
INTRODUCTION
The management of patients with adrenocortical carcinomas (ACCs) remains a challenge and patients with invasive, metastatic or recurrent disease have a poor prognosis [1, 2]). Although significant progress has been achieved in recent years both in basic and clinical research, adjuvant therapeutic options for patients with ACC remain very limited [3]. Mitotane (M), the first line adjuvant treatment, is highly toxic and its combination with etoposide, doxorubicin, and cisplatin (EDP) is generally ineffective and almost all patients will experience disease progression [4]. New molecular targeted therapies have been successfully developed for various types of cancers but not for ACCs. Thus, better treatments are urgently necessary.
Several genetic abnormalities have been found in these tumors, the most prominent being IGF2 overexpression [5–7] and mutations in TP53 and CTNNB1 (the beta-catenin gene) in both adult and pediatric adrenocortical tumors (ACTs) [8–10]. Transcriptome studies have shown that ACCs are clustered within different sets of poor prognosis for adult ACC patients according to TP53 or CTNNB1 abnormalities [10]. Accordingly, overexpression of beta-catenin in ACCs has been correlated with a worse prognosis [11]. Exon 3 CTNNB1 mutations have been found in 15–36% and 6% of adult and pediatric ACTs, respectively [8, 9, 12–15]. We previously showed that activation of both canonical and non-canonical Wnt signaling pathways are common in ACTs with or without CTNNB1 mutations [8, 9]. The hypothesis that the Wnt pathway can be activated through other mechanisms than CTNNB1 mutations has been recently reinforced. A large-scale high-resolution analysis study showed that variations in ZNRF3, which is a Wnt/beta-catenin pathway inhibitor, were the most common genetic defect found in a large number of ACC samples. ACCs presenting ZNRF3 variants showed transcriptional activation of beta-catenin target genes [16]. Thus, activation of the Wnt/beta-catenin pathway triggered by CTNNB1 and ZNRF3 mutations or down regulation of Wnt/beta-catenin inhibitors are important for ACC pathogenesis. Therefore, inhibition of the Wnt/beta-catenin signaling is a rational option and may become a promising approach.
CTNNB1 mutations found in ACCs are located at residues involved in phosphorylation, which are essential sites for beta-catenin degradation by ubiquitin/proteasome signaling. Therefore, mutations in these sites lead to beta-catenin accumulation in the nucleus, where it binds with the T cell factor (Tcf) and enhances its transcriptional activity [15]. The NCI-H295 cell line is an immortalized adrenocortical-secreting carcinoma lineage derived from an adult patient [17]. Remarkably, this cell line harbors the CTNNB1 p.S45P mutation, thus representing a good in vitro model of ACC showing Wnt/beta-catenin pathway activation [14, 15]. High-throughput screening identified small molecules that antagonize the Tcf/beta-catenin complex and inhibit the growth of tumor cell lines [18]. Among Tcf/beta-catenin antagonists, PKF115-584 has been reported to inhibit proliferation of the NCI-H295R cell line and the expression of the beta-catenin target genes cyclin D1 and c-Myc [19].
The PNU-74654 (PNU) compound is a non-FDA-approved drug which prevents that Tcf from binding to beta-catenin, acting as a Wnt/beta-catenin antagonist (Figure 1). This small molecule was found by virtual screening and confirmed by biophysical screening to interfere with protein-protein interactions [20]. Beta-catenin tightly binds to Tcf through a hot spot site. By binding to the same site, PNU can compete with Tcf. A luciferase activity assay for Tcf transactivation showed specific inhibition in the presence of PNU, confirming that this drug-like compound is an effective Wnt pathway antagonist [20].
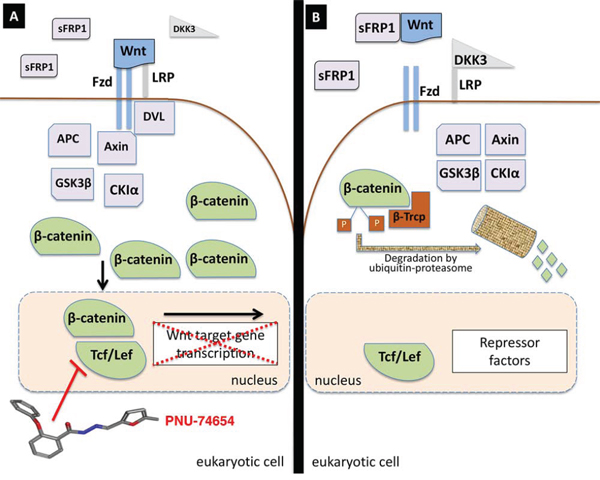
Figure 1: Wnt pathway signaling and PNU-74654 effect on the Tcf/beta-catenin complex. A. When Wnt signaling is activated, the Wnt ligand binds to the Frizzled (Fzd) receptor and LRP5/6 (LRP) co-receptor and stimulates LRP5/6 phosphorylation with the help of Dishevelled (DVL). Phosphorylated LRP recruits Axin to the membrane and disrupts the beta-catenin degradation complex. Beta-catenin accumulates in the cytoplasm and enters into the nucleus, where it binds to Tcf/Lef and co-activators triggering Wnt target gene transcription. PNU-74654, a drug-like compound, disrupts the beta-catenin/Tcf complex and arrests Wnt target gene transcription. B. When Wnt signaling is not activated (either by Wnt ligand sequestration by sFRPs and/or LRP5/6 inhibition by DKK3), cytoplasmic beta-catenin is phosphorylated by the beta-catenin degradation complex consisting of APC, Axin, GSK3beta and CKIα. Phosphorylated beta-catenin is then recognized by beta-Trcp and is degraded via the ubiquitin-proteasome pathway.
Taken together, these data suggest that the Wnt/beta-catenin pathway might be a potential targeted therapy for patients with ACC. Tcf/beta-catenin antagonism may be useful to treat patients with ACC exhibiting increased Wnt/beta-catenin signaling. In the present study, we assessed the in vitro effect of PNU on adrenocortical tumor cells. Our results showed that inhibition of the Wnt/beta-catenin signaling resulted in a significantly decreased cell viability, increased apoptosis and impaired steroidogenesis.
RESULTS
Authentication and sequencing analysis of exon 3 CTNNB1 and TP53 coding regions in cell lines
NCI-H295 and HeLa authenticity was confirmed by STR profiling (Supplementary Table 1). In our own stock of NCI-H295 cells, we confirmed the presence of the pathogenic p.S45P CTNNB1 variation. Immunofluorescence analysis showed that, under basal conditions, beta-catenin is highly expressed in the cytoplasm and nucleus of NCI-H295 cells (Figure 2a–2c). This finding confirms that the Wnt/beta-catenin pathway is activated in NCI-H295 cells. In addition, we also detected the presence of the p.P278L TP53 variation in this cell line.
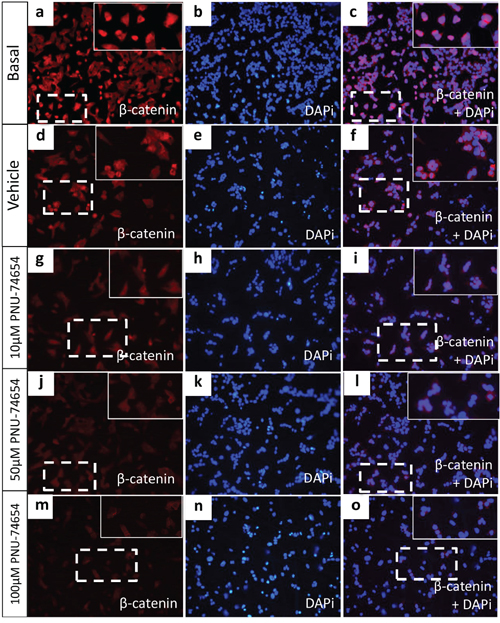
Figure 2: Beta-catenin immunofluorescence in the NCI-H295 cell line. (a-f) NCI-H295 cells displayed nuclear and cytoplasmic beta-catenin expression under basal conditions and 48 h after treatment with vehicle (DMSO). (g–i) NCI-H295 cells displayed decreased nuclear and cytoplasmic beta-catenin expression 48 h after treatment with 10 μM PNU-74654, (j–l), reduced cytoplasmic and absent nuclear beta-catenin expression after 50 μM PNU-74654 and (m–o) and very low cytoplasmic beta-catenin nuclear expression after 100 μM PNU-74654 treatment. Blue = nuclear staining (DAPi 1:25000; Cell Signaling Technology); red = beta-catenin (anti-beta-catenin 1:2000; BD Biosciences); pink = merged.
