful in defining strategies for potential clinical evaluation. In this study, we have focused most effort on the analysis of defects in DDR in case-only individuals with prostate cancer and strong family cancer history. This approach has identified variants or variant combinations in almost every patient assessed that have the potential to alter response to DNA damage, with findings further supported by direct demonstration of elevated expression of γH2AX following administration of DNA damaging stimuli. In the group of patients we examined, variants often occurred in combinations that would be predicted to interact to weaken DDR, and typically each variant was unique to the individuals investigated. These findings suggest that DDR defects may play a role in prostate cancer predisposition, similar to other hereditary cancers such as breast [87] and colorectal [88]. Two recent studies identified a high burden of germline mutations affecting DDR genes in somatic prostate cancers [89–91].
This study also suggests that non-rare variants affecting DDR may play a role in prostate cancer risk upon further study. For example, in the cohort analyzed here, 33% of patients had the same non-rare p.Q356R variant in BRCA1. BRCA1 p.Q356R is an example of a variant that has a frequency of minor allele over 5% and an increased odds ratio for breast cancer over 1.5 in carriers. In a study on association between prostate cancer risk and SNPs in a 200 kb area around the BRCA1 gene, the strongest link was for BRCA1 p.Q356R [63], with the R allele preferentially transmitted to men affected with prostate cancer before the age of 50. Mechanistically, Q356 is of particular interest as it is located in the MRE11/RAD50/NBS1 (MRN) domain, required for interaction with RAD51, p53, ZBRK1, SWI/SNF, BRAP2, ATPase, and Importin α [92]. Q356R disrupts the interaction between BRCA1 and ZBRK1, eliminating the transcriptional co-repressor function of BRCA1 [93], leading to upregulation of such BRCA1 target genes as angiopoietin-1 (ANG1), which promotes angiogenesis and proliferation [94]. In a study of 931 PCa patients, 13 independent variants of uncertain significance were identified in the MRE11/RAD50/NBS1 domain including Q356R [15]. Our data raises the idea that BRCA1 Q356R may be significant in the context of other variants that weaken DDR.
DDR genes are active in many tissues; hence, the fact that the patients analyzed developed prostate cancer as well as multiple other cancers in their families may reflect the contribution of multiple modifier variants. Our candidate approach also considered variants predicted to influence signaling by androgens and other hormones, or prostate-specific functions such as control of glycosylation and a number of damaging rare variants were also found in these genes in the sequenced probands. Our data suggests these gene pathways are worth following as genomic characterization of prostate and other hereditary tumors advances.
An important issue is how to determine if detected germline variants are indeed functional to inform cancer risk assessment. Recent studies have reported that evaluations of the germline DNA of “normal” individuals identifies a surprisingly high burden of variants that would seem to be predisposing to cancer and other diseases [11, 95]. Furthermore, variants may be detected in genes that are not expressed in the prostate or affected tissue [96–98]. These points highlight the need for functional studies to follow-up on detection of germline variants. In this study, the functional testing of patient-derived PBLs has supported the idea that responses to DNA damage in prostate cancer patients differ from those found in age and gender-matched controls. This approach can potentially be extended using systematic cell-based functional assays for phenotyping of missense alleles [99].
Ultimately, these results suggest that future strategies for risk assessment may involve a diagnostic algorithm in which high-risk patients initially are tested for well-validated, high penetrance variants, using standard panel-based screens. If initial testing fails to identify causative mutations, exome-testing focusing on cancer-relevant processes, supported by functional testing for defects in the process, may be a useful alternative approach that should be evaluated in a prospective setting. Another potentially valuable aspect of such broader testing may be implications for patient treatment. Identifying a variant that renders the cell dependent on a specific pathway may create an opportunity for synthetic lethality, as in the example of BRCA mutations and PARP inhibitors [100]. Broader approaches for functional assessment of germline variants are also needed to better inform cancer risk assessment and recommendations.
MATERIALS AND METHODS
A flowchart for the analysis in the study is provided in Figure 1.

Figure 1: Project Flow Chart. After review of family history and negative results for genetic testing for known predisposing factors, 12 DNA samples isolated from peripheral blood lymphocytes (PBLs) of 12 patients with prostate cancer were sent to Ambry Genetics for exome sequencing. Data analysis included identification of the rare variants in 826 genes selected as described in Table 2 and validation by Sanger sequencing of the variants that scored non neutral by at least 3 in silico predictors. Two variants (in AKR1C1 and PAPSS2) were further characterized by molecular modeling. PBLs were also used to assess the response to DNA damaging agents.
