
INTRODUCTION
Numerous molecularly targeted agents are now being developed for specific genomic aberrations, enabled by the efficient, rapid and accurate characterization of tumor genomes with next generation sequencing (NGS). For instance, non-small cell lung cancer (NSCLC) harboring EGFR mutations or ALK translocations and melanomas with BRAF mutations have been shown to be highly sensitive to the corresponding targeted kinase inhibition [1–3]. RAS mutations predict resistance to EGFR antibody therapy in colon cancer [4]. Subsequently, somatic mutation analysis of known or potential actionable oncogenes has now become part of the routine practice in medical oncology [5, 6]. As the number of genomic targets with matched therapies increases rapidly in the current oncology era, tissue biopsy material is now becoming an issue since genomic testing heavily relies on relatively small core or fine needle aspiration in metastatic patients [7, 8]. Until now, tumor tissue specimens have been the standard source of tumor DNA for clinical and research sequencing; however, acquisition of tumor tissue is not always feasible in patients with metastatic disease and may delay decision-making [9]. In addition, surgical or needle aspiration biopsy of visceral primary or metastatic tumors often are associated with significant medical costs and potential complications. Circulating blood biomarkers may constitute non-invasive real-time surrogates for diagnosis, prognosis, therapeutic tailoring, and resistance monitoring and mitigate needle biopsy sampling errors related to intra- or inter-tumor heterogeneity [10, 11]. For these reasons, sequencing of circulating cell-free DNA (cfDNA) has been suggested as a reasonable alternative to tumor tissue-based genomic testing [12–14].
In this study, we utilized a novel NGS panel of 54 clinically actionable genes utilizing digital sequencing of cell-free circulating tumor DNA isolated from a non-invasive blood draw (see Table S1 in the Supplementary). The test detects single nucleotide variants in all 54 genes and copy number amplifications in EGFR, ERBB2 (HER2) and MET [15]. We evaluated the concordance in genomic alterations between paired plasma cfDNA and primary tumor DNA (tDNA) samples using the same NGS method. We then conducted a prospective blinded validation of the targeted cfDNA panel via an inter-laboratory comparison of key oncogenes identified with tumor tissue using direct DNA sequencing (KRAS and BRAF) or hotspot analysis (KIT) to a second laboratory performing digital sequencing of cfDNA in corresponding plasma samples, while keeping the latter blind to the PCR reference standard results. Lastly, we tested the use of cfDNA as a follow-up monitoring of potential evolving mutations during cetuximab-based chemotherapy in metastatic colorectal cancer patients in an exploratory analysis.
RESULTS
Patient characteristics
Between February 2013 and March 2014, 75 advanced solid tumor patients were consented and enrolled in this study (clinicaltrials.gov, NCT02067754). Fourteen patients were excluded because of insufficient tissue for genomic analysis as illustrated in the STARD flowchart diagram [16] (Figure 1). Tumor section or biopsy of the primary tumor or metastasis and blood collection were conducted in all consented patients and the protocol was approved by the institutional review board. Table 1 provides the baseline patients characteristics. The most frequent cancer type was colorectal cancer (CRC) (n = 32, 52.6%), followed by melanoma (n = 13, 21.4%), gastrointestinal stromal tumor (GIST) (n = 4, 6.6%), renal cell carcinoma (RCC) (n = 3, 4.9%), gastric cancer (n = 3, 4.9%), sarcoma (n = 2, 3.2%), then 4 others with various cancer types. 87% of the patients had stage IV disease at the time of cfDNA analysis and most tDNAs (90.2%) were obtained from primary tumor sites. When dichotomized according to sampling interval between tumor tissue and blood sampling (synchronous sampling; sampling interval ≤ 6 months vs. metachronous sampling; sampling interval > 6 months), the majority of patients (71.9%) were in the synchronous sampling category. We included 14 clinical stage II colon cancer patients to compare primary tDNA and cfDNA to evaluate the concordance at the time of surgery, and also cfDNA 7-day post-surgery (10 patients) to detect the impact of surgical resection on cfDNA levels.