HeLa, a cervix carcinoma cell lineage, was employed as a non-adrenal cell line that did not carry the exon 3 CTNNB1 mutation but which also showed the p.P72R TP53 variation as well as a second intronic TP53 variation (c.97-30C > A/g.11298). Additionally, we identified an intronic TP53 variation at position c.74 + 38 (C > G rs1642785) in HeLa cells. These results are summarized in Supplementary Table 2.
Inhibition of the Tcf/beta-catenin complex induced by PNU impairs cell viability in human adrenocortical cell lines but not in mouse adrenal cell lines and human non-adrenal cell lines and prevents cell proliferation by Forskolin stimulation (Figures 3 and 4)
PNU impaired NCI-H295 cell viability. NCI-H295 cell viability was decreased 24 and 48 h after treatment with 100 and 200 μM PNU (Figure 3a–3b) and 72 h after treatment with 10, 100 and 200 μM PNU (Figure 3c). Lower doses of PNU were able to decrease cell viability 96 h after treatment: 22, 27, 50 and 97% with 10, 50, 100 and 200 μM, respectively (ANOVA: p < 0.0001; Figure 3d).
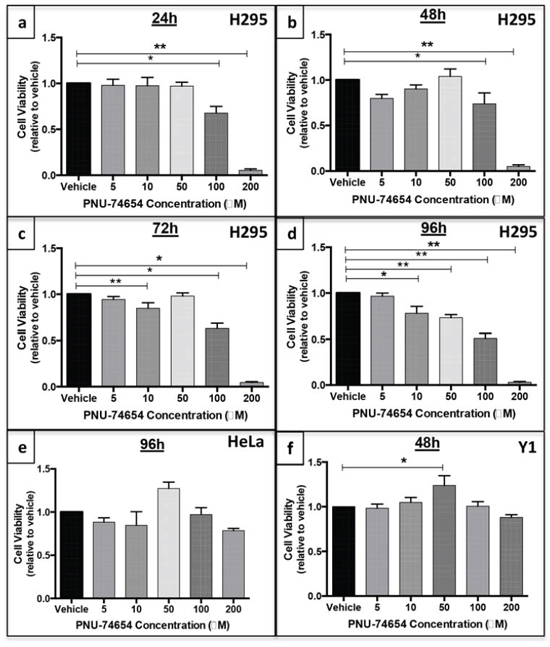
Figure 3: PNU-74654 treatment reduces cell viability in human adrenocortical cell lines but not in a mouse adrenal cell line or human non-adrenal cell line. NCI-H295 cell viability 24 a. 48 b. 72 c. and 96 d. hours after treatment with 5, 10, 50, 100 and 200 μM PNU-74654. HeLa cell viability 96 e. hours after treatment with 5, 10, 50, 100 and 200 μM PNU-74654. Y1 cell viability 48 h f. after treatment with 5, 10, 50, 100 and 200 μM PNU-74654. Cell viability was calculated by relative absorbance normalized by vehicle (DMSO). Values are reported as mean ± SEM. Statistics: ANOVA (a) *p = 0.0012; **p < 0.0001 (b) *p = 0.02; **p < 0.0001; (c) *p < 0.0001; **p = 0.03 (d) *p = 0.001, **p < 0.0001; (e) p = 0.05; (f) *p = 0.04.
PNU did not decrease HeLa cell viability 96 h after treatment compared to vehicle (p = 0.05; Figure 3e). Interestingly, PNU showed a weak effect 48 h after treatment, but this effect was observed only at a concentration of 200 μM (ANOVA: p = 0.01; Supplementary Figure 1).
PNU did not decrease Y1 cell viability 48 h after treatment (Figure 3f). We chose to evaluate these cells up to 48 h and not at 96 h because they grow much faster than NCI-H295 cells under culture.
As expected, 10 μM Forskolin increased cell proliferation by approximately 50% compared with vehicle (p = 0.0008; Figure 4). In turn, association of 5, 10 and 50 μM PNU with Forskolin prevented Forskolin-induced cell proliferation 96 h after treatment (Figure 4).
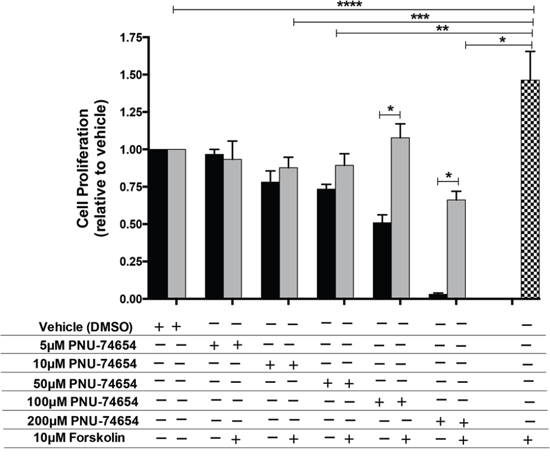
Figure 4: PNU-74654 treatment prevents Forskolin-induced cell proliferation in NCI-H295 cells. Forskolin stimulation increased cell proliferation compared with vehicle and compared with PNU treatment alone and under Forskolin stimulation (*p = 0.0007; **p = 0.03; ***p = 0.02; ****p = 0.0008). PNU treatment under Forskolin stimulation compared with PNU 100 and 200 μM (*p < 0.0001). Black bars: PNU treatment alone; Gray bars: PNU treatment under Forskolin stimulation; Black/white bar: Forskolin stimulation alone. Values are reported as mean ± SEM relative to vehicle of 3 independent experiments.
Inhibition of the Tcf/beta-catenin complex induced by PNU increased NCI-H295 cell apoptosis (Figure 5)
PNU treatment at the concentrations 10, 50 and 100 μM resulted in a dose-dependent increase (19%, 51% and 80%, respectively) of late apoptotic cells 48 h after treatment (Figure 5a and 5c–5f). PNU treatment at the concentrations of 10, 50 and 100 μM also resulted in an increase (18%, 60% and 31%, respectively) of early apoptotic cells 48 h after treatment (Figure 5a and 5d–5g). The percentage of necrotic cells was decreased after PNU treatment at the concentrations of 10, 50 and 100 μM (−10%, −15% and −4%, respectively; Figure 5a and 5c–5f). The reduction of NCI-H295 cell viability was confirmed 48 h after treatment with 50 and 100 μM by flow cytometry sorting (p = 0.04 and p = 0.01, respectively; Figure 4a). Schematic flow cytometry squares are characterized in Figure 4b.
