
INTRODUCTION
Invasive carcinoma, a common breast malignancy, is a major cause of cancer-related deaths in women worldwide [1]. Breast cancer is a heterogeneous disease. Molecularly distinct subtypes have different characters of biological behaviors and prognoses [2]. Immunohistochemical evaluation of estrogen receptor (ER), progesterone receptor (PgR), Ki-67, and human epidermal growth factor receptor 2 (HER2) is commonly used to identify the four molecular subtypes: HER2-type, luminal-type, luminal/HER2-type, and triple negative (TN)-type. Importantly, the subtypes have different clinical outcomes [3–6]. In particular, TN breast cancer (TNBC) is associated with an increased risk of early relapse, usually within 5 years from the initial diagnosis [7]. Unlike tumors that overexpress hormone receptors and HER2, TNBC cannot be treated with hormone therapy or trastuzumab, an anti-HER2 monoclonal antibody, because TNBC tumor cells lack these specific targets [8, 9]. Consequently, patients with TNBC have worse outcomes after chemotherapy than patients with other breast cancer subtypes [10]. Thus, novel therapies that target highly malignant TNBC are needed to improve breast cancer prognosis.
Cancer stem cells (CSCs) are thought to contribute to tumor initiation and maintenance, chemotherapy resistance, tumor relapse, and metastasis [11–14]. Therefore, for the successful treatment of many types of cancer, strategies that target CSCs are also needed. Multiple markers such as CD13, CD44, delta-like homologue 1, CD133, and aldehyde dehydrogenase 1 (ALDH-1) have been used to isolate and identify CSCs in various tumors [15–18]. In particular, CD44, CD133, and ALDH-1 have been used as CSC markers for the enrichment of CSCs in breast cancer [19–21].
Minichromosome maintenance (MCM) 2 is one of six related proteins that comprise the MCM complex (MCM2–7), which has an essential role in DNA replication [22]. Previous studies with human samples have established MCM2 as a proliferation marker. A high level of MCM2 expression in malignant tumors is associated with several clinicopathological parameters such as advanced tumor grade, advanced stage, and poor prognosis [23–26]. Previously, we showed that Friend leukemia virus (FLV) infection markedly enhanced the irradiation (IR)-induced apoptosis of hematopoietic cells in C3H mice expressing high levels of MCM2 via the activation of ATM, DNA-PK, and P53 [27]. Apoptosis was enhanced almost exclusively in the C3H strain [28]. We also demonstrated that the FLV-derived envelope protein gp70 enhanced cellular apoptotic signaling specifically in cells that overexpressed MCM2 [29]. Gp70 bound directly to the nuclear localization signal of MCM2 and inhibited its nuclear translocation. The cytoplasmic MCM2-gp70-complex bound to protein phosphatase 2A (PP2A), interfered with the PP2A-DNA-PK interaction, and enhanced DNA damage-induced apoptosis via the activation of p53 by DNA-PK [30]. These results suggest that regulation of the molecular dynamics of MCM2 by using gp70 offers a novel therapeutic approach by which malignant tumors that express high levels of MCM2 can be specifically eliminated. To develop an MCM2-targeted therapy, a method for efficiently introducing gp70 protein into cancer cells is required.
We focused on a protein delivery system that uses the protein transduction domain (PTD). PTD is a peptide sequence that can penetrate the cell membrane [31–33]. PTD has been widely used as a carrier for the delivery of various proteins into living cells. Protein transduction therapy using PTD for the intracellular delivery of a protein is an alternative approach to viral gene therapy [34]. Recently, a novel PTD was identified in the human transcription factor Hph-1. The Hph-1 PTD has been used to deliver immunosuppressive proteins in vitro and in vivo for the treatment of autoimmune diseases [35, 36].
In the present study, we analyzed the expression of MCM2 in HER2, luminal, luminal/HER2, and TN subtypes of human breast cancer. Furthermore, we introduced gp70 into murine breast cancer cells using PTD and investigated whether gp70 had apoptosis-enhancing effects in solid tumors such as breast cancers.
RESULTS
MCM2 is highly expressed in triple negative breast cancer
To quantify MCM2 protein expression in each breast cancer subtype, immunohistochemical staining was performed using specimens from human cases with invasive carcinoma of no special type (Figure 1A). The labeling index of MCM2 in the TN group was significantly higher than the indices in all other subtype groups (Figure 1B). The MCM2 labeling index of the HER2 and luminal/HER2 groups was higher than that of the luminal group (Figure 1B).
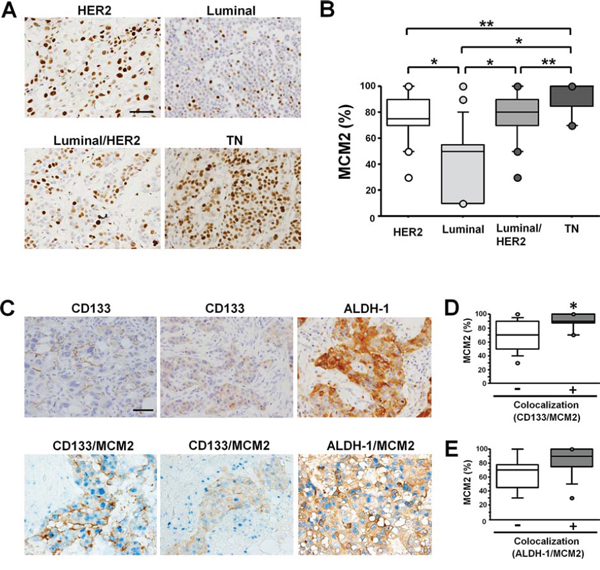
Figure 1: In samples of invasive carcinoma of no special type, MCM2 expression is highest and MCM2 frequently colocalizes with CSC markers in TNBC specimens. A. MCM2 immunohistochemical staining of invasive carcinomas of no special type showing representative features of the HER2 group (top-left), luminal group (top-right), luminal/HER2 group (bottom-left), and TN group (bottom-right). Scale bar indicates 100 μm. Note the frequent nuclear signals in the TNBC case. B. Labeling index of MCM2 in each breast cancer subtype. The index of the TN type (n = 20) of breast cancer was significantly higher than the indices of the HER2 (n = 30), luminal (n = 25), and luminal/HER2 (n = 29) types. *P < 0.0001, **P < 0.01 by Mann-Whitney U test. C. Immunostaining for CD133 in cases with invasive carcinoma of no special type. CD133 antigen localized to the cell membrane (top-left) or the cytoplasm (top-middle). Immunostaining for ALDH-1 in breast cancer (top-right). Double immunostaining for CD133 (brown) and MCM2 (blue) (bottom-left, middle). Double immunostaining for ALDH-1 (brown) and MCM2 (blue) (bottom-right). Scale bar indicates 100 μm. D. The labeling index of MCM2 in the CD133/MCM2 colocalized group (n = 17) and non-colocalized group (n = 10). *P < 0.01 by Mann-Whitney U test. E. The labeling index of MCM2 in the ALDH-1/MCM2 colocalized group (n = 15) and non-colocalized group (n = 5).
Cancer stem cell markers are frequently expressed in TNBC and colocalize with MCM2
Two patterns of CD133 expression were identified: staining of the cell membrane (Figure 1C, top-left) and staining of the cytoplasm (Figure 1C, top-middle). In contrast, ALDH-1 was exclusively expressed in the cytoplasm (Figure 1C, top-right). The frequency of CD133-positive cases was highest in the TN group (40.0%, 8 of 20), followed by the HER2 group (30.0%, 9 of 30), luminal/HER2 group (27.6%, 8 of 29), and luminal group (8.0%, 2 of 25) (Table 1). The membrane staining pattern of CD133 was most frequently observed in the TN group (37.5%), followed by the HER2 group (22.2%), luminal/HER2 group (12.5%), and luminal group (0%). However, the cytoplasmic staining pattern of CD133 was most frequently observed in the luminal group (100%), followed by the luminal/HER2 group (87.5%), HER2 group (77.8%), and TN group (62.5%) (Table 1). The percentage of ALDH-1-positive cases was highest in the TN group (25.0%, 5 of 20), followed by the HER2 group (23.3%, 7 of 30), luminal/HER2 group (17.2%, 5 of 29), and luminal group (12.0%, 3 of 25) (Table 1).
Table 1: Cancer stem cell marker was frequently expressed in TNBC and was co-localized with MCM2
CD133 positive |
Staining pattern |
CD133 & MCM2 |
ALDH-1 positive |
ALDH-1 & MCM2 |
||||
|---|---|---|---|---|---|---|---|---|
cell membrane |
cytoplasm |
Co- |
Non co-localized |
Co-localized |
Non co-localized |
|||
HER2 (n = 30) |
9 (30.0%) |
2 (22.2%) |
7 (77.8%) |
6 (66.7%) |
3 (33.3%) |
7 (23.3%) |
7 (100%) |
0 (0%) |
Luminal (n = 25) |
2 (8.0%) |
0 (0%) |
2 (100%) |
0 (0%) |
2 (100%) |
3 (12.0%) |
0 (0%) |
3 (100%) |
Luminal/HER2 (n = 29) |
8 (27.6%) |
1 (12.5%) |
7 (87.5%) |
4 (50.0%) |
4 (50.0%) |
5 (17.2%) |
5 (100%) |
0 (0%) |
TN (n = 20) |
8 (40.0%) |
3 (37.5%) |
5 (62.5%) |
7 (87.5%) |
1 (12.5%) |
5 (25.0%) |
3 (60.0%) |
2 (40.0%) |
Next, we performed double immunostaining for CD133 or ALDH-1 and MCM2 in cases that were positive for CSC markers (Figure 1C, bottom). Colocalization of MCM2 and CD133 was most frequently observed in the TN group (87.5%, 7 of 8), followed by the HER2 group (66.7%, 6 of 9), luminal/HER2 group (50.0%, 4 of 8), and luminal group (0%, 0 of 2) (Table 1). The labeling index of MCM2 was significantly higher in the CD133/MCM2 colocalized group than in the non-colocalized group (Figure 1D). Colocalization of MCM2 and ALDH-1 was most frequently observed in the HER2 (100%, 7 of 7) and luminal/HER2 (100%, 5 of 5) groups, followed by the TN group (60.0%, 3 of 5) and luminal group (0%, 0 of 3) (Table 1). The labeling index of MCM2 was higher in the ALDH-1/MCM2 colocalized group than in the non-colocalized group, although the difference was not statistically significant (Figure 1E). These results suggest that the CSC markers are highly expressed in TNBC cases and that the markers frequently colocalize with MCM2.
