
INTRODUCTION
Although mast cells (MCs) are known for rapid histamine secretion and degranulation in IgE-mediated immunity, it is now appreciated that MCs also contribute to cancer development (reviewed in [1–3]). In particular, MCs influence the polarity of TH1, TH2, and TH17 cells by regulating the maturation of antigen presenting cells [4, 5] and the release of proinflammatory cytokines [3, 6]. Proinflammatory cytokines produced by MCs may promote carcinogenesis through direct tumor stimulation as well as through their support of immune suppressive cells, such as regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSC) [6–9]. MDSCs arise from CD11b+Gr1+ immature myeloid cells (IMC) found within the tumor microenvironment. MDSCs promote carcinogenesis in part through T-cell suppression, which results in a loss in tumor immunity [10]. Recent studies suggest that MCs regulate the recruitment and immunoregulatory activity of MDSCs [11, 12]. MCs also play critical roles in epithelial cell activation, T helper cell polarization and intraepithelial stromal cell remodeling [13–16]. Given these many diverse functions, MCs could play an important role in promoting tumor development.
The intestine contains a broad range of IL-17-expressing cells, such as innate-like T cells, neutrophils, eosinophils, invariant natural killer T (iNKT) cells, αβ and γδ T cells, and lymphoid tissue inducer-like cells (LTi-like) cells [17–20]. Recently, MCs were also shown to produce IL-17 in human cancer [9, 21–23]. IL-17 is a major proinflammatory cytokine, and is known to stimulate myeloid cells, including tumor-promoting MDSCs [24, 25]. The mechanisms involved in the overproduction of IL-17 in the tumour microenvironment, however, have not been fully investigated.
Tissue histamine promotes angiogenesis, neurotransmission, gastric acid secretion, myeloid cell differentiation, and innate immunity [26, 27]. In addition, histamine has a well established role in modulating inflammatory reactions, including intestinal food allergy [31, 32]. MCs store and secret histamine yet are also regulated by histamine [33, 34]. Interestingly HDC, the enzyme generating endogenous histamine, is primarily expressed by CD11b+Gr1+ IMCs, rather than MCs [28]. IMCs are a group of innate immune cells that are often localized adjacent to MCs in the gut, and help to balance immune responses during persistent unresolved inflammation [10, 35]. In previous work, we found that knockout of HDC, the chief enzyme that generates histamine in CD11b+Gr1+ cells, increases MDSCs and tumorigenicity in the azoxymethane (AOM) and dextran sodium sulfate (DSS) model [28]. A number of studies have implicated allergy, a histamine-mediated disorder, in the modulation of cancer risk (reviewed in [29, 30]). We hypothesized that histamine would help protect patients with food allergy from developing colorectal cancer. We found that HDC gene deletion promoted colorectal carcinogenesis through increased MC production of IL-17, leading to an expansion of CD11b+Gr1+ MDSCs and suppression of CD8 cells. Interestingly, this increased tumor susceptibility was rescued by treatment with or replacement of histamine. This study identifies a histamine-dependent link between food allergy and colorectal carcinogenesis, and highlights a new pathway to target in the prevention and treatment of bowel cancer.
RESULTS
IL4/IL4Rα axis mediates WT but not HDC−/− OVA intestinal allergy
To better understand the interrelationship between histamine-secreting MC and HDC-expressing IMCs in food allergy and colorectal cancer, we treated WT C57BL/6 and HDC-EGFP mice with 30 ug i.g. and 50 ug i.p. of OVA daily for 10 consecutive days [36]. As expected, serum IL-4 levels and small intestinal mRNA expression of IL-4 receptor α chain (IL-4Rα) were significantly increased in OVA allergic mice (Figure 1A). In addition, in OVA treated HDC-EGFP mice, in which EGFP fluorescence identifies CD11b+Gr1+ HDC-expressing cells [28], we detected numerous circulating and intestinal EGFP+ myeloid cells (Figure 1B and 1C). Moreover, HDC mRNA levels were upregulated in sorted small intestinal CD11b+Gr1+ IMCs (Figure 1D).
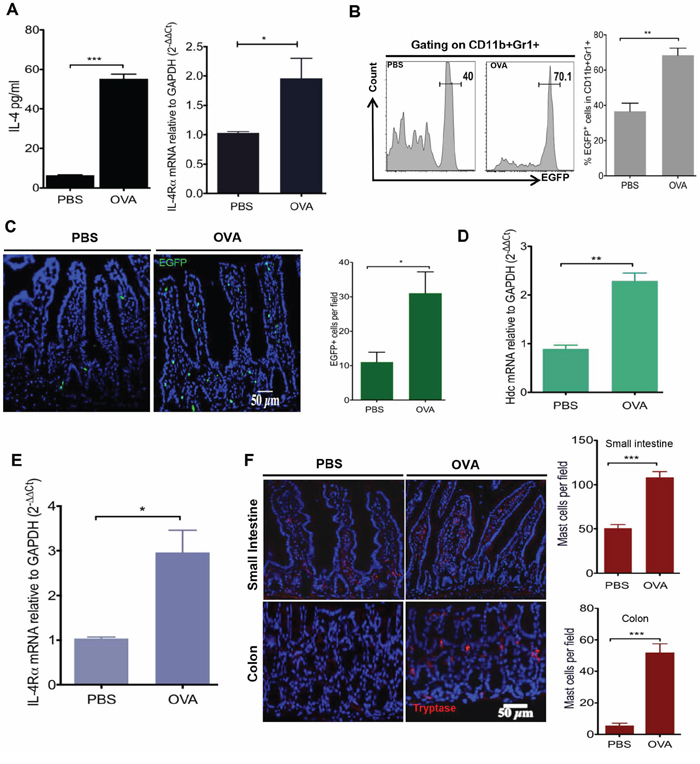
Figure 1: OVA intestinal allergy in WT mice induces the production of IL-4 and accumulation of HDC-expressed CD11b+Gr1+ cells. WT mice (n = 5/group) were immunized with OVA (30 ug i.g. and 50 ug i.p.) or PBS. A. Serum IL-4 levels were quantified by ELISA (Left); Intestinal IL-4Ra expression was exanimated by qRT-PCR (Right). B. Representative flow plots of circulating HDC-EGFP+ in blood CD11b+Gr1+ cells (Left). Bar graph (Right) illustrates the proportions of cell populations with statistics. C. Imagines show EGFP+ cells from OVA treated and PBS controls small intestine frozen sections (Left). EGFP+ cells were enumerated in each observed microscopic field (Right). Expression of Hdc mRNA D. and IL-4Rα E. from sorted small intestine CD11b+Gr1+ cells. F. Representative images of small intestine and colon immunofluorescence staining for tryptase+ MCs in HDC−/− mice treated with either OVA or PBS (Left). Tryptase+ MCs were counted per microscopic field (Right). Data are representative of at least two independent experiments.