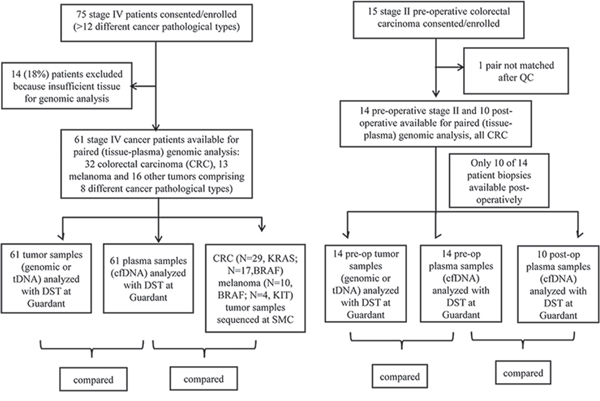
Figure 1: STARD diagram.
Table 1: Characteristics of metastatic cancer patients with genotyping analysis for paired tumor-tissue and cfDNA (N = 61)
Characteristic |
Number |
(%) |
|---|---|---|
Age (years) |
||
Median (range) |
57 |
(29–83) |
Sex |
||
Male |
39 |
(63.3) |
Female |
22 |
(37.3) |
Disease types |
||
Colorectal cancer |
32 |
52.6% |
Melanoma |
13 |
21.4% |
Gastrointestinal stromal tumor (GIST) |
4 |
6.6% |
Renal |
3 |
4.9% |
Gastric |
3 |
4.9% |
Sarcoma |
2 |
3.2% |
Bladder |
1 |
1.6% |
Neuroendocrine tumor |
1 |
1.6% |
Pancreatic cancer |
1 |
1.6% |
Thyroid cancer |
1 |
1.6% |
Total |
61 |
100% |
Pathologic stage |
||
Stage IV |
61 |
100% |
No. of metastatic sites |
||
1 |
11 |
(18.0) |
>=2 |
50 |
(82.0) |
Tumor sample origin |
||
Primary sites |
55 |
(90.2) |
Metastasis |
6 |
(9.8) |
Sampling interval between Tumor tissue and Blood |
||
Synchronous (≤ 6 months) |
34 |
(55.7) |
Metachronous (> 6 months) |
27 |
(44.3) |
Concordance between cfDNA and tDNA sequencing results
All 61 metastatic cancer patients with paired cfDNA and tDNA samples available were successfully sequenced with the DST method. For tDNA, a somatic mutation (clinically significant variants, and variants reported in COSMIC) was found in 44 samples (72.1%) while 17 samples (27.9%) had no significant genetic alterations in tDNA.
Figure 2A shows mutational profiles for cfDNA of 61 advanced cancer patients with various tumor types. For cfDNA, 29 samples (65.9%) had one or more somatic mutations with allele fractions ranging from 0.05%–53.4. The overall concordance rate between cfDNA and tDNA was 85.9%, when all detected mutations are considered (Figure 2B). Table 2 (see Table S2 in the Supplementary) shows mutational profiles of paired tumor tissue and cfDNA samples according to disease types.
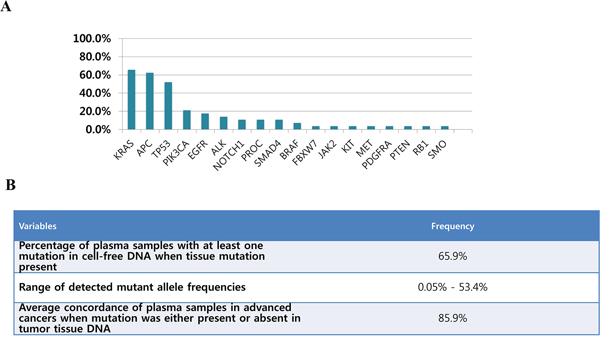
Figure 2: A. Mutational profiles (clinically significant variants, variants reported in COSMIC and other novel variants) detected in cfDNA for 61 advanced cancer patients with various tumor types and B. Details for genetic aberration analyzed in cfDNA and the concordance for comprehensive mutational profiles between tumor-tissue analysis and cfDNA.