Direct introduction of Hph-1-conjugated gp70 into breast cancer cells in vitro
To identify the breast cancer subtype of the cells, the expression of ER, PgR, and Her2 in FM3A and MTT060562 cells was examined using quantitative RT-PCR. Although their expression levels of ER, PgR, and Her2 were much lower than those of the control uterine tissue, FM3A and MTT060562 cells exhibited higher expression levels of ER and Her2 than 3T3 cells. FM3A cells had higher expression of PgR than 3T3 cells (Figure S1). To investigate the Hph-1-mediated transduction of gp70 into murine breast cancer FM3A and MTT060562 cells, we generated DNA constructs and expressed and purified control-gp70, Hph-1-gp70, and Hph-1-GFP (Figure 2A). To introduce the recombinant proteins into the cells, the FITC-conjugated recombinant proteins were incubated with FM3A, MTT060562 and 3T3 cells at a concentration of 1 μM for 2 h. Hph-1-gp70 was transduced into the living FM3A (Figure 2B), MTT060562 and 3T3 cells (Figure 2C). To determine whether lower concentrations of the recombinant proteins could be efficiently transduced into the cells, 1 × 106 FM3A and MTT060562 murine breast cancer cells and 3T3 murine fibroblast cells were incubated with the proteins at concentrations of 0, 100, 250, 500, 750, and 1000 nM for 2 h. The transduction of gp70 was then assessed using western blot analysis (Figure 2D). Hph-1-gp70 was transduced into the cytoplasm of the cell lines in a concentration-dependent manner. Transduced gp70 was detected in FM3A cells incubated with ≥ 100 nM of Hph-1-gp70 and in MTT060562 and 3T3 cells incubated with ≥ 500 nM of Hph-1-gp70 (Figure 2D). To measure the time course of Hph-1-gp70 transduction, 1 μM of Hph-1-gp70 was incubated with 1 × 106 FM3A, MTT060562, and 3T3 cells for 0, 15, 30, 60, and 120 min, and gp70 transduction was assessed with western blot analysis (Figure 2E). Hph-1-gp70 was first detected at 15 min, and the maximum concentration was reached in less than 60 min in FM3A, MTT060562, and 3T3 cells (Figure 2E). These results suggest that Hph-1-gp70 is rapidly introduced into the cytoplasm of the various cells tested.
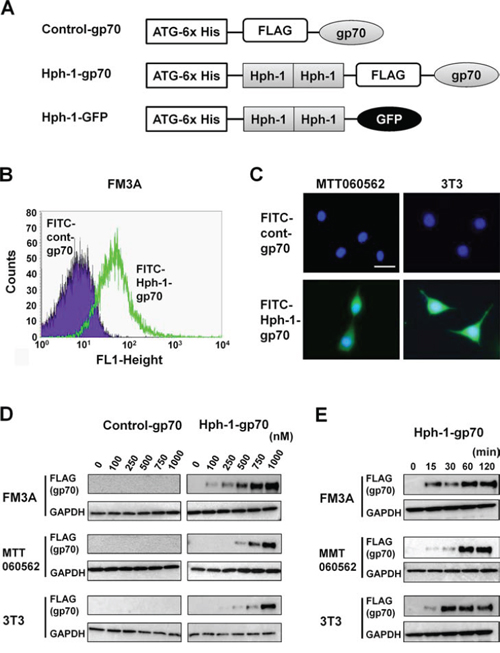
Figure 2: Protein transduction domain Hph-1 conjugated gp70 protein was directly transduced into the cell. A. Schematic illustration indicating the structure of control-gp70, Hph-1-gp70, and Hph-1-GFP. B. FITC-conjugated control-gp70 or Hph-1-gp70 were introduced into 1 × 106 FM3A at 1 μM for 2 h. FITC-positive FM3A cells were analyzed by FACS. C. FITC-conjugated control-gp70 or Hph-1-gp70 were introduced into 1 × 104 MTT060562 (left) and 3T3 (right) at 1 μM for 2 h. Hoechst-stained nuclei are shown in blue. FITC-positive cells were acquired using a BZ-9000 microscope (KEYENCE) with a 100 × objective. Scale bar indicates 100 μm. D. Control-gp70 or Hph-1-gp70 were introduced into 1 × 106 FM3A, MTT060562, and 3T3 cells at concentrations of 0, 100, 250, 500, 750, and 1000 nM for 2 h. The expression of exogenous gp70 was verified with western blot analysis. E. Hph-1-gp70 was introduced into 1 × 106 FM3A, MTT060562, and 3T3 cells at 1 μM for 0, 15, 30, 60, and 120 min, and the expression of exogenous gp70 was verified with western blot analysis.
Hph-1-gp70 binds MCM2 directly
The basal levels of endogenous MCM2 mRNA and protein were significantly higher in FM3A and MTT060562 cells than in 3T3 cells (Figure 3A). We next examined the intracellular interaction between Hph-1-gp70 and MCM2. Control-gp70, Hph-1-gp70, or Hph-1-GFP (1 μM) was incubated with 1 × 106 FM3A, MTT060562, and 3T3 cells for 2 h. Immunoprecipitation experiments revealed that Hph-1-gp70 bound to endogenous MCM2 in FM3A and MTT060562 cells (Figure 3B), but not in 3T3 cells (data not shown). However, Hph-1-gp70 bound to MCM2 in 3T3 cells when the cells were transfected with an expression plasmid encoding HA-conjugated MCM2 (Figure 3B).
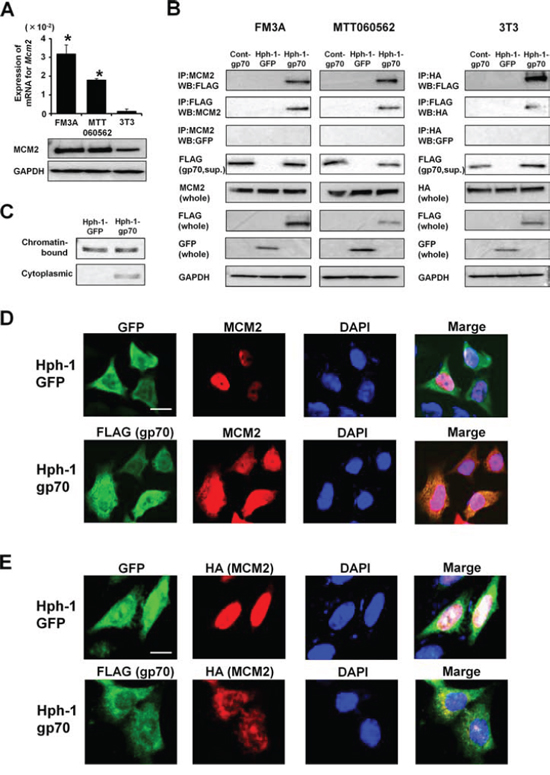
Figure 3: Hph-1-gp70 binds MCM2 directly. A. Quantitative RT-PCR analysis of Mcm2 mRNA expression (top) and western blotting analysis of MCM2 (bottom) in FM3A, MTT060562, and 3T3 cells. *P < 0.01 vs. 3T3 cells by two-tailed Student’s t-test. B. FM3A (left), MTT060562 (middle), and HA-Mcm2-expressing 3T3 cells (right) were treated with 1 μM control-gp70, Hph-1-GFP, or Hph-1-gp70 for 2 h. Cell lysates were subjected to pull-down assays to assess the binding of MCM2 to gp70. C. FM3A cells were treated with 1 μM Hph-1-GFP or Hph-1-gp70 for 2 h. Cell lysates from these cells were separated into chromatin-bound and cytoplasmic fractions. MCM2 protein was analyzed with western blotting. D. MTT060562 cells were treated with 1 μM Hph-1-GFP or Hph-1-gp70 for 2 h. FLAG-positive cells containing the gp70-derived proteins are shown in green (FITC). Endogenous MCM2-positive cells are shown in red (TRITC). DAPI-stained nuclei are shown in blue. Images were acquired using a BZ-9000 microscope (KEYENCE) with a 400 × objective. Scale bar indicates 25 μm. E. HA-Mcm2-expressing 3T3 cells were treated with 1 μM Hph-1-GFP or Hph-1-gp70 for 2 h. FLAG-positive cells containing the gp70-derived proteins are shown in green (FITC). HA-positive cells containing the MCM2-derived proteins are shown in red (TRITC). DAPI-stained nuclei are shown in blue. Images were acquired using a BZ-9000 microscope (KEYENCE) with a 400 × objective. Scale bar indicates 25 μm.