It has been shown previously that TH2 cell derived IL-4 activates CD11b+Gr1+ cells through its receptor, IL4Rα [37, 38]. Consistently, we also detected elevated IL4Ra mRNA levels in small intestinal CD11b+Gr1+ cells of OVA-treated mice compared with PBS treated controls (Figure 1E). A causal link has been suggested between allergic histamine release and cytokine production of T helper cells, given that histamine receptor(s) knockout mice exhibit similar defects in T helper cell activation on common allergic signals [39–41]. To investigate whether histamine deficiency alters myeloid cell activation in the OVA intestinal allergy model, we crossed HDC knockout mice (HDC−/−) with HDC-EGFP and subjected them to the same OVA regimen. Although HDC−/− mice showed an attenuated OVA IgE response (Supplementary Figure S1A), surprisingly HDC-EGFP+ IMCs in the small intestine remained highly abundant (Supplementary Figure S1B), despite the absence of any increase in serum IL-4 levels (Supplementary Figure S1C). The CD11b+Gr1+ cells in the HDC−/− mice functionally resembled MDSCs, with 10-fold greater expression of Arginase 1 (Arg-1) compared to CD11b+Gr1+ cells from WT mice treated with the same OVA regimen (Supplementary Figure S1D). The differences between WT and HDC−/− mice in response to OVA immunization suggested that histamine might mediate the activity of HDC−/− CD11b+Gr1+ cells. This led us to investigate MCs, the principal histamine secreting cell type. In the non-immunized (PBS control) HDC−/− mice, we found only rare tryptase+ MCs in the small intestine and colon, consistent with previous data that MC frequency is decreased in the peripheral tissues of HDC−/− mice [42, 43]. In OVA treated mice, however, there was a substantial accumulation of tryptase+ intestinal MCs. Notably, the colon MC numbers were increased over 10-fold in the OVA immunized group compared to controls (Figure 1F). This was confirmed by flow cytometry (FcεRIα and c-kit staining, Supplementary Figure S1E). Collectively, these data suggest a link between HDC deficiency and the expansion of MCs and CD11b+Gr1+ cells during OVA allergy.
MC-derived IL-17 defines HDC−/− mice OVA allergy gut immunity
MC cytokine secretion recruits other myeloid cells and is a major contributor to inflammation [44, 45]. Although genetic deficiency of HDC impairs granule synthesis and homeostasis of MCs [42, 46], it is unknown whether HDC/histamine status affects MC cytokine production in an allergic setting. To address this question, cytokine gene expression in MCs from HDC−/− mice treated with OVA was assessed by qRT-PCR, including TH1 cytokines (IFN-γ, IL-12p40, and TNFα), TH2 cytokines (IL-4, IL-5, IL10, and IL-13), and proinflammatory cytokines (IL-1β, IL-23a, IL-6, and IL-17a). Our results revealed that in HDC−/− mice, there was marked induction by OVA of a number of MC-derived proinflammatory cytokines, including several key cancer-promoting cytokines such as IL-1β and IL-6 and their downstream effector cytokine IL-17a (Figure 2A). Notably, these cytokines were not upregulated by OVA allergy in WT littermates (Supplementary Figure S2A). Thus, OVA treatment in the setting of histamine deficiency expanded and activated proinflammatory intestinal MCs.
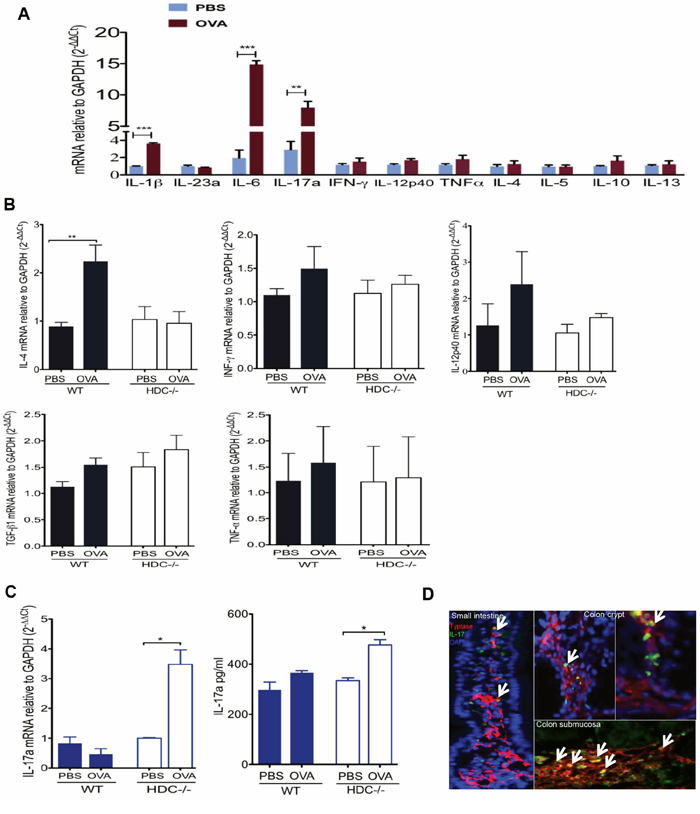
Figure 2: Accumulation of IL-17 producing MCs in HDC−/− mice OVA intestinal allergy. A. Comparison of HDC−/− intestinal MC TH1, TH2, and inflammatory cytokine genes expression between OVA and PBS treated groups (n = 4/group). B. IL-4, IFN-γ, IL-12p40, TGF-β1 and TNF-α expression in WT (n = 3) and HDC−/− (n = 5) small intestine tissue, mice treated either OVA or PBS. C. IL-17 elevated in HDC−/− mice (n = 5) but not WT mice (n = 5) treated with OVA, bar graphs show IL-17a mRNA expression in the small intestine (Left), and IL-17a protein level in sera (Right). D. Representative frozen slides co-stained with tryptase and IL-17a from OVA allergy HDC−/− mice (n = 3) in small intestine and colon. Arrows indicate tryptase+IL-17+ MCs. Data are representative of 2–3 independent experiments.