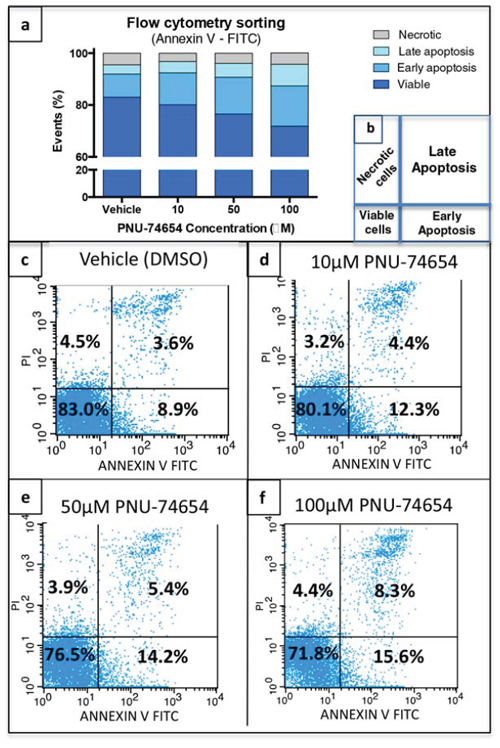
Figure 5: Tcf/Beta-catenin complex inhibition increases apoptotic cells. a, c–f. PNU-75654 induced an increase of late and early apoptotic cells 48 h after treatment with 10, 50 and 100 μM. b. Schematic figure displaying flow cytometry squares. (c–f) Representative figures of flow cytometry sorting of NCI-H295 cells 48 h after treatment with (c) vehicle, (d) 10, (e) 50, and (f) 100 μM PNU-74654. (a, c–f) Values are reported as mean percentage (%) of 3 independent experiments.
Inhibition of the Tcf/beta-catenin complex induced by PNU decreases CTNNB1 mRNA expression and nuclear beta-catenin accumulation but not TCF expression and changes beta-catenin target genes expression in NCI-H295 cells (Figures 2, 6 and 7)
Immunofluorescence analysis showed a marked decrease of nuclear and cytoplasmic beta-catenin expression 48 h after treatment with 10, 50 and 100 μM PNU compared with vehicle (Figure 2d–2o). Similarly, beta-catenin/CTNNB1 mRNA expression was decreased 48 h after PNU treatment with 50 (p = 0.04, fold change = −1.5; Figure 6b) and 100 μM (p = 0.0034, fold change = −1.8; Figure 6b) as also was beta-catenin protein expression (Figure 6a and 6b). Ctnnb1/CTNNB1 mRNA expression was not decreased by PNU treatment in Y1 or HeLa cells (Figure 6d-g and 6e-h, respectively).
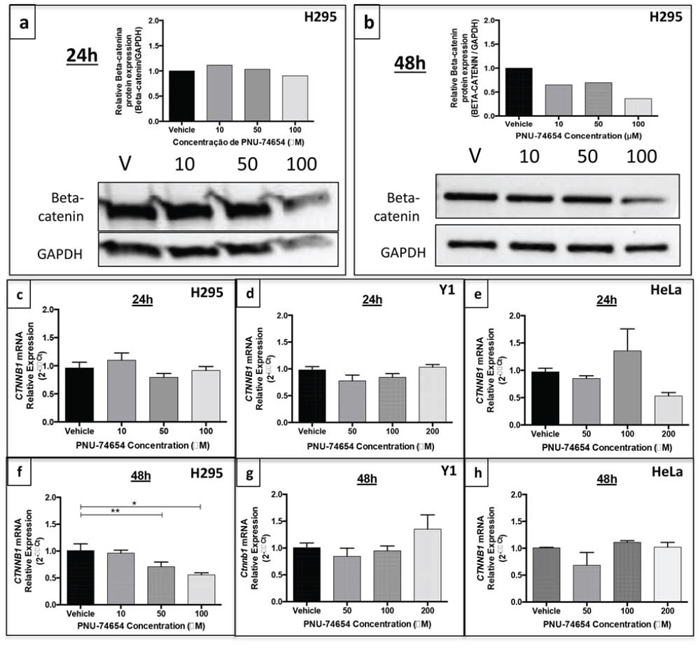
Figure 6: Beta-catenin expression in the NCI-H295 human adrenal, Y1 mouse adrenal and HeLa non-adrenal cell lines after PNU-74654 treatment. Beta-catenin protein expression after 24 a. and 48 h b. after treatment with 10, 50 and 100 μM PNU-74654 (beta-catenin antibody: 92 kDa; 1:1000; BD Biosciences/GAPDH antibody: 37kDa; 1:1000; Santa Cruz). CTNNB1/beta-catenin mRNA expression in NCI-H295 cells 24 c. and 48 h f. after treatment with 10, 50 and 100 μM PNU-74654. (f) *p = 0.003, **p = 0.04. Ctnnb1 mRNA expression in Y1 cells after 24 d. and 48 h g. after treatment with 50, 100 and 200 μM PNU-74654. CTNNB1 mRNA expression in HeLa cells after 24 e. and 48 h h. after treatment with 50, 100 and 200 μM PNU-74654. V: vehicle. Values are reported as mean ± SEM.
Immunostaining analysis showed a membranous and cytoplasmic pattern of beta-catenin accumulation in Y1 cells under basal conditions (Supplementary Figure 2). This result supports the idea that the Y1 cell line does not present Wnt/beta-catenin activation.
Regarding beta-catenin target genes, 50 μM PNU increased the mRNA expression of CCND1 (p = 0.005, fold change: 1.86; Figure 7a) and AXIN2 (p = 0.01, fold change = 2; Figure 7b) 48 h after treatment. On the other hand, MYC mRNA expression (Figure 7c) and TCF mRNA and protein expression (Figure 7d–7e) did not change after PNU treatment.
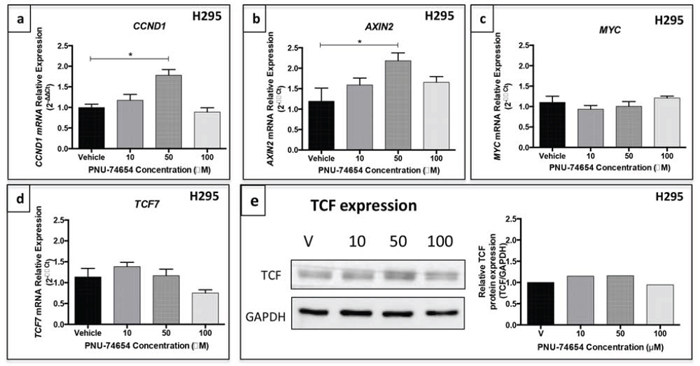
Figure 7: Gene expression of beta-catenin target genes in the NCI-H295 cell line after PNU-74654 treatment. a. CCND1 mRNA expression was increased in NCI-H295 cells 48 h after treatment with 50 μM PNU-74654. *p = 0.005. b. AXIN2 mRNA expression was increased in NCI-H295 cells 48 h after treatment with 50 μM PNU-74654. *p = 0.01. c. MYC mRNA expression was not decreased in NCI-H295 cells 48 h after treatment with 100 μM PNU-74654 (p = 0.17). TCF7 mRNA expression d. and TCF protein expression e. were not decreased in NCI-H295 cells 48 h after treatment with PNU-74654 (p = 0.02). V: vehicle. Values are reported as mean ± SEM.
Inhibition of the Tcf/beta-catenin complex induced by PNU impairs adrenal steroidogenesis in NCI-H295 and Y1 cells (Figures 8 and 9)
Under basal conditions, NCI-H295 cells retain the ability to secrete adrenal steroids [21]. Overall, PNU treatment significantly impaired adrenal steroidogenesis by decreasing steroid production and the mRNA expression of steroidogenesis-related genes in NCI-H295 cells. PNU decreased cortisol production by 72 and 78% (50 and 100 μM, respectively; ANOVA: p = 0.0002; Figure 8a) 24 h after treatment and by 60 and 78% (50 and 100 μM, respectively; ANOVA: p = 0.0001; Figure 8b) 48 h after treatment. PNU decreased testosterone production by 50 and 73% (50 and 100 μM, respectively; ANOVA: p = 0.0012; Figure 8c) 24 h after treatment and by 37 and 69% (50 and 100 μM, respectively; ANOVA: p < 0.0001; Figure 8d) 48 h after treatment. PNU decreased androstenedione production by 97.5% at 200 μM (ANOVA: p = 0.02; Figure 8e and 8f) 48 h after treatment.