In Hph-1-GFP-treated FM3A cells, MCM2 was not detected in the cytoplasmic fractions. By contrast, MCM2 was detected in the cytoplasmic fraction of FM3A cells treated with Hph-1-gp70 (Figure 3C). Furthermore, MCM2 localized to the nucleus of Hph-1-GFP-treated MTT060562 (Figure 3D, top) and HA-Mcm2-expressing 3T3 cells (Figure 3E, top). However, MCM2 remained in the cytoplasm of Hph-1-gp70-treated cells, in which Hph-1-gp70 colocalized with endogenous MCM2 in MTT060562 cells (Figure 3D, bottom) and HA-MCM2 in 3T3 cells (Figure 3E, bottom).
Hph-1-gp70 enhances DNA damage-induced apoptosis
After treatment with Hph-1-gp70, the apoptotic cell ratio increased in FM3A and MTT060562 cells, which express high levels of endogenous Mcm2 compared to control-gp70. In contrast, the apoptotic ratio did not increase when the cells were treated with Hph-1-GFP (Figure 4A and 4B). However, treatment with Hph-1-gp70 did not significantly change the apoptotic cell ratio of 3T3 cells, which express low levels of endogenous Mcm2 (Figure 4C). As expected, the apoptotic cell ratio of 3T3 cells overexpressing HA-MCM2 was higher after treatment with Hph-1-gp70 than after treatment with Hph-1-GFP (Figure 4C). Moreover, we confirmed that treatment with Hph-1-GFP or Hph-1-gp70 did not cause any significant changes in the cell cycle profiles of FM3A (Figure 4D), MTT060562 (Figure 4E), and 3T3 cells (Figure 4F). These results indicate that a certain expression level of MCM2 is required for the enhancement of apoptosis by gp70.
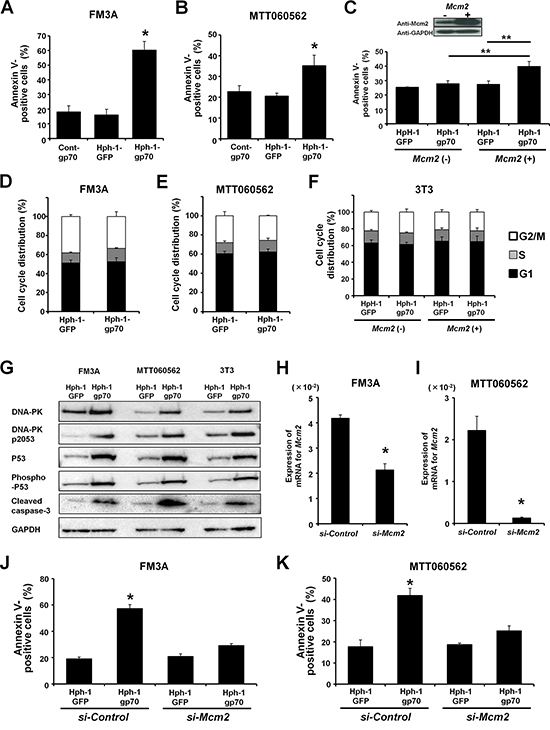
Figure 4: Hph-1-gp70 enhances doxorubicin-induced apoptosis. A. FM3A and B. MTT060562 cells were treated with 1 μM cont-gp70, Hph-1-GFP, or Hph-1-gp70 and 500 nM doxorubicin, and the apoptotic cell ratios were determined 24 h later with annexin V-staining. C. 3T3 cells transfected with HA-empty vector or HA-Mcm2 were treated with 1 μM Hph-1-GFP or Hph-1-gp70 and 500 nM doxorubicin, and the apoptotic cell ratios were determined 24 h later with annexin V-staining. D. FM3A and E. MTT060562 cells were treated with 1 μM Hph-1-GFP or Hph-1-gp70 for 24 h. The effects of treatment on the cell cycle profiles were analyzed with FACS. F. 3T3 cells transfected with HA-empty vector or HA-Mcm2 were treated with 1 μM Hph-1-GFP or Hph-1-gp70 for 24 h. The effects on the cell cycle profiles were analyzed with FACS. G. Representative western blots for DNA-PK, phospho-DNA-PK (pS2053), P53, phospho-P53, and cleaved caspase-3 in FM3A, MTT060562, and HA-Mcm2-transfected 3T3 cells. The cells were treated with 1 μM Hph-1-GFP or Hph-1-gp70 and 500 nM doxorubicin for 24 h. H. Mcm2 knockdown in FM3A and I. MTT060562 cells by using siRNA. Quantitative RT-PCR was performed to confirm the si-Mcm2-induced reduction in Mcm2 mRNA expression. J. FM3A and K. MTT060562 cells transduced with si-control or si-Mcm2 were treated with 1 μM Hph-1-GFP or Hph-1-gp70 and 500 nM of doxorubicin, and the apoptotic cell ratios were determined 24 h later with annexin V-staining. *P < 0.01, **P < 0.05 by two-tailed Student’s t-test.
As shown in Figure 4G, FM3A, MTT060562, and HA-Mcm2-expressing 3T3 cells treated with Hph-1-gp70 and doxorubicin expressed higher levels of DNA-PK, phospho-DNA-PK, P53, phospho-P53, and cleaved caspase-3 compared to cells treated with Hph-1-GFP and doxorubicin.
Treatment with siRNA (si-Mcm2) significantly reduced the expression of Mcm2 mRNA in FM3A and MTT060562 cells (Figure 4H and 4I). In contrast to the increased apoptotic cell ratios observed in FM3A and MTT060562 cells treated with si-Control, the apoptotic cell ratios in FM3A and MTT060562 cells treated with si-Mcm2 did not change markedly after Hph-1-gp70 and doxorubicin treatment (Figure 4J and 4K). Taken together, these results suggest that Hph-1-gp70 bound directly to MCM2, inhibited the translocation of MCM2 to the nucleus by forming a cytoplasmic MCM2-gp70 complex, and strongly enhanced the frequency of doxorubicin-induced apoptosis.
Hph-1-gp70 and MCM2 complex binds to PP2A and causes hyperphosphorylation of DNA-PK
To examine whether DNA-PK was exclusively required for the enhancement of apoptosis by gp70, we inhibited DNA-PK activity using the drug NU7026 in FM3A, MTT060562 and HA-Mcm2-expressing 3T3 cells treated with control-gp70, Hph-GFP, or Hph-1-gp70 and doxorubicin. Inhibition of DNA-PK activity by NU7026 substantially reduced the levels of phospho-DNA-PK (pS2053) and completely abolished apoptosis enhancement in Hph-1-gp70-treated FM3A (Figure 5A), MTT060562 (Figure 5B), and HA-Mcm2-expressing 3T3 cells (Figure 5C). These results indicate that DNA-PK activation is necessary for the enhancement of apoptosis by Hph-1-gp70. In a previous study, we showed that the gp70-MCM2 complex bound to and inhibited protein phosphatase 2A (PP2A). Therefore, we next determined whether the Hph-1-gp70 and MCM2 complex bound to PP2A. FM3A, MTT060562, and HA-Mcm2-expressing 3T3 cells were treated with control-gp70, Hph-1-GFP, or Hph-1-gp70. In Hph-1-gp70 treated cells, PP2A co-precipitated with Hph-1-gp70 and MCM2 (Figure 5D). These results suggest that the Hph-1-gp70 and MCM2 complex bound to PP2A.
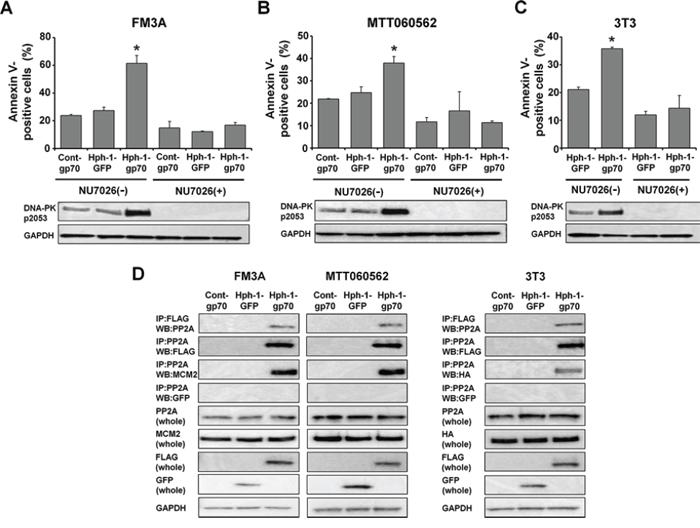
Figure 5: The Hph-1-gp70-MCM2 complex binds to PP2A and causes hyperphosphorylation of DNA-PK. A. FM3A cells and B. MTT060562 cells were pre-incubated with or without 10 μM NU7026, a DNA-PK-inhibitor, for 2 h and treated with 1 μM cont-gp70, Hph-1-GFP, or Hph-1-gp70 and 500 nM doxorubicin for 24 h. The apoptotic cell ratios were determined 24 h later with annexin V-staining (top). *P < 0.01 by two-tailed Student’s t-test. DNA-PK-pS2053 levels were analyzed by western blotting (bottom). C. HA-Mcm2-expressing 3T3 cells were pre-incubated with or without 10 μM NU7026 for 2 h and treated with 1 μM Hph-1-GFP or Hph-1-gp70 and 500 nM doxorubicin for 24 h. The apoptotic cell ratios were determined 24 h later with annexin V-staining (top). *P < 0.01 by two-tailed Student’s t-test. DNA-PK-pS2053 levels were analyzed by western blotting (bottom). D. FM3A (left), MTT060562 (middle), and HA-Mcm2-transfected 3T3 cells (right) were treated with 1 μM cont-gp70, Hph-1-GFP, or Hph-1-gp70 for 2 h. Cell lysates were subjected to pull-down assays to assess the binding of PP2A to gp70 and PP2A to MCM2.