To assess the overall contribution of MC-derived proinflammatory cytokines to the gut immune response to OVA allergy, and the role of histamine in modulating this response qRT-PCR analysis was performed on small intestinal tissue samples following OVA treatment. These studies revealed that, OVA allergy induced an up-regulation of TH2 cytokines expression in WT mice but not in HDC−/− mice (Figure 2B). Instead, we observed much greater IL-17a expression in the intestine and in the circulation of HDC−/− mice (Figure 2C). Moreover, we identified a large number of tryptase+IL17a+ MCs in the small intestine and colon of HDC−/− OVA treated mice, particularly in the colon crypts and submucosa (Figure 2D). To exclude the possibility of IL-17 production from T cells (TH17 or Tc17 cells) [47], we assessed IL-17a mRNA expression in sorted small intestinal CD3+CD4+ and CD3+CD8+ T cells in response to OVA treatment. IL-17a expression remained unchanged in both populations of T cells in HDC−/− mice following OVA sensitization (Supplementary Figure S2B). Although OVA treatment also increased MCs numbers in WT colon (Supplementary Figure S2C), only HDC−/− colon MCs showed increased IL-17a mRNA levels upon OVA induction (Supplementary Figure S2D). These data confirm that MCs are the key IL-17 producing cell in the intestine following OVA challenge in histamine deficient mice.
Differentiation of IL-17-secreting MCs is regulated by histamine
To determine if histamine deficiency was largely responsible in the OVA allergy model for the increased production of MC IL-17, we attempted to rescue the phenotype through reconstitution with WT bone marrow-derived mast cells (BMMCs) or through exogenous administration of histamine. BMMCs from WT or HDC−/− donors were infused into non-irradiated HDC−/− mice. Daily OVA administration was begun one day after adoptive transfer, and mice were sacrificed 10 days later. Mice infused with WT MCs showed a significantly lower level of IL-17 expression in the small intestine compared to mice infused with HDC−/− MCs (Figure 3A). Exogenous histamine (5 mg/kg/d i.p.×1 mo) also significantly reduced intestinal FCεRIα+c-kit+ MCs in OVA sensitized mice (Figure 3B). Histamine treatment decreased serum IL-17 levels (Figure 3C) and small intestine MC IL-17 mRNA expression compared to the control OVA treated group (Figure 3D). We also assessed the effect of histamine on MC IL-17 production by adding 5 × 10–6 M histamine to HDC−/− MC co-cultured with splenocytes and OVA323–339 peptide. Again, we found that histamine reduced IL-17 secretion in HDC−/− MC ex vivo (Figure 3E). Taken together, these data suggest that histamine deficiency promotes MC expansion and production of IL-17, leading to higher serum IL-17 levels following an allergic challenge. The replacement studies establish histamine as necessary for inhibition of allergic MC accumulation and pro-inflammatory cytokine secretion.
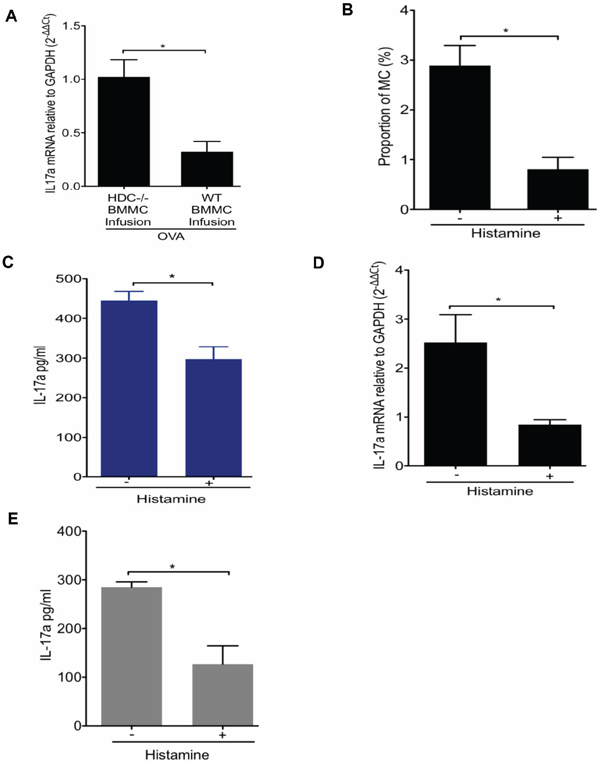
Figure 3: Effect of histamine on the regulation of IL-17-secreting MC. A. HDC−/− mice (n = 5/group) infused with either WT or HDC−/− BMMCs followed by OVA administration. OVA allergy mice infused with WT BMMCs had decreased IL-17a expression in small intestine. B. Exogenous histamine treatment lower the proportion of intestinal MC (FCεRIα+c-kit+) in HDC−/− mice (n = 3/group) OVA allergy. C. Serum IL-17a levels in HDC−/− OVA immunized mice (n = 5/group) with or without exogenous histamine injection. D. IL-17a mRNA level decreased in sorted FCεRIα+c-kit+ intestinal MCs from histamine treated, OVA immunized HDC−/− mice (n = 5/group). E. in vitro cultured HDC−/− MC and splenocytes induced with OVA323–339 peptide in the presence of histamine displayed reduced IL-17a level in supernatant. Data are representative of three independent experiments.
MC IL-17 promotes colorectal carcinogenesis in both subcutaneous and carcinogenesis models
Recent studies have suggested that proinflammatory IL-17 has a role in promoting colorectal tumorigenesis [24, 48]. Tumor-infiltrating MCs and MDSCs are commonly found in colorectal cancer (CRC) [3, 6, 12]. As we have identified MCs as a novel cellular source of IL-17 in OVA immunized HDC−/− mice, we sought to determine whether the IL-17-secreting MCs in HDC−/− mice promote colorectal carcinogenesis and whether CD11b+Gr1+ MDSCs are involved. To investigate this, we injected MC38 (C57BL/6) colon carcinoma tumor cells subcutaneously into HDC−/−;HDC-EGFP mice. One group of mice was immunized with OVA for 10d before subcutaneous inoculation of MC38 cells, followed by repeated daily doses of OVA for days 11–21 immediately prior to harvest. The control group of mice were sham immunized with PBS. We found that mice immunized with OVA had significantly larger tumors that contained a greater proportion of both MCs and HDC-GFP+ cells (Figure 4A and 4B). These findings suggested that in the setting of histamine deficiency, colorectal cancer is promoted by allergic immune response that involve accumulation of both MCs and MDSCs.