Patient selection
Case-only prostate cancer patients included in this study (n = 12) had undergone evaluation for inherited cancer risk. Each participant had a strong family cancer history as shown in Table 1, with either multiple first-degree or second-degree relatives with prostate or other cancers. The mean age at prostate cancer diagnosis was 57.8 years (range 41–68 years). Fifty-eight percent had Gleason score > = 7, and 25% had advanced stage disease (T3). Hereditary cancer syndromes evaluated in these families included hereditary breast-ovarian cancer (HBOC), Lynch syndrome (LS), and hereditary prostate cancer (HPC) [22–24]. No pathogenic mutations were identified from sequencing the following genes: BRCA1, BRCA2, MLH1, MSH2, PMS2, MSH6, and HOXB13. These patients had consented to the FCCC Risk Assessment Program Registry, which allowed further research genomic sequencing. All patients reported being white, non-Hispanic. Peripheral blood DNA from these 12 prostate cancer patients was sent to Ambry Genetics for exome sequencing.
Table 1: Family history and prostate cancer characteristics of analyzed prostate cancer patients
Patient ID |
Age at diagnosis |
Stage/Gleason |
1st degree relatives with cancer Type of cancer/age |
2nd degree relatives with cancer Type of cancer/age |
|---|---|---|---|---|
112940 |
68 |
T2aN0MX/3+4=7 |
sister-ovary 27 and colon 66 |
|
117197 |
68 |
T3bN0MX/4+3=7 |
mother-stomach 80, colon 95 |
nephew-non Hodgkin's 20′s |
117939 |
65 |
T2cNOMX/3+3=6 |
father-prostate 65 |
paternal uncle-throat 60 |
123136 |
59 |
T2cNXMX/6 |
mother-colon mid 40′s |
|
124604 |
44 |
T2cN0MX/3+3=6 |
father-melanoma 72, polyps |
paternal uncle-colon 50 |
124853 |
65 |
T2cNOMX/3+3=6 |
sister-basal cell 60 |
paternal aunt-breast |
125671 |
54 |
T2cN0MX/3+3=6 |
sister-uterine 54 |
paternal cousin-breast 36 |
126002 |
59 |
T3aN0MX/3+4=7 and 4+3=7 and 6 (multiple areas) |
maternal aunt-unknown cancer 89 |
|
129413 |
57 |
T1c/3+4=7 |
father-prostate 70, kidney 80, liver 80 |
paternal grandfather-prostate 85 |
129547 |
62 |
T2cN0MX/3+4=7 |
sister-ovary 57, cervix 57 |
paternal cousin-inflammatory breast 45 |
129748 |
41 |
T3bN1M0/4+4=8 |
father-prostate 67 |
paternal grandfather-polyps |
131534 |
52 |
T2cNO/3+4=7 |
mother-breast 68, melanoma 65 |
maternal aunt-colon 45 |
Exome sequencing
Exome sequencing of germline DNA was performed by Ambry Genetics (Aliso Viejo, CA) at 30X average coverage using a VCRome kit (Roche Nimblegen, Madison WI) for library preparation, indexing and 100 bp paired end processing using the HiSeq platform (Illumina, Hayward, CA). Human hg19 reference-guided alignment and variant calling were done using the Illumina CASAVA software pipeline. Heuristic filtering processes were applied to remove variants that fall into non-coding regions, with synonymous effect, or common variants found in the 1000 genomes, dbSNP or Exome Sequencing Project (ESP) database. The Ambry Variant Analyzer (AVA™) produced candidate mutation short lists of rare variants by restricting to variants fitting a dominant/recessive model of inheritance, as well as listing variants associated with hereditary and somatic cancers, regardless of frequency (i.e., including non-rare variants).
Development of a high value list of candidate genes
The candidate gene list (Supp Table S1) was assembled from the sources listed in Table 2. Genes collected from various sources were prescreened for possible aliases by using G-convert from G-profiler (http://biit.cs.ut.ee/gprofiler/gconvert.cgi) in batch mode to ensure use of official gene symbols with Entrez ID numbers. Manual curation using (http://www.genenames.org/) from the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) was done when G-convert did not return information. PCAP, PCA3 and HPC6, which were listed as loci relevant to prostate cancer by at least one source, were not included as they fall under the HGNC locus type “phenotype only”, indicating the causative gene has not been identified. Non-coding transcripts such as PCAT4, 5 and 6 and pseudogenes were excluded.