Treatment with Hph-1-gp70 and doxorubicin enhances cancer cell apoptosis and improves the survival of tumor-bearing mice in vivo
To determine whether breast cancer FM3A cells exhibit enhanced apoptosis in response to Hph-1-gp70 and DNA damage in vivo, FM3A cells were subcu-taneously transplanted into SCID mice. The mice were intraperitoneally injected with Hph-1-GFP or Hph-1-gp70 and treated with doxorubicin after the FM3A tumor mass was established. As expected, the FM3A tumor samples from Hph-1-gp70-injected mice exhibited strong expression of gp70 (Figure 6A, bottom-right), whereas no gp70 expression was detected in Hph-1-GFP-injected mice (Figure 6A, top-right), which exhibited strong expression of GFP in tumor cells (Figure 6A, top-left). Treatment with a low dose of doxorubicin enhanced apoptosis in Hph-1-gp70-injected SCID mice but not in Hph-1-GFP-injected mice (Figure 6B and 6C). These results indicate that Hph-1-gp70 treatment and DNA damage induction increased apoptosis in FM3A cells in vivo. Next, tumor growth was analyzed in vivo in SCID mice treated with Hph-1-GFP or Hph-1-gp70 and doxorubicin twice a week after the establishment of the FM3A tumor mass. When mice injected with Hph-1-gp70 and doxorubicin were compared with mice injected with Hph-1-GFP and doxorubicin, the former exhibited smaller tumor sizes and slower tumor expansion (Figure 6D). A survival analysis was also performed with each experimental group of mice. Mice treated with Hph-1-gp70 and doxorubicin exhibited a significant improvement in survival when compared with the other groups of mice (Figure 6E). These results show that breast cancer therapy with Hph-1-gp70 and a low-dose of doxorubicin was effective in vitro and in vivo.
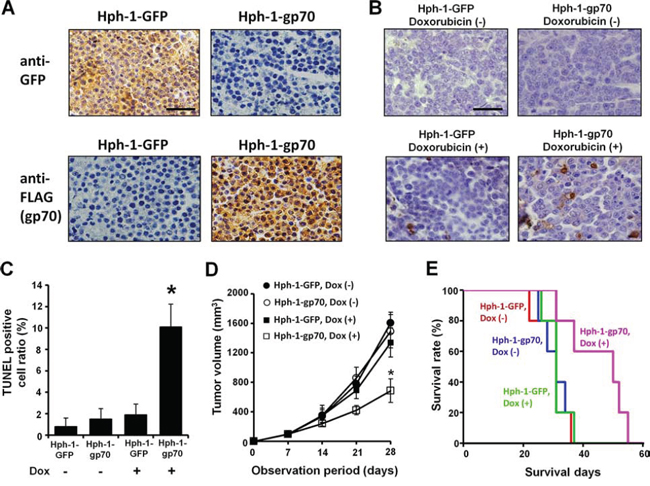
Figure 6: In vivo anti-tumor effects of Hph-1-gp70 and DNA damage in FM3A cells in SCID mice. A. Seven days after the transplantation of FM3A cells, mice were treated with Hph-1-gp70 or Hph-1-GFP and 1.5 mg/kg of doxorubicin or PBS. Expression of GFP (upper) and gp70 (lower) in FM3A tumor samples from Hph-1-gp70- or Hph-1-GFP-injected mice examined with immunohistochemistry. Images were captured with a microscope at 400 × magnification. Scale bar indicates 100 μm. B. Microscopic features of TUNEL-positive cells in an FM3A tumor. Scale bar indicates 100 μm. C. TUNEL-positive cell ratio in each group of mice. *P < 0.01 by two-tailed Student’s t-test. D. To evaluate the therapeutic effect of Hph-1-gp70 and low-dose doxorubicin, FM3A cells were transplanted into SCID mice. Seven days after the transplantation, mice were treated with 5 mg Hph-1-gp70 or Hph-1-GFP and 1.5 mg/kg of doxorubicin or PBS twice a week. The tumor size in each mouse was assessed once a week. *P < 0.05 by two-tailed Student’s t-test. E. Kaplan-Meier survival curves for FM3A-transplanted SCID mice treated with Hph-1-gp70 or Hph-1-GFP and doxorubicin. The survival time of the Hph-1-gp70, doxorubicin (+) group was longer than that of the other groups (P < 0.01 by log-rank test).
MCM2 is highly expressed in FM3A cells with high CD133 expression
Next, to evaluate the relationship between the localization of CSC markers and MCM2, we performed double immunostaining for CD133 and MCM2 in FM3A transplanted SCID mice. As shown in Figure 7A, CD133 highly expressing (CD133-high) FM3A cells frequently colocalized with MCM2 in vivo. Therefore, to examine the expression level of MCM2 in CD133-high breast cancer cells, we separated and enriched CD133-high cells from other FM3A cells. The robust expression of CD133 in the separated FM3A cells was confirmed by FACS (Figure 7B). Approximately 7% of FM3A cells were CD133-high cells (Figure S2). As expected, the mRNA and protein levels of MCM2 were significantly higher in CD133-high FM3A cells than in CD133-low FM3A cells (Figure 7C). Moreover, CD133-high or CD133-low FM3A cells exhibited no remarkable change in their cell-cycle profiles (Figure 7D). It is widely accepted that cells expressing CSC markers have drug resistance. The present study revealed that the apoptotic cell ratio after doxorubicin treatment was significantly lower in CD133-high FM3A cells than in CD133-low FM3A cells (Figure 7E). After Hph-1-gp70 treatment, doxorubicin-induced apoptosis was enhanced in CD133-low FM3A cells. Strikingly, Hph-1-gp70-induced apoptosis was at similar levels in CD133-high and low FM3A cells (Figure 7E). Furthermore, protein levels of DNA-PK, phospho-DNA-PK (pS2053), P53, phospho-P53, and cleaved caspase-3 were lower in CD133-high FM3A cells than in CD133-low FM3A cells after treatment with doxorubicin. After Hph-1-gp70 treatment, the levels of these proteins were remarkably elevated in CD133-low and CD133-high FM3A cells (Figure 7F). These results suggest that the treatment of cells with Hph-1-gp70 and a low dose of doxorubicin strongly activate apoptosis, even in CD133-high cancer cells that were originally resistant to doxorubicin-induced apoptosis.
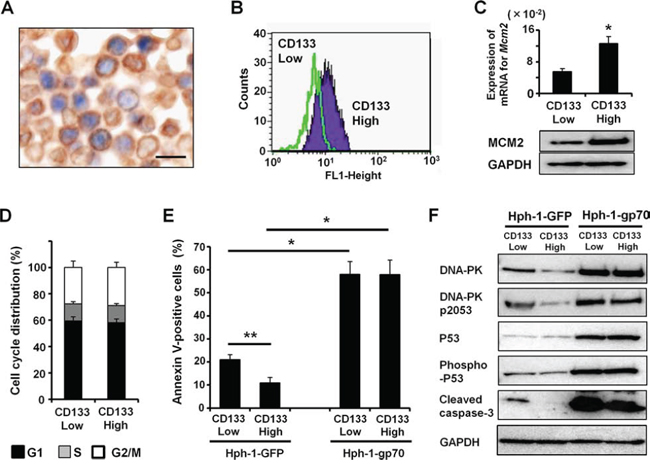
Figure 7: CD133-high cells have high MCM2 expression. A. Double immunostaining for CD133 (brown) and MCM2 (blue) in FM3A cells transplanted into SCID mice. Scale bars indicate 25 μm. B. FACS analysis of the expression of CD133 in separated FM3A cells. C. Quantitative RT-PCR analysis of Mcm2 mRNA expression (top) and western blot analysis of MCM2 (bottom) in CD133-high or -low FM3A cells. *P < 0.01 by two-tailed Student’s t-test. D. The cell cycle profiles of CD133-high or -low FM3A cells were analyzed with FACS. E. CD133-high or -low FM3A cells were treated with 1 μM Hph-1-gp70 or Hph-1-GFP and 500 nM of doxorubicin, and the apoptotic cell ratios were determined 24 h later with annexin V-staining. *P < 0.01, **P < 0.05 by two-tailed Student’s t-test. F. Representative western blots showing DNA-PK, phospho-DNA-PK (pS2053), P53, phospho-P53, and cleaved caspase-3 levels in CD133-high or -low FM3A cells treated with 1 μM Hph-1-gp70 or Hph-1-GFP and 500 nM doxorubicin for 24 h.
DISCUSSION
The MCM complex associates with origins of DNA replication to form part of the pre-replicative complex [37]. MCM proteins are usually expressed in cycling cells [38] and are frequently overexpressed in a variety of cancer and pre-cancerous cells [23–26, 39, 40]. In breast cancer, high expression of MCM2 is associated with several clinicopathological parameters, such as advanced tumor grade, advanced stage, and poor prognosis [41, 42]. Moreover, according to an NCBI GEO database (accession number: GDS2250), the expression of MCM2 is significantly higher in basal-like breast cancer than in normal breast cells and non-basal-like breast cancer cells. According to another NCBI GEO database (accession number: GDS4051), the expression of MCM2 is higher in the tamoxifen-resistant breast cancer cell line MCF7 than in tamoxifen-sensitive MCF7 cells. Among the four molecular subtypes that we analyzed, the expression of MCM2 was particularly high in TNBC. These results suggest that cancer therapy targeting MCM2 will be useful for TNBC, the subtype that has lacked a specific therapeutic target.