Table 2: Two by two comparison tables for calculation of sensitivity, specificity and diagnostic accuracy for genomic aberrations in KRAS and BRAF between tumor tissues sequenced at Samsung Medical Center and blinded cfDNA analysis at Guardant Health for advanced colorectal cancer and BRAF for advanced melanoma patients
Tumor-tissue next generation sequencing is used as the reference standard but in Table (A) for KRAS mutation status tumor-tissue NGS is used first as the reference standard then tissue NGS is compared to cfDNA NGS as the reference standard for comparison purposes
(A) Colorectal cancer |
||||||
|---|---|---|---|---|---|---|
N = 29 |
Tumor-tissue based reference standard analysis |
|||||
KRAS (Exon 12, 13) |
Mutant |
WT |
Sensitivity |
Specificity |
Accuracy |
|
cfDNA NGS |
Mutant |
5 |
3 |
83.3% |
86.9% |
86.2% |
ND |
1 |
20 |
||||
Total |
6 |
23 |
||||
N = 17 |
BRAF V600E |
Mutant |
WT |
Sensitivity |
Specificity |
Accuracy |
cfDNA NGS |
Mutant |
1 |
0 |
100% |
100% |
100% |
ND |
0 |
16 |
||||
Total |
1 |
16 |
||||
(B) Melanoma |
||||||
|---|---|---|---|---|---|---|
N = 10 |
Tumor-tissue based reference standard analysis |
|||||
BRAF V600E |
Mutant |
WT |
Sensitivity |
Specificity |
Accuracy |
|
cfDNA NGS |
Mutant |
4 |
0 |
100% |
100% |
100% |
ND |
0 |
6 |
||||
Total |
4 |
6 |
||||
N = 4 |
Tumor-tissue based reference standard analysis |
|||||
KIT |
Mutant |
WT |
Sensitivity |
Specificity |
Accuracy |
|
cfDNA NGS |
Mutant |
0 |
0 |
- |
100% |
100% |
ND |
0 |
4 |
||||
Total |
0 |
4 |
||||
(WT = wild type in tissue, ND = Not Detected in cfDNA) (A) Colorectal cancer (B) Melanoma.
Prospective inter-laboratory blinded comparison of index test (cfDNA) to reference test (direct DNA sequencing and hotspot analysis)
For the specific mutations used in the selection of matched therapy, we compared conventional direct DNA sequencing method (KRAS, BRAF, KIT) as the reference standard to cfDNA results using the Guardant360 DST panel. The DST team was completely blinded to the direct DNA sequencing results from SMC. For KRAS codons 12 and 13 mutations in 29 metastatic CRCs, 83.3% sensitivity, 86.9% specificity and 86.2% accuracy (Table 2A) were revealed between cfDNA and direct DNA sequencing of tumor FFPE specimens. For BRAF V600E mutation in 17 metastatic CRCs, sensitivity, specificity and accuracy were all 100% (Table 2B) between cfDNA and direct DNA sequencing of tumor FFPE specimens. In 10 melanoma patients, BRAF V600E mutation resulted in 100% sensitivity, specificity and accuracy between cfDNA and direct DNA sequencing of tumor FFPE specimens. KIT mutations detected in tDNA of 4 melanomas were also observed in the cfDNA DST panel.