Variant selection
By analysis through its AVA™ filtering software, Ambry produced lists of rare variants (defined as frequency less than 1% in the general population) for each patient. From these combined lists, 84 single nucleotide variations (SNVs) with more than 3 reads, Q score above 25 and leading to non-synonymous changes at the protein level in candidate genes were extracted. Variants leading to non-synonymous changes in encoded proteins were selected if they received scores indicating a protein-damaging function with at least 3 of 5 in silico predictors (PolyPhen-2 with HumDiv as model classifier [25], SIFT [26], PROVEAN [27], MutationAssessor [28], and MutationTaster [29]). The conversion of the calls made by each predictor into neutral vs. non-neutral was made using an approach that integrates different predictive algorithms [12]. For PolyPhen-2, “probably damaging” and “possibly damaging” were considered non-neutral. For MutationAssessor, “high” and “medium” were considered non-neutral. For MutationTaster, “disease causing” and “disease causing_automatic” were considered non-neutral and “polymorphism” and “polymorphism_automatic” neutral. “Damaging” (SIFT) and “deleterious” (PROVEAN) were considered non-neutral while “tolerated” (SIFT) and “neutral” (PROVEAN) were considered neutral. For MutationTaster, 0.99 was used as cut-off in the disease_causing category.
In-frame deletion (Indel) variants were characterized with PROVEAN and MutationTaster. Indels that had a length divisible by 3 and caused amino acid insertion/deletion (also called 3N indels) were also analyzed with SIFT-Indel (http://sift-dna.org) [30]. Variants associated with possible splicing defects were also selected. Mutalyzer 2.0.4 (http://mutalyzer.nl/) was used to ascertain that the variants were described according to the Human Genome Variation Society (HGVS) nomenclature, effectively matching described amino acid and nucleotide coordinates to the GRCh37/hg19 assembly [31].
Variant verification
The Exome Aggregation Consortium (ExAC) website, Cambridge, MA (version 0.3) (http://exac.broadinstitute.org/) was used to assess the frequency of the selected variants in the general population or in a particular ethnic group. The ExAC data set contains information on 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies, approximately 50% of who are of European non-Finnish ancestry. A second independent group of controls, referred to as ITMI genomes and representing an extension of the set initially described in [11], consisted of 634 white non-Hispanic individuals who denied a personal or family history of cancer. For all variants with predicted protein-damaging consequences, primers flanking the variation were designed to amplify a product of ~200 to 400 base pairs. After digestion with ExoSap-IT (Affymetrix, Santa Clara, CA), the PCR product was sent to Genewiz (South Plainfield, NJ) for Sanger sequencing.
Molecular modeling
For analysis of structural consequences of missense variants, models of PAPSS2 and AKR1C1 were generated. All molecular display figures were prepared with the UCSF Chimera software [32]. While the N-terminal kinase domain is of known structure (2AX4), a full-length model of PAPSS2 was generated with Biological Assembly Modeler [33] based on the closest homolog of known structure, PAPSS1 (PDB code 1XNJ, 77% identity, 87% similarity), and compared with a previously deposited model [34]. An alternate template structure (2QJF) was superposed to extract the placement of the substrate and product molecules, (ADP and Adenosine-5′-phosphosulfate). For the variant in AKR1C1, the structure with PDB code 1MRQ was used.
Lymphocyte cell preservation, culture, and analysis of DNA damage response (DDR)
Peripheral blood lymphocytes (PBLs) were available from 9/12 of the exome sequenced patients and 10 age-matched and gender-matched individuals without a cancer diagnosis or a family history of cancer. Control samples were obtained from the FCCC Biosample Repository Facility. For analysis of DDR, cells were cultured in RPMI-1640 containing 15% fetal bovine serum (HyClone Laboratories, Logan, UT), 2 mM L-glutamine (Life Technologies, Grand Island, NY), 50 μM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), 0.2 units human recombinant insulin (Sigma) per ml, 50 units penicillin and 50 mg streptomycin per ml (complete RPMI), and then stimulated with phytohemagglutinin (PHA)-M (Life Technologies, Grand Island, NY) and recombinant human interleukin 2 (IL-2) (NCI Preclinical Repository) for 72 hours. Cells were then treated with vehicle, 10 μM aphidicolin or 25 μM etoposide, and fixed in paraformaldehyde 2 hours later. For immunofluorescence, cells were allowed to attach to poly-d-lysine-coated 96-well plates, stained with anti-γH2AX antibody (#05–636, Millipore, Temecula, CA). Sixteen images per well were acquired at 20X (with each image acquired in 2 channels to detect γH2AX with TRITC and total DNA with DAPI) utilizing the ImageXpress micro automated microscope (Molecular Devices, Sunnyvale, CA) driven by MetaXpress software. Images were analyzed in the Multiwavelength Scoring module of MetaXpress and results were displayed and exported utilizing the AcuityXpress software package (Molecular Devices, Sunnyvale, CA).