Previous studies have suggested that the phase of the cell cycle changes after MCM2 knockdown using siRNA [43–45]. In the present study, we found that gp70 bound to with MCM2 and inhibited its nuclear translocation. However, the cell cycle profiles of the murine tumor lines were not changed by gp70 transduction (Figure 4D). In the present study, Hph-1-gp70-treated MTT060562 and 3T3 cells had MCM2 cytoplasmic fractions, yet a significant amount of MCM2 still localized to the nucleus (Figure 3D, 3E). We speculate that the cell cycle did not change because of the level of MCM2 that remained in the nucleus. However, gp70 was able to retain a significant amount of MCM2 in the cytoplasm to enhance apoptosis.
p53 is mutated or inactivated with high frequency in human breast cancer tumor cells [46]. Our study used murine breast cancer cell lines FM3A and MTT060562 that have wild-type p53 (Figure S3A). However, in 8047 cells used in our previous study, p53 has the R172H mutation (Figure S3B). Indeed, the p53 mutation (R175H) is frequently found in human breast cancers and other types of cancer [47]. Our previous study showed that therapy using gp70 enhanced apoptosis in 8047 cells [30]. In the present study, we evaluated the apoptotic cell ratio of Hph-1-gp70-treated 8047 cells after treatment with doxorubicin to induce DNA damage to determine whether this treatment is effective in a mutant p53 background. Indeed, treatment with Hph-1-gp70 increased the apoptotic index of 8047 cells (Figure S3C). Therefore, even in cells harboring p53 mutations, gp70 therapy may have similar effects as in tumors with wild-type p53. In addition, many reports have showed that anticancer agents are not as effective against breast cancer cells that have mutant p53 [48, 49]. However, other reports have indicated that high dose therapy with alkylating agents is effective against breast cancer cells that have mutant p53 [50]. Thus, cancer therapy using gp70, which enhances DNA damage-induced apoptosis, might be effective against p53-mutated breast cancer cells in combination with low doses of anticancer agents.
Previous studies have reported that activated DNA-PK stabilized MDM2 leading to the prevention of p53 degradation [51]. Our study suggests that over-activated DNA-PK is associated with p53 stabilization and activation. This result was obtained from our data showing that total p53 and phosphorylated (active) p53 levels increased to a similar extent in cells treated with Hph-1-gp70.
Because CSCs are associated with recurrence or distant metastasis after cancer therapy, it is important to develop methods that target these cells in order to improve the outcome of conventional cancer therapy [14, 52, 53]. Several reports indicate that CD133 and ALDH-1 are useful for identifying CSC-like populations in different malignancies and that CSCs are potential targets of therapy [54–57]. Furthermore, MCM2 is highly expressed in CSCs of retinoblastoma and glioblastoma [58, 59]. In the present study, CD133 and ALDH-1 were expressed in several breast cancer samples. CD133 and ALDH-1 were expressed more frequently in the TNBC group, which expressed high levels of MCM2, than in the other cancer subtypes. Immunohistochemical analysis revealed that MCM2 colocalized with CD133 in TNBC cells. Furthermore, MCM2 expression was higher in CD133-high FM3A cells than in CD133-low FM3A cells. Thus, therapies targeting MCM2-targeting may be effective against TNBC cells and CSC-like breast cancer cells.
Cancer stem cells are considered a very small fraction of cells in the cancer cell population. In the present study, CD133 was positive in about 7% of murine breast cancer cell line FM3A (Figure S2). Previous studies demonstrated that cells expressing CD133 or ALDH-1 have a stemness property [60–62]. On the other hand, our study and previous studies showed that CD133 and ALDH-1 are frequently expressed in various cancer types [63–66]. However, CD133 or ALDH-1 positive cancer cells may not necessarily possess a stemness property. Instead, these cells may have a relatively poorly differentiated character and stem cell-like phenotype. These cells may be “immature” tumor cells having stemness functions including drug resistance.
To develop a novel method for the transduction of gp70 into cancer cells, we focused on a protein delivery system that uses the PTD. The discovery of the PTD has made it possible to transduce therapeutically active agents into living cells [67, 68]. The most extensively studied PTDs are those of the HIV-1 transactivator of transcription protein (TAT), Drosophila homeodomain transcription factor Antennapedia (ANTP), and herpes simplex virus structural protein VP22 [69–73]. These PTD sequences are enriched with arginine residues, and short sequences with 6–30 consecutive arginine residues are functional PTDs [74–76]. Recently, a novel PTD was identified in the human transcriptional factor Hph-1. A previous study showed that Hph-1-PTD has good protein transduction efficiency and can deliver therapeutic protein in vitro and in vivo [35, 36, 77]. In the present study, we conjugated Hph-1-PTD to gp70 (Hph-1-gp70). In vitro, Hph-1-gp70 was rapidly and effectively introduced into breast cancer cell lines, in which it bound to MCM2 in the cytoplasm and enhanced DNA damage-induced apoptosis in cells expressing high levels of MCM2. Moreover, in an FM3A-bearing mouse model, gp70 was introduced into tumor cells through an intraperitoneal injection of Hph-1-gp70. Mice injected with Hph-1-gp70 and treated with doxorubicin exhibited a smaller tumor size and significant improvement in survival. Moreover, similar results were obtained in experiments using the ovarian cancer cell line T-Ag-MOSE (Supplementary Figure S4 and S5). These results suggest that Hph-1-gp70 offers a novel strategy for the treatment of breast cancer and other types of cancer.
In a previous study, we showed that the cytoplasmic localization of the MCM2-gp70 complex promoted the interaction of MCM2 with PP2A [30]. In this study, Hph-1-gp70 and MCM2 complex bound to PP2A (Figure 5D). PP2A has been shown to dephosphorylate DNA-PK and inhibit its function [78–80]. These results suggested that the Hph-1-gp70 and MCM2 complex bound to PP2A and inhibited the function of PP2A. Consequently, DNA-PK was hyperphosphorylated and enhanced doxorubicin-induced apoptosis via a p53/cleaved caspase 3 pathway.
Recent progress in CSC research has revealed the molecular mechanisms of resistance to chemotherapy and radiotherapy and has contributed to the development of effective chemotherapeutic regimens [81]. Several studies have demonstrated an association between CSCs and drug resistance in breast cancer cell lines [82, 83]. CD133-high FM3A cells were resistant to doxorubicin-induced apoptosis in the present study. After Hph-1-gp70 treatment, doxorubicin-induced apoptosis was enhanced not only in CD133-low FM3A cells but also in CD133-high cells. These results suggest that Hph-1-gp70 and low-dose doxorubicin treatment is an effective approach for killing cancer cells including CSCs, the treatment of which might prevent cancer relapse and metastasis. In the clinical setting for breast cancer therapy, neoadjuvant chemotherapy (NAC) regimens usually include doxorubicin or its derivatives [83, 84]. Thus, a therapeutic strategy using Hph-1-gp70-MCM2 might be a novel and effective NAC approach to treat breast cancer.
In conclusion, our data suggest that cancer therapy using Hph-1-gp70 and doxorubicin is effective for treating cancers with high MCM2 expression, including CSCs. Although several problems must be solved with regard to its application in humans, cancer therapy that exploits the apoptosis-enhancing effect of MCM2 might offer a cure for various cancers.
MATERIALS AND METHODS
Clinical samples
Formalin-fixed, paraffin-embedded (FFPE) samples of invasive carcinoma of no special type (ductal-NST) [85, 86] were used in this study. The molecular subtypes of breast cancer were defined using the immunohistochemical surrogate markers ER, PgR, and HER2. The HER2 subtype was defined as ER-, PgR-, and HER2+. The luminal subtype was defined as ER+ and/or PgR+ and HER2-. The luminal/HER2 subtype was defined as ER+ and/or PgR+ and HER2+. The TN subtype was defined as ER-, PgR-, and HER2-. In total, 104 FFPE samples of invasive carcinoma of no special type were collected from breast cancer patients who underwent surgery, including 30 patients with the HER2 subtype (median age, 56.5 [range, 33–76 years]), 25 patients with the luminal subtype (median age, 51 [range, 23–81 years]), 29 patients with the luminal/HER2 subtype (median age, 48 [range, 29–73 years]), and 20 patients with the TN subtype (median age, 58 [range, 34–80 years]). The FFPE samples were collected at the Cancer Institute Hospital, Tokyo, Japan in 2012. A list of the samples used in this study is summarized in Supplementary Table S1. The human study was approved by the ethics committees of Tokyo Medical and Dental University (No. 1458) and Cancer Institute Hospital (No. 2014–1003), and all procedures were performed in accordance with the ethical standards established by these committees. The experiments conformed to the principles set out in the WMA Declaration at Helsinki and the Department of Health and Human Services Belmont Report.
Immunohistochemistry
FFPE tissue sections (4-μm thick) were used for immunohistochemistry. Deparaffinization was followed by heat-based antigen retrieval, endogenous peroxidase blockade with 3% hydrogen peroxide, and blocking with normal sera. The primary antibodies used were as follows: MCM2, mouse monoclonal, 1:2000 (BD Biosciences, San Jose, CA, USA); CD133, mouse monoclonal, 1:100 (Miltenyi Biotec, Auburn, CA, USA); ALDH-1, rabbit monoclonal, 1:1000 (Abcam, Cambridge, UK); FLAG, mouse monoclonal, 1:250 (InvivoGen, San Diego, CA, USA); and GFP, mouse monoclonal, 1:100 (Abcam). The primary antibodies were incubated overnight at 4°C. Primary antibodies were detected using an ABC Kit (Vector Laboratories, Burlingame, CA, USA) or EnVision+ System-HRP (Dako, Glostrup, Denmark) with diaminobenzidine (DAB; Nichirei Bioscience, Japan) or the HISTOFINE simple stain AP series (Nichirei Bioscience) with Vector Blue (Vector Laboratories). A TUNEL assay was also performed using the In Situ Cell Death Detection Kit, POD (Roche Diagnostics, Tokyo, Japan) with DAB. For double immunostaining, samples were heat treated and blocked between each step.