Monitoring of molecular resistance through sequencing assay using cfDNA sequencing
A 55 year-old man was diagnosed with metastatic CRC at SMC. The major metastatic lesions were hepatic, and also involved abdominal and presacral lymph nodes. Before starting systemic chemotherapy, mutational profiles of both primary tumor tissue and plasma were evaluated. Both tumor DNA and cfDNA sequencing demonstrated TP53 mutation and KRAS wild type (Figure 3A). cfDNAs were collected before cetuximab/FOLFIRI (5-FU/irinotecan/leucovorin) treatment, and at the time of computed tomography (CT) evaluation (every 4 cycles) thereafter. After 4 cycles of cetuximab/FOLFIRI chemotherapy, the follow-up CT scan revealed tumor shrinkage corresponding to partial response based on RECIST 1.1 criteria (Figure 3A). The patient continued to receive the same regimen for 6 months without definite radiologic or clinical progression. After 12 cycles of cetuximab/FOLFIRI, new KRAS mutation emerged in the patient's plasma cfDNA without evidence of radiologic progression. After 1.5 months from the time of newly emerged KRAS mutation emergence in cfDNA, the patient exhibited radiologic progression with sacral metastasis, showing persistence of the KRAS mutation in cfDNA (Figure 3A). Another patient with KRAS Q61H mutant on both tDNA and cfDNA had stable disease and a reduction in the cfDNA KRAS Q61H mutant allele frequency from 49.9% to 0.5% after four cycles of cetuximab/irinotecan (Figure 3B). However, this patient had a newly emerged PIK3CA H1047R mutation on cfDNA analysis at 2 months of cetuximab/irinotecan chemotherapy with allele frequency of 0.8%. Because the treatment response was within stable disease per RECIST 1.1 criteria, the patient received 4 more cycles of cetuximab/irinotecan. At 4 months CT evaluation (after 8 cycles), the patient had definite radiologic progression with enlarging liver metastases and parallel increase in allele frequency of PIK3CA H1047R mutation to 2.1%.
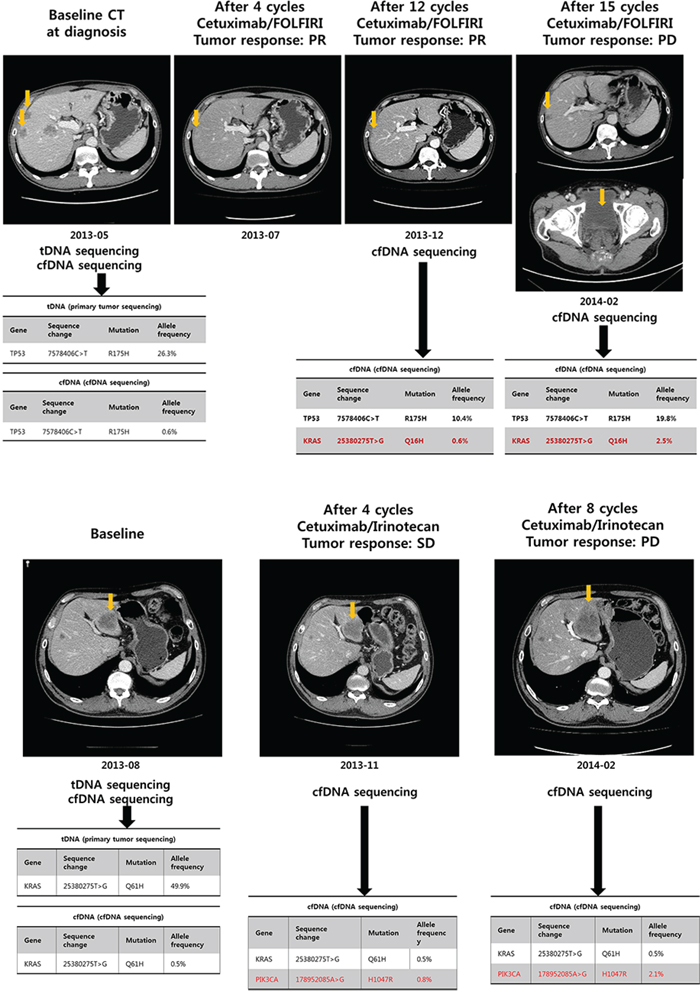
Figure 3: Patient monitoring with cfDNA during cetuximab-based treatment in metastatic colon cancer A. acquired resistance to cetuximab; B. primary resistance to cetuximab.
Analysis of cfDNA in stage II colorectal cancer patients
Fourteen patients with clinical stage II colorectal cancer received surgical treatment. Blood samples before (day 0) and after surgery (post-operative day 7) were analyzed with the cfDNA DST. The concordance rate for cfDNA to tDNA was 90.0% (95% CI, 66.7% – 98.6%) (see Figure S1A in the Supplementary). Among the ten patients with post-operative day 7 plasma samples, eight patients showed a dramatic reduction in cfDNA (see Figure S1B and Table S3 in the Supplementary). The recurrence data is being collected for this patient cohort.