ACKNOWLEDGMENTS AND GRANT SUPPORT
The Fox Chase Cancer Center Biosample Repository, Cell Culture, Genomics, Molecular Modeling, and High Throughput Facilities provided resources and expertise. Margret Einarson contributed to the γH2AX scoring; Pam Nakajima contributed to the culture of primary lymphocytes. Donna Rathgeb was responsible for preparation of the manuscript.
The authors and this work were supported by NCI Core Grant P30 CA006927; the Temple University Fund for Genomics Research; a postdoctoral fellowship from the Fox Chase Cancer Center Board of Directors (to SA); a subsidy of the Russian Government to Kazan Federal University (to I.G.S.); and Inova Health System institutional funds (to D.L.B. and J.G.V).
CONFLICTS OF INTEREST
The authors have no conflict of interest to disclose.
REFERENCES
1. American Cancer Society. Cancer Facts and Figures. 2015.
2. Brandt A, Bermejo JL, Sundquist J, Hemminki K. Age-specific risk of incident prostate cancer and risk of death from prostate cancer defined by the number of affected family members. European urology. 2010; 58:275–280.
3. Kicinski M, Vangronsveld J, Nawrot TS. An epidemiological reappraisal of the familial aggregation of prostate cancer: a meta-analysis. PloS one. 2011; 6:e27130.
4. Hunter DJ, Riboli E, Haiman CA, Albanes D, Altshuler D, Chanock SJ, Haynes RB, Henderson BE, Kaaks R, Stram DO, Thomas G, Thun MJ, Blanche H, Buring JE, Burtt NP, Calle EE, et al. A candidate gene approach to searching for low-penetrance breast and prostate cancer genes. Nature reviews Cancer. 2005; 5:977–985.
5. Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, Vaisanen MR, Szulkin R, Annala M, Yan J, Egevad LA, Zhang K, Lin R, Jolma A, Nykter M, Manninen A, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nature genetics. 2014; 46:126–135.
6. Lindstrom S, Schumacher FR, Cox D, Travis RC, Albanes D, Allen NE, Andriole G, Berndt SI, Boeing H, Bueno-de-Mesquita HB, Crawford ED, Diver WR, Gaziano JM, Giles GG, Giovannucci E, Gonzalez CA, et al. Common genetic variants in prostate cancer risk prediction—results from the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2012; 21:437–444.
7. Demichelis F, Stanford JL. Genetic predisposition to prostate cancer: Update and future perspectives. Urologic oncology. 2015; 33:75–84.
8. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121:269–275.
9. Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, Bizon C, Yan G, Gielzak M, Partin AW, Shanmugam V, Izatt T, et al. Germline mutations in HOXB13 and prostate-cancer risk. The New England journal of medicine. 2012; 366:141–149.
10. Xu J, Lange EM, Lu L, Zheng SL, Wang Z, Thibodeau SN, Cannon-Albright LA, Teerlink CC, Camp NJ, Johnson AM, Zuhlke KA, Stanford JL, Ostrander EA, Wiley KE, Isaacs SD, Walsh PC, et al. HOXB13 is a susceptibility gene for prostate cancer: results from the International Consortium for Prostate Cancer Genetics (ICPCG). Human genetics. 2013; 132:5–14.
11. Bodian DL, McCutcheon JN, Kothiyal P, Huddleston KC, Iyer RK, Vockley JG, Niederhuber JE. Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PloS one. 2014; 9:e94554.
12. Martelotto LG, Ng C, De Filippo MR, Zhang Y, Piscuoglio S, Lim R, Shen R, Norton L, Reis-Filho JS, Weigelt B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome biology. 2014; 15:484.
13. Wilson JL, Hemann MT, Fraenkel E, Lauffenburger DA. Integrated network analyses for functional genomic studies in cancer. Seminars in cancer biology. 2013; 23:213–218.