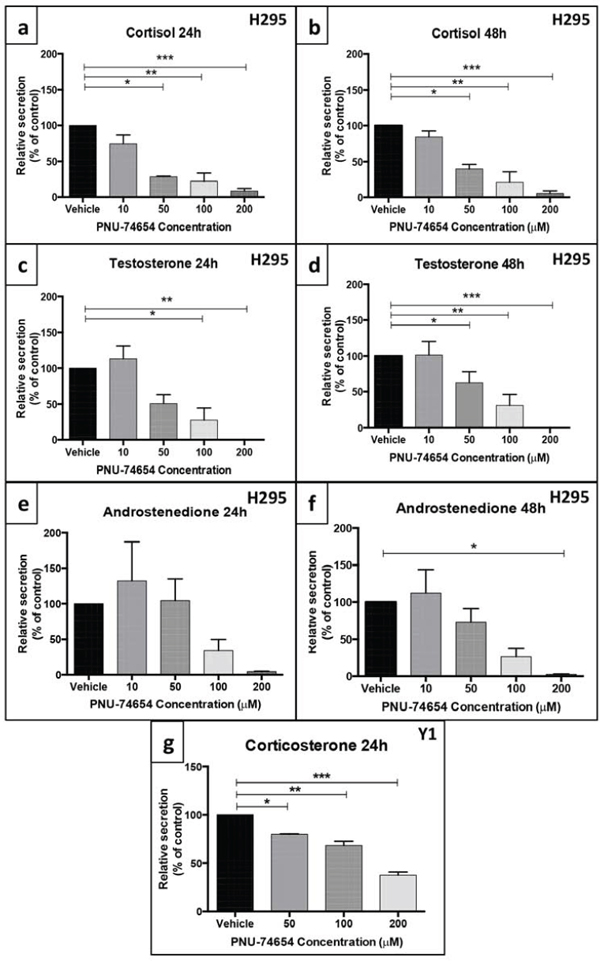
Figure 8: PNU-74654 treatment impaired adrenal steroidogenesis in the NCI-H295 and Y-1 cell lines. In NCI-H295 cells, treatment with 50, 100 and 200 μM PNU-74654 reduced cortisol secretion at 24 a. and 48 h b. after treatment. (a) *p = 0.0006; **p = 0.0003; ***p = 0.0002; (b) *p = 0.002; **p = 0.0003; ***p = 0.0002. In NCI-H295 cells, treatment with 50, 100 and 200 μM PNU-74654 reduced testosterone secretion 24 c. and 48 h d. after treatment. (c) *p = 0.01; **p = 0.003; (d) *p = 0.02; **p = 0.0005; ***p < 0.0001. In NCI-H295 cells, PNU-74654 treatment did not decrease androstenedione secretion 24 h after treatment e. but reduced androstenedione secretion at 200 μM 48 h after treatment f. *p = 0.02. g. In Y-1 cells, treatment with 50, 100 and 200 μM PNU-74654 reduced corticosterone secretion 24 h after treatment (*p = 0.02, **p = 0.003, ***p = 0.0002). Values are reported as mean ± SEM percentage relative to vehicle of at least 3 independent experiments.
PNU treatment also impaired steroid production in adrenal mouse cells. Corticosterone secretion was reduced by 20, 32 and 63% 24 h after treatment with 50, 100 and 200 μM PNU, respectively (ANOVA: p = 0.0004; Figure 8g).
PNU at 50 and 100 μM decreased NR5A1/SF1 (52 and 66%, respectively; ANOVA: p < 0.0001; Figure 9a) and CYP21A2 mRNA expression (63 and 88%, respectively ANOVA: p < 0.0001; Figure 9b) 48 h after treatment. PNU at 50 and 100 μM also decreased STAR relative protein expression 48 h after treatment (32 and 77%, respectively; Figure 9c). Additionally, PNU markedly decreased the relative protein expression of aldosterone synthase 48 h after treatment with 10, 50 and 100 μM PNU (23%, 62% and 70%, respectively; Figure 9d).
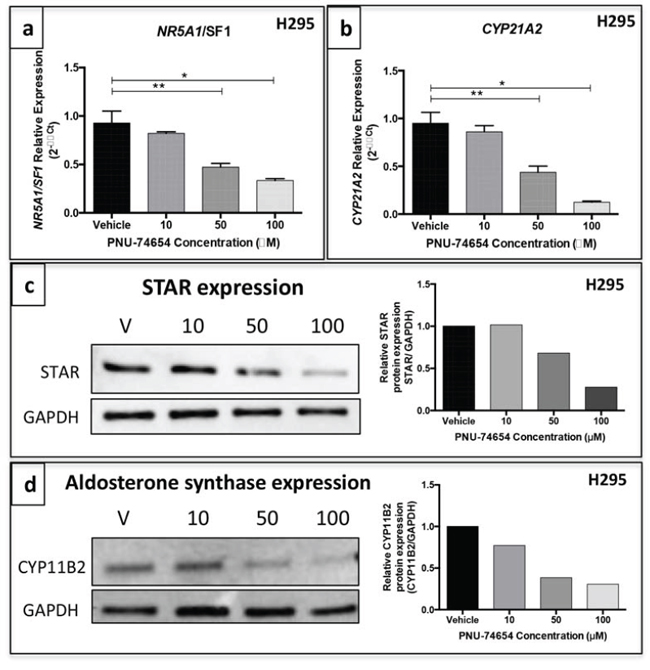
Figure 9: PNU-74654 treatment decreased the expression of steroidogenesis key regulatory enzymes in the NCI-H295 cell line. NR5A1/SF1 a. and CYP21A2 b. mRNA expression was decreased in NCI-H295 cells 48 h after treatment with 50 and 100 μM PNU-74654. *p < 0.0001; **p < 0.001. Values are reported as mean ± SEM. c. STAR protein expression in NCI-H295 cells 48 h after treatment with 10, 50 and 100 μM PNU-74654 showed by western blot. d. Aldosterone synthase protein expression in NCI-H295 cells 48 h after treatment with 10, 50 and 100 μM PNU-74654 shown by western blot. Values are plotted graphically. GAPDH was used as endogenous/loading control. V: vehicle.
DISCUSSION
In the present study we demonstrated the anti-proliferative, pro-apoptotic and anti-steroidogenic effects of the Wnt/beta-catenin pathway inhibition in ACC cell lines. In addition, we provided evidence that PNU may directly impair adrenal steroid secretion. To date, there is no effective adjuvant therapy for patients with advanced or metastatic ACCs. Very recently, a phase II clinical trial involving the suppression of the IGF system was tested in ACC patients but unfortunately the results were rather disappointing [22]. Hence, new approaches based on molecular targeted therapies are suitable.
Abnormal activation of the Wnt/beta-catenin pathway plays an important role in adrenocortical tumorigenesis [8, 10, 12, 15, 23]. Actually, Wnt/beta-catenin-related abnormalities appear to be the most common molecular signature of these tumors [8–10, 12, 14–16, 24]. In addition, beta-catenin constitutive activation in mice results in adrenal glands with malignant characteristics, such as uncontrolled neovascularization and loco-regional metastatic invasion [25]. In agreement, a previous study confirmed overexpression of Wnt target genes in ACTs harboring beta-catenin mutations as well as in the adrenal cell lineage NCI-H295. In that study, 24 h treatment with Wnt inhibitors, 10 μM PKF115-584 and 100 μM PNU, resulted in decreased beta-catenin target gene transcription [24]. However, the impact of these compounds on apoptosis, cell viability and steroidogenesis has not been assessed. Our study investigated in depth the effects of PNU on cell viability over a longer period of time (96 h). Moreover, we also determined its effects on apoptosis and adrenal steroidogenesis.