Cell lines
The FM3A cell line derived from C3H mouse breast cancer cells was purchased from the JCRB Cell Bank (Ibaraki, Osaka, Japan) and cultured in MEM (Wako, Tokyo, Japan). 3T3 cells derived from BALB/c mouse fibroblast cells were purchased from the RIKEN Cell Bank (Tsukuba, Ibaraki, Japan) and cultured in DMEM (Sigma Aldrich, St. Louis, MO, USA). The MTT060562 cell line derived from C3H mouse breast cancer cells and T-Ag-MOSE cell line derived from C3H mouse ovarian cancer cells were purchased from the JCRB Cell Bank (Ibaraki, Oosaka, Japan) and cultured in DMEM (Sigma Aldrich). The radiation-induced myeloid leukemia cell line from C3H mice, 8047, was established at the National Institute of Radiological Sciences in Chiba [29]. The cells were cultured in RPMI-1640 medium (Sigma Aldrich). The data of T-Ag-MOSE and 8047 cell lines were shown in Supplementary Figures. Medium was supplemented with 10% fetal calf serum, penicillin (50 units/mL; Invitrogen, Carlsbad, CA, USA), and streptomycin (50 μg/mL; Invitrogen). The cells were cultured at 37°C in a humidified atmosphere of 5% CO2.
Generation of Hph-1 fusion proteins to introduce proteins into cells
Gp70 was amplified from the cDNA of FLV-infected 8047 cells by using PCR primers. The primers, synthesized at a commercial laboratory (Invitrogen), were GAAAGATCTAAAAGGTCCAGCGTTCTCAAA and GAAGGTACCCTATGCAGCTATGCCGCCCATAG. GFP was amplified from the pSUPER vector (Oligoengine, Seattle, WA, USA) by using PCR primers. The primers, synthesized at a commercial laboratory (Invitrogen), were GAAAGATCTGCCACAACCATGGTGAGCAAG and CCCAAGCTTCTACTTGTACAGCTCGTCCATGCC. The PCR products were subsequently cloned into the bacterial recombinant protein expression vector pRSET C (Invitrogen). Hph-1-gp70 and Hph-1-GFP proteins contained the sequence for 2 × Hph-1-PTD (YARVRRRGPRP YARVRRRGPRP) at their N-terminus. The FLAG sequence was inserted between the Hph-1 and gp70 sequences. gp70 lacking Hph-1-PTD was also constructed and purified for use as a negative control.
The protein expression vectors were transformed into Escherichia coli BL21 Star (DE3)pLysS cells (Invitrogen) and cultured in Luria-Bertani medium containing 100 μg/mL ampicillin and 35 μg/mL chloramphenicol. Protein expression was induced for 2 h at 37°C by adding 1 mM isopropyl-β-d-thiogalactopyranoside. Cells were then sonicated in lysis buffer (6 M urea, 20 mM Tris-HCl, pH 8.0, and 500 mM NaCl). The lysates were clarified by centrifugation, and cOmplete His-tag Purification Resin (Roche) was added. Bound proteins were washed and eluted with 300 mM imidazole. Eluted proteins were desalted using PD-10 Sephadex G-25 (BD Biosciences), supplemented with 10% glycerol, and frozen at −80°C for later use.
Immunoblotting
FM3A, MTT060562, and 3T3 cells, before and after treatment with transduction proteins, were used for western blotting. The samples were loaded onto a 5%–20% gradient polyacrylamide gel (Wako, Tokyo, Japan) and electrophoretically transferred to nitrocellulose membranes (GE Healthcare, Danbury, CT, USA). The membranes were blocked with 10% skim milk in PBS. The primary antibodies were anti-FLAG M2 mouse monoclonal antibody (Sigma Aldrich), anti-HA mouse monoclonal antibody (Sigma Aldrich), anti-MCM2 mouse monoclonal antibody (BD Biosciences), anti-DNA-PKcs mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-DNA-PK S2056 (Mouse-S2053) rabbit polyclonal antibody (Assay Biotech, Sunnyvale, CA, USA), anti-P53 mouse monoclonal antibody (Merck, Darmstadt, Germany), anti-phospho-P53 (Ser 15) rabbit polyclonal antibody (Merck), anti-cleaved caspase-3 rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA, USA), anti-GFP mouse monoclonal antibody (Abcam), anti-PP2A rabbit polyclonal antibody (Cell Signaling Technology) and anti-GAPDH rabbit polyclonal antibody (Santa Cruz Biotechnology). The secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-mouse IgG (GE Healthcare) and HRP-conjugated anti-rabbit IgG (GE Healthcare). Protein expression was detected using the Clarity™ Western ECL Substrate (Bio-Rad, Hercules, CA, USA).
Immunofluorescence
To monitor the control-gp70 and Hph-1-gp70 protein in living cells, these proteins were labeled with fluorescein isothiocyanate (FITC) by using Fluorescein Labeling Kit-NH2 (Dojindo, Kumamoto, Japan). FM3A, MTT060562, and 3T3 cells were incubated with FITC-conjugated control-gp70 or Hph-1-gp70. After incubation, FM3A cells were washed PBS, and fluorescence was detected by a flow cytometer (BD FACSCanto™ Cell Analyzer; BD Biosciences). MTT060562 and 3T3 cells were washed PBS, and incubated with Hoechst 33342 (life technologies, CA, USA). Images were acquired using a BZ-9000 microscope (KEYENCE, Osaka, Japan) with a 400 × objective.
To monitor MCM2 localization after treatment with gp70, MTT060562 and 3T3 cells were fixed in 1% paraformaldehyde in PBS and permeabilized with 0.1% NP-40 in PBS at room temperature. Cells were incubated with a rabbit monoclonal anti-FLAG antibody (InvivoGen) and a mouse monoclonal anti-HA antibody (InvivoGen) or monoclonal anti-MCM2 antibody (BD Biosciences) at a 1:100 dilution in PBS for 1 h at room temperature. Cells were then stained with a FITC-conjugated anti-rabbit antibody (Dako) and a tetramethylrhodamine-5-(and-6)-isothiocyanate (TRITC)-conjugated anti-mouse antibody (Dako) at a 1:100 dilution for 20 min at room temperature. Slides were washed three times with PBS and mounted with VECTASHIELD mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Images were acquired using a BZ-9000 microscope (KEYENCE) with a 400 × objective.
Chromatin loading assay
Chromatin loading of MCM2 was performed as described previously [30]. Briefly, FM3A cells were lysed by incubating in complete cytoskeleton (CSK) buffer (20 mM HEPES, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, and 0.1% NP-40) for 15 min on ice. Cytoplasmic fractions were obtained as supernatants after low speed centrifugation (3,000 × g) at 4°C. Pellets were rinsed with complete CSK buffer for 10 min on ice and centrifuged again to obtain a chromatin-enriched fraction. Pellets were then sonicated for 5 s in CSK buffer and subjected to high-speed centrifugation (16,000 × g). The post-sonication supernatant was designated as the chromatin-bound fraction.
Immunoprecipitation
Cell lysates were prepared by incubating cell pellets on ice for 1 h in ice-cold lysis buffer containing 10 mM Tris-HCl, pH 7.5, 5 mM EDTA, 1% Nonidet P-40, 0.02% NaN3, 1 mM PMSF, 0.1% aprotinin, 100 μM leupeptin, and 100 μM TPCK (Sigma Aldrich). Cell lysates were incubated with antibody and Protein G Sepharose™ (GE Healthcare). Whole cell lysates or immunoprecipitates obtained after centrifugation were mixed with 2 × sodium dodecyl sulfate (SDS) buffer (125 mM Tris-HCl at pH 6.8, 4% SDS, 20% glycerol, 0.01% bromophenol blue, and 10% 2-mercaptoethanol) and boiled for 10 min.
Transfection of expression plasmids
The 3 × HA-conjugated MCM2 expression vector was constructed as described previously [30]. The 3 ×HA-Mcm2 construct was transfected into 3T3 cells (2 × 105 cells) using HilyMax Transfection Reagent (Nippon Gene, Tokyo, Japan). The controls were mock transfected with an empty vector.
Detection of apoptotic cells
To determine the apoptotic cell ratios in FM3A, MTT060562, and 3T3 cells after treatment with 500 nM doxorubicin for 24 h, samples were stained with propidium iodide (PI), incubated with FITC-labeled anti-annexin V antibody (BD Biosciences), and analyzed on a flow cytometer (BD FACSCanto™ Cell Analyzer; BD Biosciences).
Analysis of cell cycle distribution
Cell cycle distribution was monitored by quantifying the cellular DNA content after staining with PI. Cells were fixed with ethanol for 20 min at − 20°C. After centrifugation, cells were suspended in PBS containing PI (50 μg/mL) and RNase (0.2 mg/mL), incubated at room temperature for 30 min, and analyzed on a flow cytometer (BD FACSCanto™ Cell Analyzer).
RNA interference
The siRNA sequence for si-Mcm2 was CAGGTGACAGACTTTATCAAA. An irrelevant siRNA (si-Control; GCACACAGACTGCAATCACAGGTTA) that did not lead to the specific degradation of any cellular mRNA was used as a negative control. FM3A and MTT060562 cells (2 × 105 cells) were transfected with 120 pmol of Mcm2 or control siRNA by using the Amaxa® Cell Line Nucleofector® Kit V (Lonza, Basel, Switzerland) according to the manufacturer’s instructions.