DISCUSSION
In this prospective blinded study, the cfDNA DST panel revealed a very high sensitivity, specificity and accuracy compared with tissue-based reference standard analysis for KRAS and BRAF in CRC. Although the specificity for the DST for KRAS mutation in cfDNA compared to tissue sequencing as the reference standard analysis was 86.9%, it is likely that a cfDNA false positive reflects a tissue biopsy-based false negative, given that analytic specificity studies of DST against whole exome sequencing demonstrate near-perfect specificity. Considering DST of cfDNA as the gold standard, tissue biopsy-based KRAS mutation detection sensitivity as reference standard analysis was only 83.3%. The likely explanation for this is tumor intra- or inter-tumor heterogeneity not captured by core needle or surgical tissue biopsy sampling. However, cell-free DNA analysis did not detect the KIT mutations found in the six positive melanoma samples, perhaps reflecting that these tumors although at advanced stage, did not release tumor DNA into the circulation or false-negative results.
Analytic sensitivity for the cfDNA DST method via dilution studies has shown that a single DNA fragment with a somatic mutation can be detected in a background of 1,000 germline fragments (0.1% mutant allele fraction). Analytic specificity was also shown to be 99.9999% with a single false positive nucleotide result in nearly 1.6 million bases, covering the 54 genes in the panel (ref Lanman et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-free Circulating Tumor DNA, submitted). The negligible false positive rate for such a long (78 kbp) targeted region is the differentiating feature of this comprehensive cfDNA assay, relative to other tumor sequencing assays which typically manage the false positive rate by sequencing short regions of a small number of hotspots or hot exons in a few genes.
In metastatic CRC, 35% of patients harbored KRAS exon 2 mutations and 12% of patients harbored BRAF exon 15 mutations. Currently, KRAS and BRAF mutations are routinely tested in tumor tissue by various methods for selection of anti-EGFR therapy in metastatic CRC patients [17–22]. Although these tissue-based methods have proven clinical utility, they depend on the availability of tumor samples, as well as quality and the quantity of the tumor specimen. Tissue specimens must first undergo pathologic review to assure adequate tumor cell content and this process plus the sequencing workflow itself may have a long data turnaround (2–3 weeks at SMC). In contrast, 100% of cfDNA samples were successfully sequenced within 10 days using DST despite the transport distance between Korea and California. It has been shown that patients with tumor mutations in KRAS exon 2 as well as KRAS exons 3 or 4 or NRAS exon 2, 3, or 4 are likely to show resistance to anti-EGFR agents [23]. Thus, in order to select only the wild type RAS CRC population for anti-EGFR treatment, more rapid and efficient methods of genomic assessment are needed. Comprehensive cfDNA analysis using sequencing assay with DST might be a useful candidate method that could meet these needs.
The emergence of clinical resistance to previously effective anti-neoplastic therapy results from the acquisition of molecular alterations in genes or pathways that govern the resistant mechanisms. Defining these mechanisms of resistance to targeted agents is difficult because it is extremely difficult to acquire serial tumor biopsies in patients with advanced disease at multiple time points. In this study, we demonstrated a potential clinical application of cfDNA genomics which may allow detection of emergence of genomic alterations at acquired resistance to targeted therapy. Although it is an exploratory analysis, we attempted to monitor serial changes of mutational profiles for cfDNA in a KRAS wild-type CRC patient receiving cetuximab-based chemotherapy. In this patient, we observed that the emergence of KRAS mutation was associated with secondary resistance to cetuximab-based chemotherapy. Detection of the KRAS variant in cfDNA of this patient was ascertained before radiologic relapse. This finding is consistent to those of previous studies [24, 25]. Nevertheless, DST allows sequencing of 54 genes rather than hotspot mutations in cfDNA which provide broader opportunities to detect newly emerged genomic alterations
There have been limited studies utilizing NGS technologies for the detection of tumor somatic mutations in body fluids [26–29]. Narayan et al. reported that a deep sequencing assessment can be a useful strategy for the detection of low abundance point mutations in surrogate tissues [30]. Our study, assessing mutations in a comprehensive panel of genes and performed on patients with a large variety of tumor types, further extends the applicability of the NGS for analysis of aberrant genomic events in cfDNA.