A previous study had shown that PKF115-584, currently called Calphostin C, dose-dependently inhibited beta-catenin-dependent transcription and H295R proliferation, and induced entry of cells into the S phase [19]. However the topological polar surface of PKF115-584 is 194 square angstrom (Å2), higher than recommended for good absorption (less than 140 Å2), characterizing this compound as poorly absorbed by cells [26]. On the other hand, the topological surface of PNU-74654 is 63.8 Å2 causing this compound to be very well absorbed by cells for a better effect on Wnt signaling inhibition (National Center for Biotechnology Information. PubChem Compound Database; CID = 60119583, https://pubchem.ncbi.nlm.nih.gov/compound/60119583 (accessed Aug. 12, 2015).) [27]. In our study, PNU was able to decrease cell viability since 24 h after treatment at higher but apparently non-cytotoxic concentrations (100 μM). After a longer time of treatment (72 h and 96 h) this effect was achieved even with a lower dose (10 μM). Additionally, we analyzed the impact of this drug under conditions of stimulation with Forskolin, which has a potent effect on adrenal cell proliferation. Forskolin alone increased cell proliferation by approximately 50%. In contrast, PNU prevented the cell proliferation stimulated by Forskolin. The effects of PNU appear to be partially specific for cells carrying Wnt/beta-catenin activation. Our results showed that PNU does not decrease cell proliferation in cells that do not carry exon 3 beta-catenin mutations, such as HeLa, a non-adrenal cell line, as previously reported, and Y1, a mouse adrenal tumor cell line [19].
In NCI-H295 cells, the analysis of apoptosis 48 h after PNU treatment showed a dose-dependent increase of apoptotic cells. These data support the proposal that inhibition of Wnt/beta-catenin signaling impairs cell proliferation mostly by increasing early and late apoptosis. In agreement, Doghman et al. showed that Wnt/beta-catenin inhibition by PKF115-584 repressed the entry of H295R cells into the S phase and induced their apoptosis [19]. In addition, an increase of TUNEL-positive cells, which indicates the presence of apoptotic cells, was described in Sf1/Crelowbeta-catenin knockout mice, supporting the idea that the loss of beta-catenin in the adrenal cortex progressively contributes to the loss of adrenocortical tissue via apoptosis [28].
We found reduced expression of beta-catenin at the mRNA and protein level 48 h after PNU treatment. In addition, we also found a reduction of nuclear beta-catenin accumulation in PNU-treated cells, as shown by immunofluorescence. Similarly, to previous data, our results show that shorter exposure to PNU (24 h) did not affect beta-catenin expression [24]. In agreement with our results, it has been reported that PNU was able to decrease beta-catenin protein expression in juvenile zebrafish after 10 days of exposure [29]. Taken together, these data show that PNU might have a later effect and appears to require a prolonged exposure to affect gene and protein expression. Thus, PNU may directly decrease beta-catenin expression as well as its nuclear localization by directly binding to beta-catenin. Interestingly, this effect occurred 48 h after treatment with 10 and 50 μM PNU, even before the reduction of cell viability. This finding suggests that increased cell proliferation in adrenal tumor cells is driven, at least in part, by increased Wnt/beta-catenin signaling, as suggested by transgenic mouse model studies [25]. In addition, silencing beta-catenin reduced cell proliferation in H295R in vitro and in a xenograft mouse model [30].
We also observed that PNU led to increased mRNA expression of the beta-catenin target genes CCND1 and AXIN2. In agreement, it has been shown that the Wnt/beta-catenin pathway inhibition resulted in increased AXIN2 mRNA expression in endometrial cells [31]. Furthermore, our data suggest a potential effect on beta-catenin-dependent transcription, a mechanism of action that has been reported for other inhibitors of the Tcf/beta-catenin complex [18, 32]. The absence of an effect of PNU on MYC mRNA, a well-known beta-catenin target gene, supports the hypothesis that MYC overexpression is common in many cancers but not in ACTs [8, 33]. TCF7 mRNA expression did not change in response to PNU treatment, supporting the hypothesis that this drug directly interferes with the Tcf/beta-catenin interaction [20].
The majority of ACCs secrete excessive amounts of cortisol, androgens or both, resulting in significant morbidity associated with excess hormone production [3]. Thus, an adjuvant therapy that, in addition to having antitumoral effects, is also able to directly decrease excessive hormone secretion is highly desirable [34]. Therefore, we also investigated the effects of inhibiting the Wnt/beta-catenin signaling on steroid production and steroidogenic enzyme gene expression. PNU treatment resulted in decreased cortisol, testosterone and, to a lesser extent, androstenedione secretion. Interestingly, adrenal steroidogenesis was impaired as early as 24 h after treatment, a time point where no decreased cell viability was yet observed. Hence, these results point to a potential direct effect of PNU on adrenal steroid secretion. This hypothesis is supported by the fact that PNU elicited reduction of corticosterone secretion but not of cell viability in Y1 cells. At higher doses it is also likely that the effect of PNU on cell decrease may contribute in part to decreased hormone production. On the other hand, we found that PNU treatment impaired the mRNA expression of SF1 and CYP21A2, both important genes required for steroidogenesis, suggesting that PNU directly impairs adrenal steroid secretion by inhibiting the initial steps of steroidogenesis.
SF1 is required for StAR gene expression, which is an early and limiting step in normal steroidogenesis. Accordingly, the finding of impaired StAR protein levels after PNU treatment in NCI-H295 cells reinforces this hypothesis [35, 36]. In addition, PNU treatment also decreased the protein levels of aldosterone synthase, encoded by the CYP11B2 gene. This result agrees with data reported by Berthon et al (2014), who showed that the inhibition of the Wnt pathway decreased hormone secretion, CYP11B2 and CYP21 expression after angiotensin stimulation [37]. Our findings support previous data regarding the importance of the Wnt/beta-catenin signaling in adrenal cortex homeostasis [28]. Mouse studies have suggested a critical role for beta-catenin in the maintenance of adrenal cortex progenitor cells. Partial knockout of beta-catenin in the adrenal cortex resulted in depletion of adrenocortical cells upon aging [30]. On the other hand, it has been recently suggested that the Wnt/beta-catenin signaling may control adrenal homeostasis by inhibiting fasciculata differentiation and promoting the undifferentiated state of progenitor cells, resulting in suppression of steroidogenesis [38].
There are some possible limitations to the present study. We evaluated the effects of PNU only on immortalized tumor cell lines, which are known to have possibly accumulated additional genetic defects and chromosomal instability over time. However, the NCI-H295 lineage is a good surrogate model for the analysis of the effect of the Wnt/beta-catenin signaling inhibition on ACC since it harbors the p.S45P beta-catenin mutation, as confirmed in our own stock of cells. This mutation leads to the activation of the constitutive Wnt/beta-catenin pathway [14, 15]. In addition, this lineage retains the ability to produce adrenal steroids, permitting the assessment of the effects of experimental treatments on hormonal secretion [17, 21]. One could argue that the PNU doses used were excessively high, namely the dose of 200 μM and possibly the dose of 100 μM, resulting in cytotoxic effects. However, these high doses did not result in cytotoxic effects in non-adrenal cells, such as HeLa, or in Y1 mouse adrenal cells. Furthermore, lower doses (10 and 50 μM) were able to impair H295 cell viability 96 h after treatment. These findings suggest a specific effect on cells exhibiting increased Wnt/beta-catenin signaling. In addition, we only evaluated in vitro effects and the results of this study do not ensure that the same effect will be obtained in in vivo models. The use of agents suppressing this pathway in clinical practice must be very cautious in view of toxicity and side effects since the Wnt signaling is a ubiquitous signaling pathway [32].
In conclusion, the results of this in vitro study demonstrated that blocking the Tcf/beta-catenin complex inhibited the Wnt/beta-catenin signaling as shown by decreased beta-catenin expression and nuclear accumulation, and impaired expression of beta-catenin target genes. These effects resulted in increased apoptosis, decreased tumor cell viability, and impairment of adrenal steroidogenesis. These promising findings may pave the way for further experiments inhibiting this pathway in pre-clinical models of ACCs. Taken together, these lines of evidence indicate that inhibitors of the Wnt/beta-catenin complex may become useful for the individualized treatment of patients with ACC presenting increased Wnt/beta-catenin signaling.