14. Wu L, Schaid DJ, Sicotte H, Wieben ED, Li H, Petersen GM. Case-only exome sequencing and complex disease susceptibility gene discovery: study design considerations. Journal of medical genetics. 2015; 52:10–16.
15. Leongamornlert D, Saunders E, Dadaev T, Tymrakiewicz M, Goh C, Jugurnauth-Little S, Kozarewa I, Fenwick K, Assiotis I, Barrowdale D, Govindasami K, Guy M, Sawyer E, Wilkinson R, Antoniou AC, Eeles R, et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. British journal of cancer. 2014; 110:1663–1672.
16. Davis JD, Lin SY. DNA damage and breast cancer. World journal of clinical oncology. 2011; 2:329–338.
17. Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, Easton D, Kote-Jarai Z. The genetic epidemiology of prostate cancer and its clinical implications. Nature reviews Urology. 2014; 11:18–31.
18. Pauty J, Rodrigue A, Couturier A, Buisson R, Masson JY. Exploring the roles of PALB2 at the crossroads of DNA repair and cancer. The Biochemical journal. 2014; 460:331–342.
19. Tung N, Silver DP. Chek2 DNA damage response pathway and inherited breast cancer risk. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011; 29:3813–3815.
20. Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, Carver BS, Chen Y, Watson PA, Shah NP, Fujisawa S, Goglia AG, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer discovery. 2013; 3:1245–1253.
21. Sun T, Oh WK, Jacobus S, Regan M, Pomerantz M, Freedman ML, Lee GS, Kantoff PW. The impact of common genetic variations in genes of the sex hormone metabolic pathways on steroid hormone levels and prostate cancer aggressiveness. Cancer prevention research. 2011; 4:2044–2050.
22. Daly MB, Pilarski R, Axilbund JE, Buys SS, Crawford B, Friedman S, Garber JE, Horton C, Kaklamani V, Klein C, Kohlmann W, Kurian A, Litton J, Madlensky L, Marcom PK, Merajver SD, et al. Genetic/familial high-risk assessment: breast and ovarian, version 1. 2014. Journal of the National Comprehensive Cancer Network: JNCCN. 2014; 12:1326–1338.
23. Carter BS, Bova GS, Beaty TH, Steinberg GD, Childs B, Isaacs WB, Walsh PC. Hereditary prostate cancer: epidemiologic and clinical features. The Journal of urology. 1993; 150:797–802.
24. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. 2004; 96:261–268.
25. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010; 7:248–249.
26. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003; 31:3812–3814.
27. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015.
28. Reva B, Antipin Y, Ser C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids research. 2011; 39:e118.
29. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods. 2014; 11:361–362.
30. Hu J, Ng PC. SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PloS one. 2013; 8:e77940.
31. Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Human mutation. 2008; 29:6–13.
32. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004; 25:1605–1612.
33. Shapovalov MV, Wang Q, Xu Q, Andrake M, Dunbrack RL Jr. BioAssemblyModeler (BAM): user-friendly homology modeling of protein homo- and heterooligomers. PloS one. 2014; 9:e98309.
34. Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic acids research. 2009; 37:D387–392. (Database issue).
35. Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science (New York NY). 2001; 291:1284–1289.
36. Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome research. 2003; 13:2363–2371.
37. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012; 44:685–689.
38. Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, McKenna A, Sivachenko A, Rosenberg M, Kiezun A, Voet D, Lawrence M, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nature medicine. 2014; 20:682–688.
39. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
40. Dragani TA, Canzian F, Pierotti MA. A polygenic model of inherited predisposition to cancer. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1996; 10:865–870.
41. Mavaddat N, Pharoah PD, Michailidou K, Tyrer J, Brook MN, Bolla MK, Wang Q, Dennis J, Dunning AM, Shah M, Luben R, Brown J, Bojesen SE, Nordestgaard BG, Nielsen SF, Flyger H, et al. Prediction of breast cancer risk based on profiling with common genetic variants. Journal of the National Cancer Institute. 2015; 107.
42. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:7155–7160.
43. Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO reports. 2009; 10:990–996.
44. Li Z, Xiao W, McCormick JJ, Maher VM. Identification of a protein essential for a major pathway used by human cells to avoid UV-induced DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:4459–4464.