Analytic sensitivity should not be confused with clinical sensitivity, however. Although the DST panel has high analytic sensitivity when DNA from cell lines with known mutations is spiked into plasma, it is limited by biology, i.e. the assay cannot measure cfDNA in patients whose tumors do not shed significant DNA into the circulation, such as stage I cancers or primary brain tumors that are isolated from systemic circulation by the blood-brain barrier. These factors limit clinical sensitivity of any cfDNA method. Despite this potential limitation, the concordance rate for all mutations found across 54 driver genes in various tumor types was 85.9% between tDNA and cfDNA.
This is the first blinded, prospective, external validation study to compare NGS of a comprehensive 54-gene panel using matched cfDNA and tDNA samples. We demonstrate a high concordance rate between cfDNA and tDNA and showed that cfDNA can be utilized as a monitoring tool for newly emerged mutations. The concordance rate for all mutations found across 54 genes in solid tumors was 85.9% between tDNA and cfDNA. In an exploratory analysis, we monitored serial changes of mutational profiles for cfDNA in two KRAS wild-type CRC patients receiving cetuximab-based chemotherapy. Detection of the KRAS variant in cfDNA of this patient was detected before radiologic relapse. Our study suggests the potential utility of cfDNA cancer panel as an alternative genomic test obviating the need for tumor biopsy at diagnosis and at resistance. Currently, we are testing the impact of cfDNA in refractory cancer patients on progression-free survival in the NEXT-2 trial (NCT#02140463).
MATERIALS AND METHODS
Patients
The institutional review board of the Samsung Medical Center (SMC) approved the study. All study participants provided written informed consent before study entry. Briefly, consented patients with metastatic cancer eligible for clinical trial enrollment or chemotherapy based on genomic biomarkers were eligible to enter the study. Patients with pathologically confirmed cancer and who had either archived tissue or fresh tissues were eligible for genomic analysis. The STARD flow diagram for in this study was summarized in Figure 1 [15]. For stage II colorectal cancer patients, baseline blood for cfDNA at the time of surgery in the operation room and at postoperative 7-day follow-up sample were drawn. For this patient cohort, surgical specimens were procured at the time of surgery for tDNA analysis.
Tumor samples
All tumor specimens (except for stage II colon) were paraffin embedded tumor tissues. Tumor areas (> 60%) were dissected under microscopy from 4-μm-thick unstained sections by comparison with an H&E stained slide, and genomic DNA was extracted using a Qiagen DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. After extraction, we measured concentrations and 260/280 and 260/230 nm ratios using a spectrophotometer (ND1000, Nanodrop Technologies, ThermoFisher Scientific, MA, USA). Each sample was then quantified with a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). Tumor tissue DNA (tDNA) was analyzed with direct DNA sequencing (KRAS and BRAF) and hotspot analysis (KIT) at SMC when quantity was sufficient. Simultaneously, at least 100 ng of tumor DNA (tDNA) was sent for next-generation sequencing utilizing Digital Sequencing™ technology (as described below) at Guardant Health, Inc. (Guardant).
Blood samples and circulating cell-free DNA isolation and quantification
For each enrolled patient, blood was collected during routine phlebotomy as part of standard cancer care. Blood samples were immediately processed upon receipt to isolate plasma. Plasma was isolated from EDTA tubes by centrifugation at 1,600 g during 10 minutes at 4°C. Plasma was aliquoted and stored at −70°C. CfDNA was extracted from aliquots (1 mL) of plasma using the QIAamp circulating nucleic acid kit (Qiagen) with the QIAvac 24 Plus vacuum manifold, following the manufacturer's instructions, and quantified by Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). All cfDNA analysis was performed at Guardant.