MATERIALS AND METHODS
This study was approved by the local Ethics Committee (HCFMRP-USP #43758/2013).
Adrenocortical tumor cell lines
The NCI-H295 and Y1 adrenocortical cell lines were kindly provided by Professor Claudimara Lotfi (University of Sao Paulo) [39]. HeLa cells were kindly provided by Dr. Paulo Peitl Jr, PhD and Dr. Beatriz Paixao, PhD (DNAapta®). The NCI-H295 human adrenal cell line was cultured in RPMI 1640 medium (GIBCO, Life Technologies, Foster City, CA) supplemented with 2% fetal bovine serum (Sigma Aldrich, St. Louis, MO, USA), 1% ITS Premix (BD Biosciences) and 1% penicillin/streptomycin (100 mg/mL; GIBCO Life Technologies) and harvested weekly. The Y1 mouse adrenal and HeLa human non-adrenal cell lines were cultured in DMEM medium (Sigma Aldrich) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and harvested every 2–3 days. All cell lines were cultured in monolayer and maintained under standard conditions at 37°C with 5% CO2. For all experiments, cell lines were harvested at the third passage. NCI-H295 and HeLa cell lines were authenticated by analyzing the following STR markers: CSF1PO, D13S317, D16S539, D5S818, D7S820, THO1, TPOX and vWA (Supplementary Table 1).
DNA extraction and genetic analysis of the CTNNB1 and TP53 genes
The analysis of exon 3 CTNNB1 and TP53 coding gene was performed to confirm the presence of previously described TP53 and CTNNB1 genetic variations in our own stock of NCI-H295 and HeLa cells. Genomic DNA was extracted from the cell lines using the QIAamp DNA Minikit (QIAGEN Inc., Valencia, CA). Exon 3 CTNNB1 and the TP53 coding gene were amplified by PCR and fragments were sequenced using the ABI Prism Big Dye Terminator v3.1 Cycle Sequencing Kit on an ABI 3130 instrument (Applied Biosystems, Life Technologies). PCR primers and conditions are available upon request.
PNU-74654 (PNU)
The PNU-74654 compound (Sigma Aldrich) was resuspended in dimethyl sulfoxide (DMSO; Sigma Aldrich) at stock concentrations of 31.2 mM. For working solutions, PNU-74654 was diluted 100X in growth medium with no serum deprivation and then diluted according to the required concentrations. The half-maximal inhibitory concentration (IC50) of PNU-74654 for each cell line is shown in Supplementary Table 3.
Treatment of cell lines with PNU-74654
NCI-H295 cells were plated at 200,000 cells per well in 24-well plates for gene expression, protein analysis and adrenal steroid measurements. After 48 h, cells were treated with vehicle (0.1%-0.4% DMSO) or 10, 50, 100 and 200 μM PNU-74654. After 24 and 48 h, medium supernatants were collected for adrenal steroid measurements. Adherent cells were fixed for immunofluorescence or harvested for RNA and protein isolation. At least three independent experiments were performed.
Y1 cells were plated at 200,000 cells per well in 24-well plates for gene expression and corticosterone measurement. After 24 h, cells were treated with vehicle or 10, 50, 100 and 200 μM PNU-74654. After 24 and 48 h, medium supernatants were collected for corticosterone measurements and adherent cells were harvested for RNA isolation. At least two independent experiments were performed.
HeLa cells were plated at 50,000 cells per well in 24-well plates for gene expression and protein analysis. After 24 h, cells were treated with vehicle or 50, 100 and 200 PNU-74654. After 24 and 48 h, adherent cells were harvested for RNA and protein isolation. At least two independent experiments were performed.
Protein isolation and western blot
For protein isolation, the cells were lysed after treatment by homogenization in 100 μL IP Lysing Buffer (Pierce, Thermo Scientific) and 1 μL of Halt Protease and Phosphatase inhibitor cocktail (Thermo Scientific). Protein concentration was measured by BCA protein assay (Pierce, Thermo Scientific). Equal amounts (20 μg) of protein were subjected to SDS-PAGE, transferred to nitrocellulose membranes, blocked in TBST-T containing 5% skim milk and probed with anti-beta-catenin (dilution: 1:1000; #610154, BD Biosciences), anti-TCF (dilution: 1:1000, #sc-101170; Santa Cruz Biotechnology), anti-STAR (dilution: 1:200, #sc-25806; Santa Cruz Biotechnology) and anti-CYP11B2 (dilution: 1:500, provided by Dr. Celso Gomez-Sanchez, University of Mississipi Medical Center [40]). Anti-GAPDH (dilution: 1:1000, #sc-47724; Santa Cruz Biotechnology) was used as endogenous/loading control. Complexes were visualized with HRP-conjugated anti-mouse (dilution: 1:3000; #sc-2005; Santa Cruz Biotechnology) and anti-rabbit (dilution: 1:2000; #sc-; Santa Cruz Biotechnology) secondary antibodies and developed by enhanced chemiluminescence (Immun-Star™ WesternC™ Chemiluminescence Kit) on a ChemiDoc XRS+ System (Bio-Rad™, Hercules, CA, USA). Acquired bands were analyzed using the Image Lab™ software (Bio-Rad).
Immunofluorescence
After treatment with PNU-74654, medium was collected and cells were fixed in methanol for 3 minutes, washed 3 times with 0.01 M PBS, and incubated with 10% normal horse serum for 1 h for blocking. Sections were then incubated overnight at room temperature with anti-beta-catenin primary antibody (dilution: 1:2000, #610154, BD Biosciences). Next, cells were washed 3 times with 0.01 M PBS, incubated with secondary antibody Cy™5-conjugated AffiniPure donkey anti-mouse antibody (Jackson Immuno Research, #715-175-150, dilution: 1:250; red color) for 4 h and washed 3 times with 0.01 M PBS. For nuclear counterstaining, cells were incubated with 4′,6-diamidino-2-phenylidone (DAPi; Cell Signaling Technology, #4083, dilution: 1:25,000) for 2 minutes, washed in 1X PBS, and slides were set with Fluoromount (Sigma-Aldrich). Expression and localization of beta-catenin were observed with a Leica TCS SP5 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) with fixed exposure time for all samples.
Immunostaining
To assess the beta-catenin cellular localization in the Y1 cell line, cells were seeded over a coverslip in a 24-well plate for 24 h and fixed in methanol for 3 minutes, washed 3 times with 0.01 M PBS, and incubated with Hydrogen Peroxide Block for 10 minutes followed by Super Block (REVEAL Biotin-Free Polyvalent HRP, AMS Biotechnology, Abingdon, UK) solution for 30 minutes for blocking. Sections were then incubated overnight 4°C with anti-beta-catenin primary antibody (dilution: 1:200, #610154, BD Biosciences). Next, cells were washed 3 times with 0.01 M PBS, incubated with Complement (REVEAL Biotin-Free Polyvalent HRP, AMS Biotechnology) for 10 min and washed twice with 0.01 M PBS. Cells were then incubated with HRP conjugate (REVEAL Biotin-Free Polyvalent HRP, AMS Biotechnology) for 15 min, washed 4 times with 0.01 M PBS and incubate with peroxidase substrate chromogen (DAB; brown color). Expression and localization of beta-catenin was observed with a Zeiss microscope (Axio observer inverted microscope, Carl Zeiss, Germany).
RNA isolation and qPCR
Total RNA from NCI-H295 cells was isolated using TRIzol® according to the manufacturer's protocol (Life Technologies). RNA concentrations were quantified by spectrometry (Nanodrop 2000, Thermo Fisher Scientific Inc., Waltham, MA, USA). RNA integrity was checked according to the 28S/18S ratio, with an acceptable range of 1.6 to 2.0 and confirmed by 1.2% agarose gel electrophoresis. mRNA was reverse transcribed from 500 ng of total RNA using the High Capacity cDNA Reverse Transcription kit and MultiScribe® enzyme (Life Technologies).