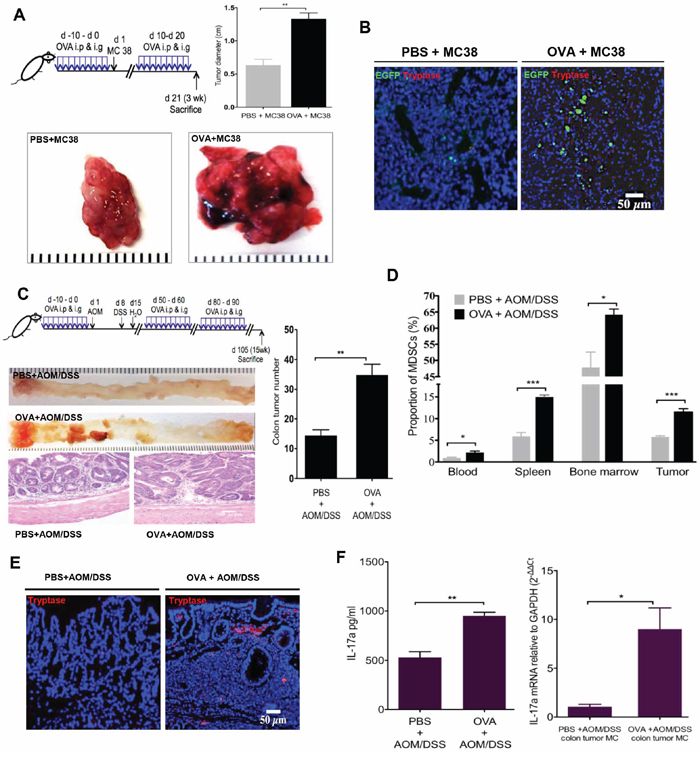
Figure 4: OVA intestinal allergy promote colorectal carcinogenesis in HDC−/− mice. A. Tumor diameter from MC38 colon carcinoma tumor cells injected HDC−/−;HDC-GFP mice. Mice (n = 8/group) treated with OVA exhibited bigger tumor diameter than PBS controls. B. Increased accumulation of MCs and HDC-EGFP+ myeloid cells within MC38 carcinoma in the group of mice treated with OVA. C. Schematic representation of the OVA immunization plus AOM/DSS carcinogenesis protocol (Upper panel). Representative macroscopic pictures and H&E staining images from paraffin embedded colon tissue of tumor mice (n = 10/group, Lower panel). The number of tumors with statistics presented by bar picture (Right). D. Flow cytometry analysis of CD11b+Gr1+ MDSCs proportion in CD45+ cells in OVA or PBS plus AOM/DSS treated mice (n = 10/group) in blood, spleen, bone marrow, and colon tumor. E. Representative tryptase immunofluorescence staining from tumor-bearing mice colon frozen sections. F. OVA immunization increases serum IL-17a level and IL-17a mRNA expression in sorted tumor MCs from AOM/DSS colorectal carcinogenesis mice (n = 10/group). Data are representative from two independent experiments.
We extended these tumorigenicity studies to a primary colorectal carcinogenesis model. We injected HDC−/− mice with 10 mg/kg of AOM, followed by 7 d of exposure to 2.5% DSS in the drinking water in order to generate colorectal tumors [28]. Before and after AOM/DSS treatment, we immunized mice with three 10d cycles of OVA or PBS. After the last cycle of OVA or PBS administration, IgE to OVA was only detectable in OVA treated group (Supplementary Figure S3A). Although both groups of mice developed colorectal tumors, OVA treated mice had significantly more colorectal tumors that exhibited higher grades of dysplasia and evidence of intramucosal carcinoma (Figure 4C). Furthermore, OVA-treated mice had a greater proportion of MDSCs in their bone marrow, spleen and circulation (Figure 4D). Notably, the number of infiltrating MCs within colorectal tumors was significantly higher in the OVA group (Figure 4E). Finally, serum IL-17a and tumor MC IL-17a mRNA levels were significantly increased after OVA plus AOM/DSS versus AOM/DSS alone (Figure 4F). Collectively, our results suggest that in histamine deficient mice, IL-17-expressing allergic MCs are capable of promoting CRC in part through recruitment of CD11b+Gr1+ cells.
HDC−/− MCs augment CD11b+Gr1+ immunosuppressive function
Given the marked expansion of CD11b+Gr1+ cells in OVA-induced tumors above, we wondered whether these cells are the immune-suppressive MDSCs known to promote cancer. To address this, we isolated splenocytes from OVA-induced HDC−/− mice and treated the splenocytes ex vivo with OVA323–339 peptide with or without the addition of HDC−/− BMMCs. After 96 h, CD11b+Gr1+ cells were flow sorted, and the expression of cyclooxygenase-2 (Cox-2), Arg-1, and Ki67 were determined by real-time qRT-PCR. Our data confirm that in the presence of OVA, HDC−/− BMMCs were able to induce proliferation and increase expression of Cox-2 and Arg-1 in CD11b+Gr1+ cells (Figure 5A), consistent with an MDSC phenotype. Additionally, secreted IL-17 was increased in the supernatant of OVA-treated splenocytes (Figure 5B). These results confirm that IL-17 expressing MCs are able to promote the proliferation of CD11b+Gr1+ cells and enhance their immunosuppressive function through upregulation of Cox-2 and Arg-1. These data also support a potential role for OVA-induced CD11b+Gr1+ myeloid cells in mediating suppressive effects on T cells, contributing to the observed increase in colorectal cancer.
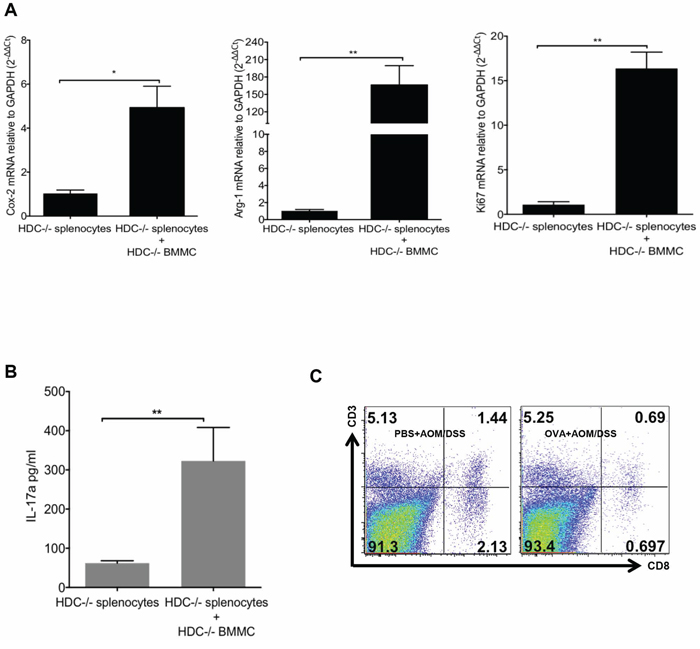
Figure 5: MCs support CD11b+Gr1+ MDSCs immunosuppression. A. Co-culture of BMMCs and splenocytes from HDC−/− mice in the presence of OVA323–339 peptide. 96 h after culture, CD11b+Gr1+ cells were sorted, expressions of Cox-2, Arg-1 and Ki67 were determined by qRT-PCR. B. Supernatant IL-17 was determined by ELISA. C. Representative flow plots show the frequency of CD3+CD8+ cytotoxic T cell in tumor margin colon tissue (n = 3). Data are representative from 3 independent experiments.