SYBR Green real-time RT-PCR
RNA was extracted from FM3A, MTT060562 and 3T3 cell lines by using TRIzol (Invitrogen) according to the manufacturer’s instructions. Briefly, the liquid phase was incubated with chloroform for phase separation. Total RNA was extracted using one isopropanol precipitation step and one ethanol wash. The RNA pellet was diluted in RNase- and DNase-free water (Qiagen, Hilden, Germany). cDNA was generated from the RNA using TaqMan® Reverse Transcription Reagents (Applied Biosystems [ABI], Foster, CA, USA), and quantitative RT-PCR was performed. For quantitative RT-PCR, specific primers were used with the LightCycler SYBR Green master mix (Roche, Basel, Switzerland). The sequences of the primers are as follows: for Mcm2, GAGGATGGAGAGGAACTCATTG and ATCTTCCTCGCTGCTGTCA; for estrogen receptor (ER), GCGCAAGTGTTACGAAGTGG and AGCACCCATTTCATTTCGGC; for progesterone receptor (PgR), CAGAAAGGGGTTGTCCCCAG and TTCCGGAAATTCCACAGCCA; and for Her2, CCTGTCGACATGGACACCAA and GGACTCTCACCCCAACAGTG. The PCR product was detected using an ABI Prism 7900HT Sequence Detection System (ABI, Carlsbad, CA, USA). The primers and TaqMan probes for Gapdh were purchased from ABI. Mcm2, ER, PgR and Her2 RNA levels were normalized to the level of Gapdh.
Separation of CD133-high cells among FM3A cells
CD133-positive cells were separated from FM3A cells using a pluriBead kit (pluriSelect, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, the total population of FM3A cells was incubated with 50 μL of beads directly conjugated to rabbit anti-mouse CD133 antibody (Abnova, Taipei, Taiwan) at room temperature for 30 min. The suspended cells were added to a cell strainer. The labeled CD133-high cells were retained on the cell strainer, and the unlabeled cells were eluted. Subsequently, the separated cells were incubated with FITC-labeled anti-mouse CD133 antibody (eBioscience, San Diego, CA USA) and analyzed on a flow cytometer (BD FACSCanto™ Cell Analyzer).
Cancer-bearing mouse model
Six-week-old male specific-pathogen-free SCID mice (C.B.17scid/scid, H-2d) were purchased from CLEA Japan Inc. (Tokyo, Japan). To evaluate the therapeutic effect of Hph-1-gp70 and low-dose doxorubicin, FM3A cells (2 × 106 cells/50 μL PBS) were inoculated subcutaneously into the left flank of mice. One week after transplantation, mice were treated twice a week with an intraperitoneal injection of 5 mg of Hph-1-gp70 or Hph-1-GFP and with 1.5 mg/kg of doxorubicin. The tumor volumes were calculated weekly as (width)2 × length × 0.52. The animal experiments were conducted and carried out in strict accordance with the Act on Welfare and Management of Animals of the government of Japan and the Guidelines for the Care and Use of Laboratory Animals of the Tokyo Medical and Dental University. All experiments were approved by the Animal Experiment Committee of Tokyo Medical and Dental University (No. 100115). The animals were housed in standard cage at 25°C, in a 12/12 light-dark cycle in a clean room. All efforts were made to minimize suffering in animal experiments.
Statistical analysis
Data are presented in the figures as the mean ± SD. For each figure, statistical tests are justified as appropriate. The Mann-Whitney U-test was used to analyze the immunohistochemistry data obtained using human invasive breast cancer samples. For Kaplan-Meier analysis of SCID mice transplanted with FM3A cells, a log-rank test was performed. The statistical significance in in vitro experiments was determined using a two-tailed Student’s t-test. P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
We thank Ms. Sachiko Ishibashi and Ms. Miori Inoue of the Tokyo Medical and Dental University for technical assistance. This work was supported by a grant-in-aid (24590476) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
Authors’ contributions
The overall study was conceived and designed by SA and MK (Kitagawa). SA, KY, MK (Kurata), and SA-S performed the experiments and analyzed the data. RH and FA provided clinical samples. SA and MK (Kitagawa) wrote the manuscript.
REFERENCES
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics. CA Cancer J Clin. 2010; 60:277–300.
2. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001; 98:10869–10874.
3. Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009; 101:736–750.
4. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006; 295:2492–2502.
5. Gómez HL, Castañeda CA, Vigil CE, Vidaurre T, Velarde RG, Cruz WR, Pinto JA, Suazo JF, Garcés MR, Neciosup SP, Vallejos CS. Prognostic effect of hormone receptor status in early HER2 positive breast cancer patients. Hematol Oncol Stem Cell Ther. 2010; 3:109–115.
6. Castaneda CA, Andrés E, Barcena C, Gómez HL, Cortés-Funés H, Ciruelos E. Behaviour of breast cancer molecular subtypes through tumour progression. Clin Transl Oncol. 2012; 14:481–485.
7. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007; 13:4429–4434.
8. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009; 9 Suppl 2:S73–81.
9. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007; 13:2329–2334.
10. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010; 363:1938–1948.
11. D’Amour KA, Gage FH. Are somatic stem cells pluripotent or lineage-restricted? Nat Med. 2002; 8:213–214.
12. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432:396–401.
13. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66:9339–9344.
14. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105–111.
15. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, Barnard GF, Doki Y, Mori M. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010; 120:3326–3339.
16. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006; 25:1696–1708.
17. Xu X, Liu RF, Zhang X, Huang LY, Chen F, Fei QL, Han ZG. DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther. 2012; 11:629–638.
18. Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, Lo CM, Guan XY, Chan KW. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012; 55:807–820.
19. Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, Yao Z, Dong XY, Zhao N, Liu N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013; 32:544–553.
20. Kim YS, Jung MJ, Ryu DW, Lee CH. Clinicopathologic characteristics of breast cancer stem cells identified on the basis of aldehyde dehydrogenase 1 expression. J Breast Cancer. 2014; 17:121–128.
21. Olsson E, Honeth G, Bendahl PO, Saal LH, Gruvberger-Saal S, Ringnér M, Vallon-Christersson J, Jönsson G, Holm K, Lövgren K, Fernö M, Grabau D, Borg A, Hegardt C. CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer. 2011; 11:418.
22. Bochman ML, Schwacha A. The Mcm2–7 complex has in vitro helicase activity. Mol Cell. 2008; 31:287–293.
23. Dudderidge TJ, Stoeber K, Loddo M, Atkinson G, Fanshawe T, Griffiths DF, Williams GH. Mcm2, Geminin, and KI67 define proliferative state and are prognostic markers in renal cell carcinoma. Clin Cancer Res. 2005; 11:2510–2517.
24. Going JJ, Keith WN, Neilson L, Stoeber K, Stuart RC, Williams GH. Aberrant expression of minichromosome maintenance proteins 2 and 5, and Ki-67 in dysplastic squamous oesophageal epithelium and Barrett’s mucosa. Gut. 2002; 50:373–377.
25. Davies RJ, Freeman A, Morris LS, Bingham S, Dilworth S, Scott I, Laskey RA, Miller R, Coleman N. Analysis of minichromosome maintenance proteins as a novel method for detection of colorectal cancer in stool. Lancet. 2002; 359:1917–1919.
26. Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, Zaman MS, Hirata H, Yamamura S, Ueno K, Tanaka Y, Dahiya R. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010; 70:2809–2818.
27. Kitagawa M, Yamaguchi S, Hasegawa M, Tanaka K, Sado T, Hirokawa K, Aizawa S. Friend leukemia virus infection enhances DNA damage-induced apoptosis of hematopoietic cells, causing lethal anemia in C3H hosts. J Virol. 2002; 76:7790–7798.
28. Tanaka K, Watanabe K, Yamaguchi S, Hasegawa M, Kitagawa M, Aizawa S. Cytological basis for enhancement of radiation-induced mortality by Friend leukaemia virus infection. Int J Radiat Biol. 2004; 80:673–681.
29. Hasegawa M, Kurata M, Yamamoto K, Yoshida K, Aizawa S, Kitagawa M. A novel role for acinus and MCM2 as host-specific signaling enhancers of DNA-damage-induced apoptosis in association with viral protein gp70. Leuk Res. 2009; 33:1100–1107.
30. Abe S, Kurata M, Suzuki S, Yamamoto K, Aisaki K, Kanno J, Kitagawa M. Minichromosome maintenance 2 bound with retroviral Gp70 is localized to cytoplasm and enhances DNA-damage-induced apoptosis. PLoS One. 2012; 7:e40129.
31. Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000; 97:13003–13008.
32. Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001; 276:5836–5840.
33. Mai JC, Shen H, Watkins SC, Cheng T, Robbins PD. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem. 2002; 277:30208–30218.
34. Yamada S, Kanno H, Kawahara N. Trans-membrane peptide therapy for malignant glioma by use of a peptide derived from the MDM2 binding site of p53. J Neurooncol. 2012; 109:7–14.
35. Choi JM, Ahn MH, Chae WJ, Jung YG, Park JC, Song HM, Kim YE, Shin JA, Park CS, Park JW, Park TK, Lee JH, Seo BF, Kim KD, Kim E S, Lee DH, Lee SK. Intranasal delivery of the cytoplasmic domain of CTLA-4 using a novel protein transduction domain prevents allergic inflammation. Nat Med. 2006; 12:574–579.
36. Choi JM, Kim SH, Shin JH, Gibson T, Yoon BS, Lee DH, Lee SK, Bothwell AL, Lim JS. Transduction of the cytoplasmic domain of CTLA-4 inhibits TcR-specific activation signals and prevents collagen-induced arthritis. Proc Natl Acad Sci U S A. 2008; 105:19875–19880.
37. Wu PY, Nurse P. Establishing the program of origin firing during S phase in fission Yeast. Cell. 2009; 136:852–864.
38. Madine MA, Swietlik M, Pelizon C, Romanowski P, Mills AD, Laskey RA. The roles of the MCM, ORC, and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells. J Struct Biol. 2000; 129:198–210.