45. Santarpia L, Iwamoto T, Di Leo A, Hayashi N, Bottai G, Stampfer M, Andre F, Turner NC, Symmans WF, Hortobagyi GN, Pusztai L, Bianchini G. DNA repair gene patterns as prognostic and predictive factors in molecular breast cancer subtypes. The oncologist. 2013; 18:1063–1073.
46. Ali AM, Kirby M, Jansen M, Lach FP, Schulte J, Singh TR, Batish SD, Auerbach AD, Williams DA, Meetei AR. Identification and characterization of mutations in FANCL gene: a second case of Fanconi anemia belonging to FA-L complementation group. Human mutation. 2009; 30:E761–770.
47. Jafary F, Salehi M, Sedghi M, Nouri N, Jafary F, Sadeghi F, Motamedi S, Talebi M. Association between mismatch repair gene MSH3 codons 1036 and 222 polymorphisms and sporadic prostate cancer in the Iranian population. Asian Pacific journal of cancer prevention: APJCP. 2012; 13:6055–6057.
48. Duraturo F, Liccardo R, Cavallo A, De Rosa M, Grosso M, Izzo P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. International journal of cancer Journal international du cancer. 2011; 129:1643–1650.
49. Huang M, Kennedy R, Ali AM, Moreau LA, Meetei AR, D’Andrea AD, Chen CC. Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA repair. 2011; 10:1203–1212.
50. Peng M, Xie J, Ucher A, Stavnezer J, Cantor SB. Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses. The EMBO journal. 2014; 33:1698–1712.
51. Guillemette S, Branagan A, Peng M, Dhruva A, Scharer OD, Cantor SB. FANCJ localization by mismatch repair is vital to maintain genomic integrity after UV irradiation. Cancer research. 2014; 74:932–944.
52. Zhang FP, Domanskyi A, Palvimo JJ, Sariola H, Partanen J, Janne OA. An adenosine triphosphatase of the sucrose nonfermenting 2 family, androgen receptor-interacting protein 4, is essential for mouse embryonic development and cell proliferation. Molecular endocrinology. 2007; 21:1430–1442.
53. Rouleau N, Domans’kyi A, Reeben M, Moilanen AM, Havas K, Kang Z, Owen-Hughes T, Palvimo JJ, Janne OA. Novel ATPase of SNF2-like protein family interacts with androgen receptor and modulates androgen-dependent transcription. Molecular biology of the cell. 2002; 13:2106–2119.
54. Mirzaei H, Schmidt KH. Non-Bloom syndrome-associated partial and total loss-of-function variants of BLM helicase. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:19357–19362.
55. Frank B, Hoffmeister M, Klopp N, Illig T, Chang-Claude J, Brenner H. Colorectal cancer and polymorphisms in DNA repair genes WRN, RMI1 and BLM. Carcinogenesis. 2010; 31:442–445.
56. Silvestri V, Rizzolo P, Zanna I, Falchetti M, Masala G, Bianchi S, Papi L, Giannini G, Palli D, Ottini L. PALB2 mutations in male breast cancer: a population-based study in Central Italy. Breast cancer research and treatment. 2010; 122:299–301.
57. Litim N, Labrie Y, Desjardins S, Ouellette G, Plourde K, Belleau P, BRCAs I, Durocher F. Polymorphic variations in the FANCA gene in high-risk non-BRCA1/2 breast cancer individuals from the French Canadian population. Molecular oncology. 2013; 7:85–100.
58. Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS. Aprataxin resolves adenylated RNA-DNA junctions to maintain genome integrity. Nature. 2014; 506:111–115.
59. Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006; 443:713–716.
60. Dopeso H, Mateo-Lozano S, Elez E, Landolfi S, Ramos Pascual FJ, Hernandez-Losa J, Mazzolini R, Rodrigues P, Bazzocco S, Carreras MJ, Espin E, Armengol M, Wilson AJ, Mariadason JM, Ramon YCS, Tabernero J, et al. Aprataxin tumor levels predict response of colorectal cancer patients to irinotecan-based treatment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010; 16:2375–2382.
61. Rolseth V, Krokeide SZ, Kunke D, Neurauter CG, Suganthan R, Sejersted Y, Hildrestrand GA, Bjoras M, Luna L. Loss of Neil3, the major DNA glycosylase activity for removal of hydantoins in single stranded DNA, reduces cellular proliferation and sensitizes cells to genotoxic stress. Biochimica et biophysica acta. 2013; 1833:1157–1164.