We also investigated the influence of IL-17-secreting MCs on MDSCs in the tumor microenvironment in vivo. We compared Cox-2, Arg-1, and Ki67 mRNA expression in tumor MDSCs in OVA immunized versus PBS-treated HDC−/− mice. Again, OVA treatment led to up-regulation of Cox-2, Arg-1 and Ki67 expression in MDSCs (Supplementary Figure S3B). Furthermore, OVA treated mice had fewer CD8+ T cells within the tumor margin compared to PBS treated controls (Figure 5C), suggesting that loss of cytotoxic T cells may be related to the promotion of carcinogenesis.
DISCUSSION
In this study, we found in a food allergy model an expansion of IL-17 producing MCs in the intestine. In the absence of histamine-mediated negative feedback, these primed MCs promoted the expansion of MDSCs, which in turn suppressed CD8+ T cells and enhanced colorectal carcinogenesis. The production and release of histamine by MDSC and MCs, respectively, represents an important brake on MDSCs and MC in intestinal allergy (Figure 6). This may be a central mechanism in atopic patients developing colorectal cancer, and thus a potential target for prevention and therapy of intestinal malignancy.
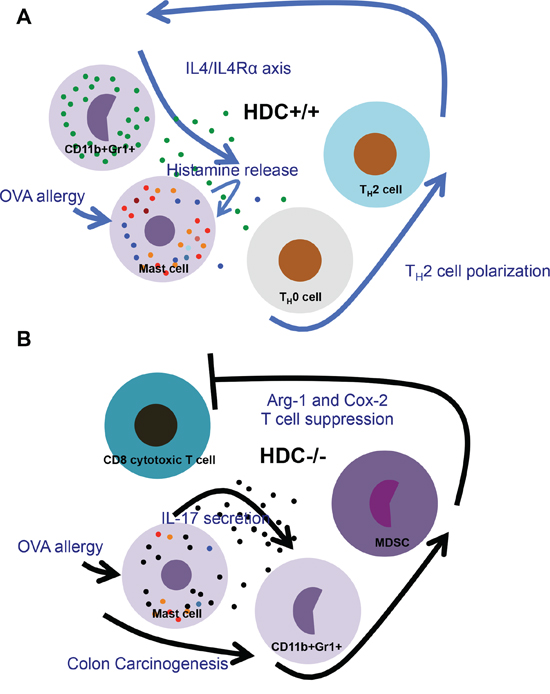
Figure 6: Schematic comparison of changes in HDC−/− vs WT intestine immunity during OVA allergy and following carcinogenesis. A. In WT gut, releasing of histamine supports the normal innate allergic reactions and adoptive T helper cell activation, therefore regulates MC function through feedback manner, which maintains the homeostasis of the gut immunity. B. Under histamine deficiency, proinflammatory IL-17 produced by MCs augments MDSCs, in particular, upregulates Arg-1 and Cox-2 in MDSCs, thus promotes colorectal carcinogenesis.
Previous studies have demonstrated that HDC expression defines the majority of CD11b+Gr1+ MDSCs. HDC-EGFP is also expressed in the majority of IMCs in normal (non tumor-bearing) mice, and these myeloid cells represent the primary site of histamine synthesis. While MCs do not express HDC-EGFP or synthesize histamine, they take up and store histamine, and release it in response to acute allergic stimuli [28]. MCs, however, do express histamine receptors and are thought to be tightly regulated by histamine [34, 42, 43, 46]. Thus, in HDC deficient mice that have low levels of circulating histamine, we show that OVA immunization leads to an expansion of intestinal IL-17 expressing MCs. This abnormal accumulation of IL-17-secreting MCs was clearly due to histamine deficiency, since adding exogenous histamine in vivo reduced MC numbers and serum IL-17 levels, and also reduced IL-17 secretion in ex vivo cultures. In addition, these abnormalities could be rescued by adoptive transfer of MCs from HDC competent hematopoietic cells.
The inflammatory cytokine IL-17, which is significantly elevated in gastrointestinal inflammation and cancer, was originally attributed primarily to a T cell (TH17) response [49, 50]. However, more recently, several types of innate immune cells have been found to be additional sources of IL-17 production in the regulation of gut immunity and gastrointestinal cancer [51, 52]. In human esophageal squamous cell carcinoma (ESCC), MCs were identified as the predominant IL-17 expressing cell type by immune microscopy [23]. In the intestine of OVA immunized HDC−/− mice, the expression level of IL-17 was exclusively elevated in sorted MCs but not in either CD4 or CD8 T cells. Thus, in the setting of histamine deficiency, MC cells are polarized into IL-17 secreting cells.
IL-17 production in MCs was associated with an accumulation of MDSCs. Previous studies have suggested a causal relationship between IL-17 production and the level of circulating MDSCs [18, 50, 53]. During parasitic infection, MCs are thought to stimulate responses by immature myeloid cells, helping to mobilize CD11b+Gr1+ cells to assist with the clearance of parasites [11]. Thus, it appears that the IL-17-expressing subset of MCs expanded in the setting of histamine deficiency is particularly effective in mobilizing and recruiting cancer-promoting MDSCs to the intestinal mucosa. OVA immunization of HDC−/− mice implanted with MC38 colon cancer cells led to significantly larger tumors with increased numbers of MCs and MDSCs. In the AOM/DSS colon carcinogenesis model, OVA immunization led to increased MCs and MDSCs, and increased colorectal tumors, which correlated with a reduction in CD8+ T cells.
Tumor-infiltrating MCs have been implicated in CRC pathogenesis and are associated with a poor prognosis in human colon cancer patients [54, 55]. In mouse models of inflammation-associated colonic cancer, MCs are abundant in the tumor tissue; in contrast, MC-deficient mice are protected from colitis-associated colorectal carcinogenesis [3]. In our study, we observed an accumulation of tumor-associated MCs in response to immunization and intestinal allergy in HDC−/− mice, which supports the notion that MC acts as an important modulator in mouse enteric allergy and prolonged allergic inflammation-associated carcinogenesis.