39. Freeman A, Morris LS, Mills AD, Stoeber K, Laskey RA, Williams GH, Coleman N. Minichromosome maintenance proteins as biological markers of dysplasia and malignancy. Clin Cancer Res. 1999; 5:2121–2132.
40. Meng MV, Grossfeld GD, Williams GH, Dilworth S, Stoeber K, Mulley TW, Weinberg V, Carroll PR, Tlsty TD. Minichromosome maintenance protein 2 expression in prostate: characterization and association with outcome after therapy for cancer. Clin Cancer Res. 2001; 7:2712–2718.
41. Loddo M, Kingsbury SR, Rashid M, Proctor I, Holt C, Young J, El-Sheikh S, Falzon M, Eward KL, Prevost T, Sainsbury R, Stoeber K, Williams GH. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. Br J Cancer. 2009; 100:959–970.
42. Wojnar A, Pula B, Piotrowska A, Jethon A, Kujawa K, Kobierzycki C, Rys J, Podhorska-Okolow M, Dziegiel P. Correlation of intensity of MT-I/II expression with Ki-67 and MCM-2 proteins in invasive ductal breast carcinoma. Anticancer Res. 2011; 31:3027–3033.
43. Lau KM, Chan QK, Pang JC, Li KK, Yeung WW, Chung NY, Lui PC, Tam YS, Li HM, Zhou L, Wang Y, Mao Y, Ng HK. Minichromosome maintenance proteins 2, 3 and 7 in medulloblastoma: overexpression and involvement in regulation of cell migration and invasion. Oncogene. 2010; 29:5475–5489.
44. Liu Y, He G, Wang Y, Guan X, Pang X, Zhang B. MCM-2 is a therapeutic target of Trichostatin A in colon cancer cells. Toxicol Lett. 2013; 221:23–30.
45. Zhang X, Teng Y, Yang F, Wang M, Hong X, Ye LG, Gao YN, Chen GY. MCM2 is a therapeutic target of lovastatin in human non-small cell lung carcinomas. Oncol Rep. 2015; 33:2599–2605.
46. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
47. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014; 25:304–317.
48. Kandioler-Eckersberger D, Ludwig C, Rudas M, Kappel S, Janschek E, Wenzel C, Schlagbauer-Wadl H, Mittlböck M, Gnant M, Steger G, Jakesz R. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res. 2000; 6:50–56.
49. Chrisanthar R, Knappskog S, Løkkevik E, Anker G, Ostenstad B, Lundgren S, Risberg T, Mjaaland I, Skjønsberg G, Aas T, Schlichting E, Fjösne HE, Nysted A, Lillehaug JR, Lønning PE. Predictive and prognostic impact of TP53 mutations and MDM2 promoter genotype in primary breast cancer patients treated with epirubicin or paclitaxel. PLoS One. 2011; 6:e19249.
50. Lehmann-Che J, André F, Desmedt C, Mazouni C, Giacchetti S, Turpin E, Espié M, Plassa LF, Marty M, Bertheau P, Sotiriou C, Piccart M, Symmans WF, Pusztai L. de Thé H: Cyclophosphamide dose intensification may circumvent anthracycline resistance of p53 mutant breast cancers. Oncologist. 2010; 15:246–252.
51. Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A. 2008; 105:7785–7790.
52. Takahashi RU, Takeshita F, Fujiwara T, Ono M, Ochiya T. Cancer stem cells in breast cancer. Cancers (Basel). 2011; 3:1311–1328.
53. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006; 355:1253–1261.
54. Wu Y, Wu PY. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev. 2009; 18:1127–1134.
55. Cho DY, Lin SZ, Yang WK, Hsu DM, Lin HL, Lee HC, Lee WY, Chiu SC. The role of cancer stem cells (CD133(+)) in malignant gliomas. Cell Transplant. 2011; 20:121–125.
56. Alison MR, Guppy NJ, Lim SM, Nicholson LJ. Finding cancer stem cells: are aldehyde dehydrogenases fit for purpose? J Pathol. 2010; 222:335–344.
57. Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, Wang H, Liu Z, Su Y, Stass SA, Katz RL. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009; 7:330–338.
58. Mohan A, Kandalam M, Ramkumar HL, Gopal L, Krishnakumar S. Stem cell markers: ABCG2 and MCM2 expression in retinoblastoma. Br J Ophthalmol. 2006; 90:889–893.
59. Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, Reynolds BA. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011; 134:1331–1343.
60. Singh SK, Venugopal C, Hallett R, Vora P, Manoranjan B, Mahendram S, Qazi M, McFarlane N, Subapanditha M, Nolte S, Singh M, Bakhshinyan D, Garg N, Vijayakumar T, Lach B, Provias JP, Reddy K, Murty N, Doble B, Bhatia M, Hassell JA. Pyrvinium targets CD133 in human glioblastoma brain tumor-initiating cells. Clin Cancer Res. 2015.
61. Fitzgerald TL, Rangan S, Dobbs L, Starr S, Sigounas G. The impact of Aldehyde dehydrogenase 1 expression on prognosis for metastatic colon cancer. J Surg Res. 2014; 192:82–89.
62. Yang L, Ren Y, Yu X, Qian F, Bian BS, Xiao HL, Wang WG, Xu SL, Yang J, Cui W, Xiong G, Yang K, Qian C, Zhang X, Zhang P, Cui YH, Bian XW. ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod Pathol. 2014; 27:775–783.
63. Zhang M, Song T, Yang L, Chen R, Wu L, Yang Z, Fang J. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J Exp Clin Cancer Res. 2008; 27:85.
64. Wu S, Yu L, Wang D, Zhou L, Cheng Z, Chai D, Ma L, Tao Y. Aberrant expression of CD133 in non-small cell lung cancer and its relationship to vasculogenic mimicry. BMC Cancer. 2012; 12:535.
65. Deng S, Yang X, Lassus H, Liang S, Kaur S, Ye Q, Li C, Wang LP, Roby KF, Orsulic S, Connolly DC, Zhang Y, Montone K, Bützow R, Coukos G, Zhang L. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS One. 2010; 5:e10277.
66. Tsang JY, Huang YH, Luo MH, Ni YB, Chan SK, Lui PC, Yu AM, Tan PH, Tse GM. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012; 136:407–417.
67. Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997; 88:223–233.
68. Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999; 285:1569–1572.
69. Guo Q, Zhao G, Hao F, Guan Y. Effects of the TAT peptide orientation and relative location on the protein transduction efficiency. Chem Biol Drug Des. 2012; 79:683–690.
70. Tanaka M, Kamata R, Yanagihara K, Sakai R. Suppression of gastric cancer dissemination by ephrin-B1-derived peptide. Cancer Sci. 2010; 101:87–93.
71. Planel S, Salomon A, Jalinot P, Feige JJ, Cherradi N. A novel concept in antiangiogenic and antitumoral therapy: multitarget destabilization of short-lived mRNAs by the zinc finger protein ZFP36L1. Oncogene. 2010; 29:5989–6003.
72. Sugita T, Yoshikawa T, Mukai Y, Yamanada N, Imai S, Nagano K, Yoshida Y, Shibata H, Yoshioka Y, Nakagawa S, Kamada H, Tsunoda SI, Tsutsumi Y. Comparative study on transduction and toxicity of protein transduction domains. Br J Pharmacol. 2008; 153:1143–1152.
73. Fan YF, Lu CZ, Xie J, Zhao YX, Yang GY. Apoptosis inhibition in ischemic brain by intraperitoneal PTD-BIR3-RING (XIAP). Neurochem Int. 2006; 48:50–59.
74. Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000; 10:290–295.
75. Wadia JS, Dowdy SF. Protein transduction technology. Curr Opin Biotechnol. 2002; 13:52–56.
76. Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med. 2007; 13:443–448.
77. Sato R, Iiizumi S, Kim ES, Honda F, Lee SK, Adachi N, Koyama H, Mizutani S, Morio T. Impaired cell adhesion, apoptosis, and signaling in WASP gene-disrupted Nalm-6 pre-B cells and recovery of cell adhesion using a transducible form of WASp. Int J Hematol. 2012; 95:299–310.
78. Douglas P, Moorhead GB, Ye R, Lees-Miller SP. Protein phosphatases regulate DNA-dependent protein kinase activity. J Biol Chem. 2001; 276:18992–18998.
79. Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005; 20:801–809.
80. Wang Q, Gao F, Wang T, Flagg T, Deng X. A nonhomologous end-joining pathway is required for protein phosphatase 2A promotion of DNA double-strand break repair. Neoplasia. 2009; 11:1012–1021.
81. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res. 2006; 66:1883–1890. discussion 1895–1886.
82. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008; 10:R25.
83. Aomatsu N, Yashiro M, Kashiwagi S, Takashima T, Ishikawa T, Ohsawa M, Wakasa K, Hirakawa K. CD133 is a useful surrogate marker for predicting chemosensitivity to neoadjuvant chemotherapy in breast cancer. PLoS One. 2012; 7:e45865.
84. Gianni L, Eiermann W, Semiglazov V, Manikhas A, Lluch A, Tjulandin S, Zambetti M, Vazquez F, Byakhow M, Lichinitser M, Climent MA, Ciruelos E, Ojeda B, Mansutti M, Bozhok A, Baronio R, Feyereislova A, Barton C, Valagussa P, Baselga J. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet. 2010; 375:377–384.
85. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006; 10:529–541.
86. Adélaïde J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, Charafe-Jauffret E, Cervera N, Desplans J, Parzy D, Schoenmakers E, Viens P, Jacquemier J, Birnbaum D, Bertucci F, Chaffanet M. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007; 67:11565–11575.