Digital sequencing technology (see Method in the Supplementary)
DNA sequencing for KRAS and BRAF and hotspot analysis (KIT)
KRAS mutation tests were performed at the designated central laboratory of SMC as described previously [31]. Mutations in codons 12 and 13 of the KRAS gene were detected by direct sequencing of polymerase chain reaction (PCR) products amplified from DNA extracted from representative tumor tissue. BRAF V600E direct sequencing and KIT hotspot mutations were tested according to our previous work [32]. Briefly, tumor-rich areas (>80%) were extracted from paraffin–embedded tissue sections, and 10 4-μm-thick sections containing a representative portion of each tumor block were subjected to DNA isolation using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Deeply pigmented samples were incubated with Chelex-100 (Bio-Rad Laboratories) to prevent PCR inhibition by melanin [33]. Purified DNA was incubated for 10 min at room temperature with an equal volume of a 5% Chelex-100 solution equilibrated in Qiagen AE buffer, heated to 95°C for 2 min, and allowed to cool. The Chelex-100 resin was pelleted in a microfuge, and the supernatant DNA used for PCR reactions. PCR products were processed for the DNA sequencing reaction using the ABI-PRISM BigDye Terminator version 3.1 (Applied Biosystems, Foster, CA, USA) with both forward and reverse sequence-specific primers. Sequence data were generated using the ABI PRISM 3100 DNA Analyzer (Applied Biosystems).
Statistics
First, sequencing of plasma cfDNA was compared to sequencing of tDNA, and the concordance rate between sequencing of cfDNA and tDNA was defined as the percentage agreement for all mutations found in a patient when both cfDNA and tDNA were both positive for mutations or both negative (tissue wild type or cfDNA “not detected”), then averaged for all 61 patients with matched plasma-tissue sample pairs. Secondly, clinical validity was evaluated for specific key oncogenic mutations according to tumor types such as KRAS and/or BRAF in colorectal cancer, and BRAF or KIT in melanoma. Sensitivity, specificity and diagnostic accuracy were calculated by comparing sequencing results for cfDNA to direct DNA sequencing (KRAS and BRAF) and hotspot analysis (KIT) performed at our laboratory (SMC) [34]. The Guardant Health laboratory remained blinded to the SMC reference standard results for these key oncogenic mutations and SMC conducted the unblinding and statistical analysis of results.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C1951, HI14C2188, HI14C3418). Support was also provided by a grant from the 20 by 20 project of Samsung Medical Center (GF01140111).
CONFLICTS OF INTEREST
None declared except for RBL, SM, HE, AT who are employed by Guardant Health Inc.
ROLE OF THE FUNDER/SPONSOR
The funders had no role in the design and conduct of the study.
Author contributions
Seung Tae Kim and Won-Suk Lee served as co-first authors, each with equal contribution to the manuscript
Study concept and design : Jeeyun Lee and AmirAli Talasaz
Acquisition, analysis or interpretation of data : Seung Tae Kim, Won-Suk Lee, Richard B. Lanman, Stefanie Mortimer, Oliver A. Zill, Kyoung-Mee Kim, Kee Taek Jang, Seok-Hyung Kim, Se Hoon Park, Joon Oh Park, Young Suk Park, Ho Yeong Lim, Helmy Eltoukhy, Won Ki Kang, Woo Yong Lee, Hee-Cheol Kim, and Keunchil Park.
Draft of the manuscript : Seung Tae Kim, Won-Suk Lee, Richard B. Lanman, Jeeyun Lee and AmirAli Talasaz
Administrative, technical, or material support : Richard B. Lanman, Stefanie Mortimer, Oliver A. Zill, Kyoung-Mee Kim, Kee Taek Jang, Seok-Hyung Kim, Helmy Eltoukhy, Jeeyun Lee and AmirAli Talasaz.
Study supervision : Richard B. Lanman, Jeeyun Lee and AmirAli Talasaz
Jeeyun Lee and AmirAli Talasaz had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis
REFERENCES
1. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
2. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809–819.
3. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363:1693–1703.
4. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008; 359:1757–1765.
5. Cardarella S, Ortiz TM, Joshi VA, Butaney M, Jackman DM, Kwiatkowski DJ, Yeap BY, Janne PA, Lindeman NI, Johnson BE. The introduction of systematic genomic testing for patients with non-small-cell lung cancer. J Thorac Oncol. 2012; 7:1767–1774.
6. Nowak F, Soria JC, Calvo F. Tumour molecular profiling for deciding therapy-the French initiative. Nat Rev Clin Oncol. 2012; 9:479–486.
7. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, Ishikawa Y, Wistuba I, Flieder DB, Franklin W, Gazdar A, Hasleton PS, Henderson DW, et al. Diagnosis of lung adenocarcinoma in resected specimens: implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch Pathol Lab Med. 2013; 137:685–705.
8. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, Ishikawa Y, Wistuba I, Flieder DB, Franklin W, Gazdar A, Hasleton PS, Henderson DW, et al. Diagnosis of lung cancer in small biopsies and cytology: implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch Pathol Lab Med. 2013; 137:668–684.
9. Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME, Abbruzzese JL, Tam AL. Use of research biopsies in clinical trials: are risks and benefits adequately discussed?. J Clin Oncol. 2013; 31:17–22.
10. Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013; 59:110–118.
11. Yun J, Lee J, Jang J, Lee EJ, Jang KT, Kim JH, Kim KM. KIT amplification and gene mutations in acral/mucosal melanoma in Korea. APMIS. 2011; 119:330–335.
12. Perkins G, Yap TA, Pope L, Cassidy AM, Dukes JP, Riisnaes R, Massard C, Cassier PA, Miranda S, Clark J, Denholm KA, Thway K, Gonzalez De Castro D, et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS One. 2012; 7:e47020.
13. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–1209.
14. Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, Higashiyama M, Kodama K, Imamura F, Kato K. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011; 17:7808–7815.
15. Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, Lijmer JG, Moher D, Rennie D, de Vet HC. Toward complete and accurate reporting of studies of diagnostic accuracy. The STARD initiative. Am J Clin Pathol. 2003; 119:18–22.
16. Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, Lijmer JG, Moher D, Rennie D, de Vet HC. Towards complete and accurate reporting of studies of diagnostic accuracy: The STARD Initiative. Radiology. 2003; 226:24–28.
17. Bando H, Yoshino T, Tsuchihara K, Ogasawara N, Fuse N, Kojima T, Tahara M, Kojima M, Kaneko K, Doi T, Ochiai A, Esumi H, Ohtsu A. KRAS mutations detected by the amplification refractory mutation system-Scorpion assays strongly correlate with therapeutic effect of cetuximab. Br J Cancer. 2011; 105:403–406.
18. Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011; 60:116–129.
19. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011; 29:2011–2019.
20. Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009; 27:5924–5930.
21. Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frebourg T, Michel P, Sabourin JC, Boissiere-Michot F. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009; 27:1122–1129.
22. Pinto P, Rocha P, Veiga I, Guedes J, Pinheiro M, Peixoto A, Pinto C, Fragoso M, Sanches E, Araujo A, Alves F, Coutinho C, Lopes P, et al. Comparison of methodologies for KRAS mutation detection in metastatic colorectal cancer. Cancer Genet. 2011; 204:439–446.
23. Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmuller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014; 15:1065–1075.
24. Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537–540.
25. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532–536.
26. Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, Siu SS, Chan WC, Chan SL, Chan AT, Lai PB, Chiu RW, Lo YM. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013; 59:211–224.
27. Tran B, Brown AM, Bedard PL, Winquist E, Goss GD, Hotte SJ, Welch SA, Hirte HW, Zhang T, Stein LD, Ferretti V, Watt S, Jiao W, et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer. 2013; 132:1547–1555.
28. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108–112.
29. Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O’Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B, Diaz LA Jr., Velculescu VE. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012; 4: 162ra154.
30. Narayan A, Carriero NJ, Gettinger SN, Kluytenaar J, Kozak KR, Yock TI, Muscato NE, Ugarelli P, Decker RH, Patel AA. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012; 72:3492–3498.
31. Kim ST, Lim do H, Jang KT, Lim T, Lee J, Choi YL, Jang HL, Yi JH, Baek KK, Park SH, Park YS, Lim HY, Kang WK1, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther. 2011; 10:1993–1999.
32. Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012; 30:2008–2014.
33. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008; 14:6821–6828.
34. Altman DG, Bland JM. Diagnostic tests 1: Sensitivity and specificity. BMJ. 1994; 308:1552.