A number of questions remain regarding the relationships between food allergy and cancer, with contrasting observations noted in epidemiologic studies [29, 30, 56–59]. On the one hand, allergic reactions strengthen host immunity that provides a strong defense against attack from invading pathogens and aberrant cells, which likely enhances cancer immune surveillance [60]. In this sense, allergic responses support protective innate immune responses and T helper cell polarization [61]. On the other hand, some studies have suggested that prolonged host allergic immunity associated with the accumulation of chronic pro-inflammatory myeloid cells might predispose to tumor development [59]. We and others have previously shown a regulatory role for histamine in the differentiation of immature innate myeloid cells. In addition, mice deficient in histamine exhibit accelerated tumor progression, and epigenetic downregulation of HDC expression in CD11b+Gr1+ cells is associated with carcinogenesis [28]. Given the pivotal role of histamine in both allergy and cancer, our results suggest that histamine-mediated MC differentiation may critically link the two disorders.
In histamine deficient gut immunity, OVA allergic MCs promote tumorigenesis through the production of the proinflammatory cytokine IL-17, expansion of MDSCs and reduced activity of tumoricidal CD8+ T cells. Restoring histamine downregulates this pathway (Figure 6). Further studies will be needed to address whether histamine or histamine analogues could be used as a novel immune modulating cancer therapy, or if histamine receptor antagonism may be detrimental in certain patients at high risk for colorectal cancer.
MATERIALS AND METHODS
Mice
All animals were maintained under specific pathogen-free (SPF) conditions. 6–8 week age female mice: C57BL/6 WT mice, HDC-EGFP (C57BL/6 background, HDC+/+), HDC−/− mice (C57BL/6 background) in which intron 5 and exon 6 were replaced with a neomycin cassette, and HDC−/−;HDC-EGFP (C57BL/6 background, generated by crossing C57BL/6 HDC-EGFP mice with C57BL/6 HDC−/− mice) were involved in this study [28, 42]. In OVA immunization experiment, mice were administrated orally (i.g., 30 μg) and intraperitoneally (i.p., 50 μg) without adjuvant each day for 10 consecutive days. All experiment protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University.
Antibodies and reagents
OVA, histamine, and AOM were purchased from Sigma-Aldrich. OVA peptide (OVA323–339: ISQAVHAAHAEINEAGR) was obtained from AnaSpec. DSS was obtained from MP Biomedical. Anti-mast cell tryptase mouse monoclonal antibody was obtained from Abcam. Antibodies used for flow cytometry analysis and cell sorting and IL-17 immunofluorescence staining were obtained from Biolegend, including FCεRIα (MAR-1), c-kit (2B8), CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7), CD11b (M1/70), Gr1 (8C5), and IL17a (TC11–18H10.1).
Real-time quantitative RT-PCR and primers
PureLink® RNA Mini Kit or RNAqueous®-Micro Total RNA Isolation Kit were used for tissue RNA or sorted cell RNA isolation. cDNA was synthesized by using SuperScript® III First-Strand Synthesis System (Life Technologies, Grand Island, NY). Gene expression was determined using FastStart Universal SYBR Green or Probe Real-time qRT-PCR (Roche Applied Science, Indianapolis, IN). PrimeTime qRT-PCR Assays (Integrated DNA Technologies, Coralville, Iowa) were performed to measure mouse IL-17a (Mm.PT.56a.6531092) and Ki67 (Mm.PT.51.13130770) gene expression. Mouse GAPDH (Mm.PT.39a.1) was used as internal control. The sequences of primers for SYBR Green qRT-PCR are listed in Supplementary Table S1. Applied Biosystems Prism 9700 PCR machine (Applied Biosystems) was used for all real-time PCR experiments.
Cell culture
Mouse BMMCs were differentiated in vitro from isolated WT or HDC−/− mice tibia and femur bone marrow cells in RPMI 1640 medium (Invitrogen) supplemented with 1 mM sodium pyruvate (Gibco), 1 mM HEPES (Invitrogen), 2 mM L-glutamine, 1% penicillin-streptomycin (Gibco), and 10% fetal calf serum (FCS, Gibco), with the presence of 1 ng/ml recombinant mouse IL-3 (Biolegend) and 10 ng/ml mouse SCF (Biolegend). The percentage of MCs was determined by flow cytometry after 4 weeks of differentiation. Purity >95% FCεRIα+c-kit+ BMMCs were subjected to further experiments. In some experiments, BMMCs were co-cultured with HDC−/− splenocytes in the presence of 10 ng/ml GM-CSF (Biolegend); 5 × 10−6 M histamine and/or 5 μg/ml OVA323–339 peptide was added respectively for additional 96 h culture.
BMMC adoptive transfer
Differentiated BMMCs from WT or HDC−/− mice bone marrow cells were injected intravenously into syngeneic HDC−/− mice. OVA was administrated orally (30 μg) and intraperitoneally (50 μg) one day after MC transfer. 10 d after, mice were killed and exanimated form IL-17 expression.
Flow cytometry
Intraepithelial lymphocytes (IELs) were prepared from small intestine jejunum by twice 15 min mechanical vibrating in HBSS (Gibco) medium supplemented with 5% FBS, 2 mM EDTA (Invitrogen), 0.15 mg/ml DTT (Sigma-Aldrich), and 10 mM HEPES (Invitrogen). Small intestine Lamina propria (LP), colon and colon tumor tissue were digested respectively with collagenase IV (Worthington Biochemical Corp, Lakewood, NJ), dispase II (Gibco), and DNAse I (Roche). The resulting cells were subjected to 40% and 80% Percoll gradient (GE Healthcare, Pittsburgh, PA) centrifugation to enrich the mononuclear leucocytes. Blood and spleen cells were treated with red blood cell (RBC) lysis solution (Biolegend). Cells from pooled LP and IEL, blood, spleen, and bone marrow cells were passed through a 70 μm cell strainer to prepare monolayer cell suspension for multi-color flow cytometry antibodies staining and analysis. Flow cytometry was performed on LSRII flow cytometer (BD biosciences). Data were analyzed and presented using Flowjo 9 software (Tree Star, Ashland, OR). In some experiments, FCεRIα+c-kit+, CD3+CD4+, CD3+CD8+, or CD11b+Gr1+ cells were sorted from FACSAria cell sorter (BD bioscience) for further experiments.
ELISA
Mice were bled from the submandibular veins. Sera were separated by Microtainer tube with serum separator (BD, Franklin Lakes, NJ). The concentration of serum IL-4 or IL-17, and cell culture supernatant IL-17 were determined using mouse IL-4 or IL-17a ELISA MAX™ Deluxe kit (Biolegend) following the manufacturer's protocol. Serum OVA specific IgE was evaluated with Biolegend LEGEND MAX™ Mouse OVA Specific IgE ELISA Kit.