62. Barry KH, Koutros S, Berndt SI, Andreotti G, Hoppin JA, Sandler DP, Burdette LA, Yeager M, Freeman LE, Lubin JH, Ma X, Zheng T, Alavanja MC. Genetic variation in base excision repair pathway genes, pesticide exposure, and prostate cancer risk. Environmental health perspectives. 2011; 119:1726–1732.
63. Douglas JA, Levin AM, Zuhlke KA, Ray AM, Johnson GR, Lange EM, Wood DP, Cooney KA. Common variation in the BRCA1 gene and prostate cancer risk. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007; 16:1510–1516.
64. Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA repair. 2014; 19:169–175.
65. Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Molecular and cellular biology. 2014; 34:1380–1388.
66. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell. 2013; 49:872–883.
67. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nature reviews Molecular cell biology. 2014; 15:7–18.
68. Chang SW, Su CH, Chen HH, Huang CW, Tsao LP, Tsao YP, Chen SL. DDB2 is a novel AR interacting protein and mediates AR ubiquitination/degradation. The international journal of biochemistry & cell biology. 2012; 44:1952–1961.
69. Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, Rapic-Otrin V. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:E2737–2746.
70. Rizner TL, Penning TM. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids. 2014; 79:49–63.
71. Liu C, Lou W, Zhu Y, Yang JC, Natiminty N, Gaikwad N, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer research. 2015.
72. Wang R, Asangani IA, Chakravarthi BV, Ateeq B, Lonigro RJ, Cao Q, Mani RS, Camacho DF, McGregor N, Schumann TE, Jing X, Menawat R, Tomlins SA, Zheng H, Otte AP, Mehra R, et al. Role of transcriptional corepressor CtBP1 in prostate cancer progression. Neoplasia. 2012; 14:905–914.
73. Moiola CP, De Luca P, Zalazar F, Cotignola J, Rodriguez-Segui SA, Gardner K, Meiss R, Vallecorsa P, Pignataro O, Mazza O, Vazquez ES, De Siervi A. Prostate tumor growth is impaired by CtBP1 depletion in high-fat diet-fed mice. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014; 20:4086–4095.
74. Imberg-Kazdan K, Ha S, Greenfield A, Poultney CS, Bonneau R, Logan SK, Garabedian MJ. A genome-wide RNA interference screen identifies new regulators of androgen receptor function in prostate cancer cells. Genome research. 2013; 23:581–591.
75. Zhang Q, Nottke A, Goodman RH. Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:2802–2807.
76. Carascossa S, Gobinet J, Georget V, Lucas A, Badia E, Castet A, White R, Nicolas JC, Cavailles V, Jalaguier S. Receptor-interacting protein 140 is a repressor of the androgen receptor activity. Molecular endocrinology. 2006; 20:1506–1518.
77. Ghoussaini M, Fletcher O, Michailidou K, Turnbull C, Schmidt MK, Dicks E, Dennis J, Wang Q, Humphreys MK, Luccarini C, Baynes C, Conroy D, Maranian M, Ahmed S, Driver K, Johnson N, et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nature genetics. 2012; 44:312–318.
78. Ferreira AM, Tuominen I, Sousa S, Gerbens F, van Dijk-Bos K, Osinga J, Kooi KA, Sanjabi B, Esendam C, Oliveira C, Terpstra P, Hardonk M, van der Sluis T, Zazula M, Stachura J, van der Zee AG, et al. New target genes in endometrial tumors show a role for the estrogen-receptor pathway in microsatellite-unstable cancers. Human mutation. 2014; 35:1514–1523.
79. Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. The Journal of biological chemistry. 2003; 278:5052–5061.
80. Sitz JH, Tigges M, Baumgartel K, Khaspekov LG, Lutz B. Dyrk1A potentiates steroid hormone-induced transcription via the chromatin remodeling factor Arip4. Molecular and cellular biology. 2004; 24:5821–5834.
81. Niki T, Takahashi-Niki K, Taira T, Iguchi-Ariga SM, Ariga H. DJBP: a novel DJ—binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-antagonizes this inhibition by abrogation of this complex. Molecular cancer research: MCR. 2003; 1:247–261.
82. Kosari F, Asmann YW, Cheville JC, Vasmatzis G. Cysteine-rich secretory protein-3: a potential biomarker for prostate cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2002; 11:1419–1426.