For quantitative Real-Time PCR (qPCR), TaqMan® assays (Life Technologies, Foster City, CA, USA) were used according to the manufacturer's recommendation using cDNA (diluted 1:5) as template. TaqMan® assays for NCI-H295 and HeLa cells: CTNNB1 (Hs00170025_m1), CCND1 (Hs00765553_m1), AXIN2 (Hs00610344_m1), MYC (Hs00153408_m1), TCF7 (Hs00175273_m1), NR5A1/SF1 (Hs00610436_m1) and CYP21A2 (Custom: AIVI3LV). TaqMan® assay for Y1 cells: Ctnnb1 (Mm01350391_m1). Relative expression values were determined by the 2−ΔΔCt method using GUSB (ID: 4326320E) as endogenous control.
Adrenal steroid measurement
Cortisol, testosterone and androstenedione concentrations were measured in the supernatant of the NCI-H295 cell medium and corticosterone concentrations were measured in the supernatant of the Y1 cell medium by radioimmunoassay (RIA) as previously described [41]. The effect of PNU effect on hormone secretion was analyzed as previously reported by Fassnacht et al. [42].
Cell proliferation assay
The CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corporation, Madison, WI, USA), an MTS-based assay, was used to determine cell viability before and after treatment with PNU. Briefly, cells were plated on 96-well plates at a density of 20,000 cells/well (NCI-H295) or 10,000 cells/well, incubated for 24 h (Y1 and HeLa) and 48 h (NCI-H295) and then treated with 5, 10, 50, 100 and 200 μM PNU in triplicate. After 24, 48, 72 and 96 h of incubation for NCI-H295 cells, 24 and 48 h of incubation for Y1 cells and 48 and 96 h of incubation for HeLa cells, 20 μL of CellTiter 96® Aqueous Solution was added to each well and incubated for 1–3 h at 37°C with 5% CO2 according to the manufacturer's datasheet. Absorbance at 490 nm was obtained from a microplate reader (BioRad). At least three independent experiments were performed for each cell line.
Stimulation of cell proliferation with forskolin
To analyze the impact of PNU under conditions of stimulation with Forskolin, NCI-H295 cells were plated on 96-well plates at a density of 20,000 cells/well, incubated for 48 h and then stimulated with 10 μM Forskolin (Sigma Aldrich) and treated with 5, 10, 50, 100 and 200 μM PNU in triplicate. After 96 h of stimulation/treatment, 20 μL of CellTiter 96® Aqueous Solution was added to each well and incubated for 2 h at 37°C with 5% CO2 according to the manufacturer's datasheet. Absorbance at 490 nm was obtained from a microplate reader. Three independent experiments were performed.
Detection of apoptosis with annexin V-FITC (flow cytometry)
NCI-H295 cells were plated at 350,000 cells per well in 6-well plates in triplicate. After 48 h, cells were treated with vehicle (0.08%, 0.16% and 0.32% DMSO) or with 10, 50 and 100 μM PNU. After 48 h, cells were harvested, triplicates were pooled, washed once in 0.01 M PBS and suspended in binding buffer (Sigma Aldrich) at a minimum of 1.106 total cells and assayed according to manufacturer's recommendations (Annexin V-FITC Apoptosis Detection Kit, Sigma Aldrich).
The procedure basically consists of the binding of annexin V-FITC to phosphatidylserine in the cell membranes, which are beginning the apoptotic process, and the binding of propydium iodide to cellular DNA in cells where the cell membrane has been totally compromised. The cells were incubated with annexin V-FITC and propydium iodide at room temperature for 10 min and then analyzed by flow cytometry. Annexin V-FITC is detected as green fluorescence and propydium iodide as red fluorescence. Results were analyzed with the BD CellQuest Prosoftware considering 10,000 events.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 software (GraphPad, San Diego, CA). One-way ANOVA followed by Dunnett's multiple comparison test was used to determine differences before and after treatment with all PNU concentrations. For analysis of hormone secretion, all values are reported as mean of percentage and standard error (SEM) relative to vehicle. The level of significance was set at p ≤ 0.05 in all analyses. Continuous variables are reported as mean and standard error (SEM).
ACKNOWLEDGMENTS
We thank Dr. Celso Gomez-Sanches (University of Mississipi Medical Center) for kindly providing the aldosterone synthase antibody. We thank Professor Claudimara Lotfi (Institute of Biomedical Sciences, University of Sao Paulo, ICB/USP) for providing the NCI-H295 and Y1 adrenal cell lines and Dr. Paulo Peitl Jr, PhD and Dr. Beatriz Paixao, PhD (DNAapta®) for providing the Hela cell line and Dr. Silvia G. Ruginsk and Dr. Rodrigo C. Rorato for their kind help with the Leica microscope. We acknowledge Glauco Ferro Leal, MChem (Brazilian Synchrotron Light Laboratory/Brazilian Center for Research in Energy and Materials - LNLS/CNPEM, Campinas, SP, Brazil; Institute of Chemistry of São Carlos/University of São Paulo - IQSC/USP, São Carlos, SP, Brazil), for providing support in Chemical physics. We thank Jose Roberto Silva, Wendy Turatti and Rogerio Zuliani (Laboratory of Endocrinology-Neuroendocrinology and Molecular Biology - HC-FMRP-USP) for technical support. We also acknowledge Camila Cristina Oliveria Menezes and Patrícia Vianna Bonini Palma (Flow citometry Laboratory, Hemotherapy Center of Ribeirao Preto) for technical support with the flow cytometer.
CONFLICTS OF INTEREST
The authors have nothing to declare.
GRANT SUPPORT
This research was supported by the Sao Paulo Research Foundation (FAPESP; LFL fellowship 2011/10512-3; Grants 2007/58365-3 and 2011/13807-4) and the National Council of Technological and Scientific Development (CNPq; Grant 474273-2013-3).
REFERENCES
1. Fassnacht M, Libe R, Kroiss M, Allolio B. Adrenocortical carcinoma: a clinician’s update. Nat Rev Endocrinol. 2011; 7:323–335.
2. Giordano TJ. Classification of adrenal cortical tumors: promise of the ‘molecular’ approach. Best Pract Res Clin Endocrinol Metab. 2010; 24:887–892.
3. Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ, Hammer GD. Adrenocortical carcinoma. Endocr Rev. 2014; 35:282–326.
4. Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, Sorbye H, Torpy DJ, Stepan V, Schteingart DE, Arlt W, Kroiss M, et al. Combination chemotherapy in advanced adrenocortical carcinoma. The New England journal of medicine. 2012; 366:2189–2197.
5. Gicquel C, Bertagna X, Schneid H, Francillard-Leblond M, Luton JP, Girard F, Le Bouc Y. Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. J Clin Endocrinol Metab. 1994; 78:1444–1453.
6. Almeida MQ, Fragoso MC, Lotfi CF, Santos MG, Nishi MY, Costa MH, Lerario AM, Maciel CC, Mattos GE, Jorge AA, Mendonca BB, Latronico AC. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab. 2008; 93:3524–3531.
7. Wilkin F, Gagne N, Paquette J, Oligny LL, Deal C. Pediatric adrenocortical tumors: molecular events leading to insulin-like growth factor II gene overexpression. The Journal of clinical endocrinology and metabolism. 2000; 85:2048–2056.
8. Leal LF, Mermejo LM, Ramalho LZ, Martinelli CE Jr, Yunes JA, Seidinger AL, Mastellaro MJ, Cardinalli IA, Brandalise SR, Moreira AC, Tone LG, Scrideli CA, Castro M, Antonini SR. Wnt/beta-catenin pathway deregulation in childhood adrenocortical tumors. J Clin Endocrinol Metab. 2011; 96:3106–3114.
9. Mermejo LM, Leal LF, Colli LM, Fragoso MC, Latronico AC, Tone LG, Scrideli CA, Tucci S, Martinelli CE, Yunes JA, Mastellaro MJ, Seidinger AL, Brandalise SR, Moreira AC, Ramalho LN, Antonini SR, et al. Altered expression of noncanonical Wnt pathway genes in paediatric and adult adrenocortical tumours. Clin Endocrinol (Oxf). 2014; 81:503–510.