Immunofluorescence staining and histological analysis
Mice small intestinal and colonic tissue samples were collected and fixed with 4% (w/v) paraformaldehyde (PFA) and cryoprotected with 30% (w/v) sucrose for making tissue frozen sections. For paraffin sections, tissue samples were fixed with 10% (w/v) formalin and dehydrated in 70% ethanol. For mast cell tryptase staining, frozen sections were first treated with Mouse on Mouse (M.O.M.) Fluorescein Kit (Vector), and then stained with primary antibody (mouse tryptase, Abcam) and secondary antibody (Goat anti-Mouse IgG, Alexa Fluor® 594 conjugate, Life technologies). IL-17a was stained by a FITC-conjugated monoclonal antibody (Biolegend). Tumor paraffin sections were stained with hematoxylin and Eosin (H&E).
Statistical analysis
Data were presented as means ± SEMs. Statistic differences between different groups were assessed by two-tailed Student's t test with the GraphPad Prism 5 program (GraphPad Software, San Diego, CA). P < 0.05 was considered to be statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
ACKNOWLEDGMENTS AND FUNDING
We thank Kristie Gordon and Chenhong Liu for assisting in the flow cytometry analysis and cell sorting. We also appreciate Pavel Tishchenko, Yanan Ding, and Dr. Siu-Hong Ho from CCTI Flow Cytometry Core (supported in part by the Office of the Director, National Institutes of Health under awards S10OD020056 and S10RR027050).
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest.
GRANT SUPPORT
This work was supported by the grant from NIH to T.C.W (5R01DK048077-19).
Author contributions
X.C. and T.C.W. designed the studies, developed experimental method, and wrote the manuscript. X.C., M.J.C., T.C., Z.J., E.B.C.W., B.W.R., H.W., M.C.F. were involved in performing experiments, acquiring data, and interpreting result. K.K.N. and Y.T. maintained transgenic mouse colonies and supplied reagents. M.J.C., B.S.R., D.L.W., and S.M. were involved in experimental design, manuscript drafting and editing. T.C.W. supervised all studies and received funding.
REFERENCES
1. Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. European journal of immunology. 2010; 40:1843–1851.
2. Liu J, Zhang Y, Zhao J, Yang Z, Li D, Katirai F, Huang B. Mast cell: insight into remodeling a tumor microenvironment. Cancer and Metastasis Reviews. 2011; 30:177–184.
3. Tanaka T, Ishikawa H. Mast cells and inflammation-associated colorectal carcinogenesis. Semin Immunopathol. 2013; 35:245–254.
4. Mazzoni A, Siraganian RP, Leifer CA, Segal DM. Dendritic cell modulation by mast cells controls the Th1/Th2 balance in responding T cells. J Immunol. 2006; 177:3577–3581.
5. Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M. Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol. 2011; 41:1883–1893.
6. Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, Weitz J, Koch M, Halverson AL, Bentrem DJ, Khazaie K. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:6430–6435.
7. Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, Ogawa H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. 2002; 109:1351–1359.
8. Saleem SJ, Martin RK, Morales JK, Sturgill JL, Gibb DR, Graham L, Bear HD, Manjili MH, Ryan JJ, Conrad DH. Cutting edge: mast cells critically augment myeloid-derived suppressor cell activity. J Immunol. 2012; 189:511–515.
9. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011; 187:490–500.
10. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews Immunology. 2012; 12:253–268.
11. Martin RK, Saleem SJ, Folgosa L, Zellner HB, Damle SR, Nguyen GK, Ryan JJ, Bear HD, Irani AM, Conrad DH. Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells. J Leukoc Biol. 2014; 96:151–159.
12. Danelli L, Frossi B, Gri G, Mion F, Guarnotta C, Bongiovanni L, Tripodo C, Mariuzzi L, Marzinotto S, Rigoni A, Blank U, Colombo MP, Pucillo CE. Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer immunology research. 2015; 3:85–95.
13. Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. 2010; 5:e11944.
14. Heger K, Fierens K, Vahl JC, Aszodi A, Peschke K, Schenten D, Hammad H, Beyaert R, Saur D, van Loo G, Roers A, Lambrecht BN, Kool M, Schmidt-Supprian M. A20-deficient mast cells exacerbate inflammatory responses in vivo. PLoS Biol. 2014; 12:e1001762.
15. Kenna TJ, Brown MA. The role of IL-17-secreting mast cells in inflammatory joint disease. Nature reviews Rheumatology. 2013; 9:375–379.
16. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013; 121:4930–4937.
17. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, Dekruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014; 20:54–61.
18. Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, Strieter RM, Rosin DL, Okusa MD. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010; 120:331–342.
19. Kim JS, Jordan MS. Diversity of IL-17-producing T lymphocytes. Cell Mol Life Sci. 2013; 70:2271–2290.
20. Jones SA, Sutton CE, Cua D, Mills KH. Therapeutic potential of targeting IL-17. Nat Immunol. 2012; 13:1022–1025.
21. Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond). 2012; 122:487–511.
22. Jain P, Javdan M, Feger FK, Chiu PY, Sison C, Damle RN, Bhuiya TA, Sen F, Abruzzo LV, Burger JA, Rosenwald A, Allen SL, Kolitz JE, Rai KR, Chiorazzi N, Sherry B. Th17 and non-Th17 interleukin-17-expressing cells in chronic lymphocytic leukemia: delineation, distribution, and clinical relevance. Haematologica. 2012; 97:599–607.
23. Wang B, Li L, Liao Y, Li J, Yu X, Zhang Y, Xu J, Rao H, Chen S, Zhang L, Zheng L. Mast cells expressing interleukin 17 in the muscularis propria predict a favorable prognosis in esophageal squamous cell carcinoma. Cancer Immunol Immunother. 2013; 62:1575–1585.
24. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W, Ferrara N. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nature medicine. 2013; 19:1114–1123.
25. Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, Chakravarty P, Thompson RG, Kollias G, Smyth JF, Balkwill FR, Hagemann T. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009; 119:3011–3023.
26. Wood JD. Histamine, mast cells, and the enteric nervous system in the irritable bowel syndrome, enteritis, and food allergies. Gut. 2006; 55:445–447.
27. Xie H, He SH. Roles of histamine and its receptors in allergic and inflammatory bowel diseases. World J Gastroenterol. 2005; 11:2851–2857.
28. Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G, Park H, Shykind B, Diacovo TG, Falus A, Wang TC. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med. 2011; 17:87–95.
29. Merrill RM, Isakson RT, Beck RE. The association between allergies and cancer: what is currently known? Ann Allergy Asthma Immunol. 2007; 99:102–116. quiz 117–109, 150.