83. Ribeiro FR, Paulo P, Costa VL, Barros-Silva JD, Ramalho-Carvalho J, Jeronimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, Teixeira MR. Cysteine-rich secretory protein-3 (CRISP3) is strongly up-regulated in prostate carcinomas with the TMPRSS2-ERG fusion gene. PloS one. 2011; 6:e22317.
84. Drake RR, Jones EE, Powers TW, Nyalwidhe JO. Altered glycosylation in prostate cancer. Advances in cancer research. 2015; 126:345–382.
85. DePrimo SE, Diehn M, Nelson JB, Reiter RE, Matese J, Fero M, Tibshirani R, Brown PO, Brooks JD. Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome biology. 2002; 3:RESEARCH0032.
86. Oostdijk W, Idkowiak J, Mueller JW, House PJ, Taylor AE, O’Reilly MW, Hughes BA, de Vries MC, Kant SG, Santen GW, Verkerk AJ, Uitterlinden AG, Wit JM, Losekoot M, Arlt W. PAPSS2 Deficiency Causes Androgen Excess via Impaired DHEA Sulfation-In Vitro and in Vivo Studies in a Family Harboring Two Novel PAPSS2 Mutations. The Journal of clinical endocrinology and metabolism. 2015; 100:E672–680.
87. Fachal L, Dunning AM. From candidate gene studies to GWAS and post-GWAS analyses in breast cancer. Current opinion in genetics & development. 2015; 30C:32–41.
88. Lynch PM. When and how to perform genetic testing for inherited colorectal cancer syndromes. Journal of the National Comprehensive Cancer Network: JNCCN. 2013; 11:1577–1583.
89. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228.
90. Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, Tait JF, Corey E, Vessella RL, Walsh T, Shendure J, Nelson PS. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nature communications. 2014; 5:4988.
91. Stratifying prostate patients for olaparib. Cancer discovery. 2015; 5:568–569.
92. Tommasi S, Pilato B, Pinto R, Monaco A, Bruno M, Campana M, Digennaro M, Schittulli F, Lacalamita R, Paradiso A. Molecular and in silico analysis of BRCA1 and BRCA2 variants. Mutation research. 2008; 644:64–70.
93. Zheng L, Pan H, Li S, Flesken-Nikitin A, Chen PL, Boyer TG, Lee WH. Sequence-specific transcriptional corepressor function for BRCA1 through a novel zinc finger protein, ZBRK1. Molecular cell. 2000; 6:757–768.
94. Furuta S, Wang JM, Wei S, Jeng YM, Jiang X, Gu B, Chen PL, Lee EY, Lee WH. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer cell. 2006; 10:13–24.
95. Johnston JJ, Lewis KL, Ng D, Singh LN, Wynter J, Brewer C, Brooks BP, Brownell I, Candotti F, Gonsalves SG, Hart SP, Kong HH, Rother KI, Sokolic R, Solomon BD, Zein WM, et al. Individualized Iterative Phenotyping for Genome-wide Analysis of Loss-of-Function Mutations. American journal of human genetics. 2015; 96:913–925.
96. MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012; 335:823–828.
97. Grant RC, Al-Sukhni W, Borgida AE, Holter S, Kanji ZS, McPherson T, Whelan E, Serra S, Trinh QM, Peltekova V, Stein LD, McPherson JD, Gallinger S. Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Human genomics. 2013; 7:11.
98. Elsayed SM, Phillips JB, Heller R, Thoenes M, Elsobky E, Nurnberg G, Nurnberg P, Seland S, Ebermann I, Altmuller J, Thiele H, Toliat M, Korber F, Hu XJ, Wu YD, Zaki MS, et al. Non-manifesting AHI1 truncations indicate localized loss-of-function tolerance in a severe Mendelian disease gene. Human molecular genetics. 2015; 24:2594–2603.
99. Thormaehlen AS, Schuberth C, Won HH, Blattmann P, Joggerst-Thomalla B, Theiss S, Asselta R, Duga S, Merlini PA, Ardissino D, Lander ES, Gabriel S, Rader DJ, Peloso GM, Pepperkok R, Kathiresan S, et al. Systematic Cell-Based Phenotyping of Missense Alleles Empowers Rare Variant Association Studies: A Case for LDLR and Myocardial Infarction. PLoS genetics. 2015; 11:e1004855.
100. Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2014; 25:32–40.
101. Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nature reviews Cancer. 2011; 11:467–480.
102. Couture JF, Legrand P, Cantin L, Luu-The V, Labrie F, Breton R. Human 20alpha-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. Journal of molecular biology. 2003; 331:593–604.