10. Ragazzon B, Libe R, Gaujoux S, Assie G, Fratticci A, Launay P, Clauser E, Bertagna X, Tissier F, de Reynies A, Bertherat J. Transcriptome analysis reveals that p53 and {beta}-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res. 2010; 70:8276–8281.
11. Heaton JH, Wood MA, Kim AC, Lima LO, Barlaskar FM, Almeida MQ, Fragoso MC, Kuick R, Lerario AM, Simon DP, Soares IC, Starnes E, Thomas DG, Latronico AC, Giordano TJ, Hammer GD. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and beta-catenin. The American journal of pathology. 2012; 181:1017–1033.
12. Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, Libe R, Rene-Corail F, Audebourg A, Vacher-Lavenu MC, Groussin L, Bertagna X, Dousset B, Bertherat J, Tissier F. Wnt/beta-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. The Journal of clinical endocrinology and metabolism. 2011; 96:E419–426.
13. Gaujoux S, Grabar S, Fassnacht M, Ragazzon B, Launay P, Libe R, Chokri I, Audebourg A, Royer B, Sbiera S, Vacher-Lavenu MC, Dousset B, Bertagna X, Allolio B, Bertherat J, Tissier F. beta-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res. 2011; 17:328–336.
14. Tadjine M, Lampron A, Ouadi L, Bourdeau I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf). 2008; 68:264–270.
15. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, Rene-Corail F, Jullian E, Gicquel C, Bertagna X, Vacher-Lavenu MC, Perret C, Bertherat J. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005; 65:7622–7627.
16. Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, et al. Integrated genomic characterization of adrenocortical carcinoma. Nature genetics. 2014; 46:607–612.
17. Gazdar AF, Oie HK, Shackleton CH, Chen TR, Triche TJ, Myers CE, Chrousos GP, Brennan MF, Stein CA, La Rocca RV. Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer Res. 1990; 50:5488–5496.
18. Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer cell. 2004; 5:91–102.
19. Doghman M, Cazareth J, Lalli E. The T cell factor/beta-catenin antagonist PKF115-584 inhibits proliferation of adrenocortical carcinoma cells. J Clin Endocrinol Metab. 2008; 93:3222–3225.
20. Trosset JY, Dalvit C, Knapp S, Fasolini M, Veronesi M, Mantegani S, Gianellini LM, Catana C, Sundstrom M, Stouten PF, Moll JK. Inhibition of protein-protein interactions: the discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins. 2006; 64:60–67.
21. Rainey WE, Saner K, Schimmer BP. Adrenocortical cell lines. Mol Cell Endocrinol. 2004; 228:23–38.
22. Lerario AM, Worden FP, Ramm CA, Hesseltine EA, Stadler WM, Else T, Shah MH, Agamah E, Rao K, Hammer GD. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Hormones & cancer. 2014; 5:232–239.
23. El Wakil A, Lalli E. The Wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol. 2011; 332:32–37.
24. Durand J, Lampron A, Mazzuco TL, Chapman A, Bourdeau I. Characterization of differential gene expression in adrenocortical tumors harboring beta-catenin (CTNNB1) mutations. The Journal of clinical endocrinology and metabolism. 2011; 96:E1206–1211.
25. Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, Taketo MM, Tissier F, Bertherat J, Lefrancois-Martinez AM, Martinez A, Val P. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet. 2010; 19:1561–1576.
26. Palm K, Stenberg P, Luthman K, Artursson P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharmaceutical research. 1997; 14:568–571.
27. Information NCf B. PubChem Compound Database. NCBI, pp. PNU-74654 compound.
28. Kim AC, Reuter AL, Zubair M, Else T, Serecky K, Bingham NC, Lavery GG, Parker KL, Hammer GD. Targeted disruption of beta-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development. 2008; 135:2593–2602.
29. Pradhan A, Olsson PE. Juvenile ovary to testis transition in zebrafish involves inhibition of ptges. Biology of reproduction. 2014; 91:33.
30. Gaujoux S, Hantel C, Launay P, Bonnet S, Perlemoine K, Lefevre L, Guillaud-Bataille M, Beuschlein F, Tissier F, Bertherat J, Rizk-Rabin M, Ragazzon B. Silencing mutated beta-catenin inhibits cell proliferation and stimulates apoptosis in the adrenocortical cancer cell line H295R. PLoS One. 2013; 8:e55743.
31. Chandra V, Fatima I, Manohar M, Popli P, Sirohi VK, Hussain MK, Hajela K, Sankhwar P, Dwivedi A. Inhibitory effect of 2-(piperidinoethoxyphenyl)-3-(4-hydroxyphenyl)-2H-benzo(b)pyran (K-1) on human primary endometrial hyperplasial cells mediated via combined suppression of Wnt/beta-catenin signaling and PI3K/Akt survival pathway. Cell Death Dis. 2014; 5:e1380.
32. Sukhdeo K, Mani M, Zhang Y, Dutta J, Yasui H, Rooney MD, Carrasco DE, Zheng M, He H, Tai YT, Mitsiades C, Anderson KC, Carrasco DR. Targeting the beta-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc Natl Acad Sci U S A. 2007; 104:7516–7521.
33. Szabo PM, Racz K, Igaz P. Underexpression of C-myc in adrenocortical cancer: a major pathogenic event? Horm Metab Res. 2011; 43:297–299.
34. Daniel E, Newell-Price J. THERAPY OF ENDOCRINE DISEASE: Steroidogenesis enzyme inhibitors in Cushing’s syndrome. European journal of endocrinology / European Federation of Endocrine Societies. 2015; 172:R263–80.
35. Caron KM, Ikeda Y, Soo SC, Stocco DM, Parker KL, Clark BJ. Characterization of the promoter region of the mouse gene encoding the steroidogenic acute regulatory protein. Mol Endocrinol. 1997; 11:138–147.
36. Hu Y, Dong C, Chen M, Lu J, Han X, Qiu L, Chen Y, Qin J, Li X, Gu A, Xia Y, Sun H, Li Z, Wang Y. Low-dose monobutyl phthalate stimulates steroidogenesis through steroidogenic acute regulatory protein regulated by SF-1, GATA-4 and C/EBP-beta in mouse Leydig tumor cells. Reprod Biol Endocrinol. 2013; 11:72.
37. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro MC, Plouin PF, Skah S, Plateroti M, Lefebvre H, Sahut-Barnola I, Batisse-Lignier M, Assie G, Lefrancois-Martinez AM, et al. WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet. 2014; 23:889–905.
38. Walczak EM, Kuick R, Finco I, Bohin N, Hrycaj SM, Wellik DM, Hammer GD. Wnt signaling inhibits adrenal steroidogenesis by cell-autonomous and non-cell-autonomous mechanisms. Mol Endocrinol. 2014; 28:1471–1486.
39. Franca MM, Ferraz-de-Souza B, Santos MG, Lerario AM, Fragoso MC, Latronico AC, Kuick RD, Hammer GD, Lotfi CF. POD-1 binding to the E-box sequence inhibits SF-1 and StAR expression in human adrenocortical tumor cells. Molecular and cellular endocrinology. 2013; 371:140–147.
40. Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, Satoh F, Maekawa T, Nakamura Y, Sasano H, Gomez-Sanchez EP. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol. 2014; 383:111–117.
41. Moreira AC, Elias LL. Pituitary-adrenal responses to corticotropin-releasing hormone in different degrees of adrenal 21-hydroxylase deficiency. The Journal of clinical endocrinology and metabolism. 1992; 74:198–203.
42. Fassnacht M, Hahner S, Beuschlein F, Klink A, Reincke M, Allolio B. New mechanisms of adrenostatic compounds in a human adrenocortical cancer cell line. European journal of clinical investigation. 2000; 30:76–82.