30. Engkilde K, Thyssen JP, Menne T, Johansen JD. Association between cancer and contact allergy: a linkage study. BMJ open. 2011; 1:e000084.
31. Smolinska S, Jutel M, Crameri R, O’Mahony L. Histamine and gut mucosal immune regulation. Allergy. 2014; 69:273–281.
32. Santos J, Bayarri C, Saperas E, Nogueiras C, Antolin M, Mourelle M, Cadahia A, Malagelada JR. Characterisation of immune mediator release during the immediate response to segmental mucosal challenge in the jejunum of patients with food allergy. Gut. 1999; 45:553–558.
33. Wiener Z, Buzas E, Kovacs P, Csaba G, Szabo D, Kittel A, Pallinger E, Watanabe T, Ohtsu H, Ichikawa A, Nagy A, Falus A. Highly reduced peritoneal mast cell number and decreased c-kit expression in histidine decarboxylase knock out mice. Inflamm Res. 2001; 50:S55–56.
34. Mirzahosseini A, Dalmadi B, Csutora P. Histamine receptor H4 regulates mast cell degranulation and IgE induced FcepsilonRI upregulation in murine bone marrow-derived mast cells. Cell Immunol. 2013; 283:38–44.
35. Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, Blatner NR, Hix LM, Zhang M, Dennis KL, Salabat MR, Heiferman M, Grippo PJ, Munshi HG, Gounaris E, Bentrem DJ. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCDelta468 mice. Cancer Res. 2011; 71:1627–1636.
36. Yang PC, Xing Z, Berin CM, Soderholm JD, Feng BS, Wu L, Yeh C. TIM-4 expressed by mucosal dendritic cells plays a critical role in food antigen-specific Th2 differentiation and intestinal allergy. Gastroenterology. 2007; 133:1522–1533.
37. Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001; 166:2173–2177.
38. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003; 170:270–278.
39. Saligrama N, Case LK, Krementsov DN, Teuscher C. Histamine H(2) receptor signaling x environment interactions determine susceptibility to experimental allergic encephalomyelitis. FASEB J. 2014; 28:1898–1909.
40. Dunford PJ, O’Donnell N, Riley JP, Williams KN, Karlsson L, Thurmond RL. The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J Immunol. 2006; 176:7062–7070.
41. Noubade R, Milligan G, Zachary JF, Blankenhorn EP, del Rio R, Rincon M, Teuscher C. Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J Clin Invest. 2007; 117:3507–3518.
42. Ohtsu H, Tanaka S, Terui T, Hori Y, Makabe-Kobayashi Y, Pejler G, Tchougounova E, Hellman L, Gertsenstein M, Hirasawa N, Sakurai E, Buzas E, Kovacs P, Csaba G, Kittel A, Okada M, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001; 502:53–56.
43. Wiener Z, Andrasfalvy M, Pallinger E, Kovacs P, Szalai C, Erdei A, Toth S, Nagy A, Falus A. Bone marrow-derived mast cell differentiation is strongly reduced in histidine decarboxylase knockout, histamine-free mice. Int Immunol. 2002; 14:381–387.
44. Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, Strom TB, Zheng XX, Noelle RJ. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006; 442:997–1002.
45. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, Huang B. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One. 2010; 5:e8922.
46. Nakazawa S, Sakanaka M, Furuta K, Natsuhara M, Takano H, Tsuchiya S, Okuno Y, Ohtsu H, Nishibori M, Thurmond RL, Hirasawa N, Nakayama K, Ichikawa A, Sugimoto Y, Tanaka S. Histamine synthesis is required for granule maturation in murine mast cells. Eur J Immunol. 2014; 44:204–214.
47. Zhao Y, Balato A, Fishelevich R, Chapoval A, Mann DL, Gaspari AA. Th17/Tc17 infiltration and associated cytokine gene expression in elicitation phase of allergic contact dermatitis. Br J Dermatol. 2009; 161:1301–1306.
48. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012; 491:254–258.
49. Ericksen RE, Rose S, Westphalen CB, Shibata W, Muthupalani S, Tailor Y, Friedman RA, Han W, Fox JG, Ferrante AW Jr., Wang TC. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut. 2014; 63:385–394.
50. Yazawa T, Shibata M, Gonda K, Machida T, Suzuki S, Kenjo A, Nakamura I, Tsuchiya T, Koyama Y, Sakurai K, Shimura T, Tomita R, Ohto H, Gotoh M, Takenoshita S. Increased IL-17 production correlates with immunosuppression involving myeloid-derived suppressor cells and nutritional impairment in patients with various gastrointestinal cancers. Molecular and clinical oncology. 2013; 1:675–679.
51. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature reviews Immunology. 2010; 10:479–489.
52. Wu D, Wu P, Huang Q, Liu Y, Ye J, Huang J. Interleukin-17: a promoter in colorectal cancer progression. Clinical & developmental immunology. 2013; 2013:436307.
53. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, Xia W, Chen Z, Wang K, Zhang T, Xu J, Han Y, Zhang T, et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014; 40:785–800.
54. Wu X, Zou Y, He X, Yuan R, Chen Y, Lan N, Lian L, Wang F, Fan X, Zeng Y, Ke J, Wu X, Lan P. Tumor-infiltrating mast cells in colorectal cancer as a poor prognostic factor. International journal of surgical pathology. 2013; 21:111–120.
55. Malfettone A, Silvestris N, Saponaro C, Ranieri G, Russo A, Caruso S, Popescu O, Simone G, Paradiso A, Mangia A. High density of tryptase-positive mast cells in human colorectal cancer: a poor prognostic factor related to protease-activated receptor 2 expression. Journal of cellular and molecular medicine. 2013; 17:1025–1037.
56. Vesterinen E, Pukkala E, Timonen T, Aromaa A. Cancer incidence among 78,000 asthmatic patients. International journal of epidemiology. 1993; 22:976–982.
57. Prizment AE, Folsom AR, Cerhan JR, Flood A, Ross JA, Anderson KE. History of allergy and reduced incidence of colorectal cancer, Iowa Women’s Health Study. Cancer Epidemiol Biomarkers Prev. 2007; 16:2357–2362.
58. Rittmeyer D, Lorentz A. Relationship between Allergy and Cancer: An Overview. International Archives of Allergy and Immunology. 2012; 159:216–225.
59. Shadman M, White E, De Roos AJ, Walter RB. Associations between allergies and risk of hematologic malignancies: Results from the VITamins and lifestyle cohort study. American Journal of Hematology. 2013; 88:1050–1054.
60. Wang H, Diepgen TL. Atopic dermatitis and cancer risk. Br J Dermatol. 2006; 154:205–210.
61. Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012; 484:465–472.