
INTRODUCTION
Cancer is a complex pathology manifested by uncontrolled growth of cells that have undergone various transformations from physiologically normal cells. Several hallmarks provide a methodical and rational approach in studying this disease, namely the sustaining of proliferative signaling, evasion of growth suppressors, resistance to cell death, replicative immortality, angiogenesis, activation of invasion, and metastasis [1]. In recent years, however, strong evidence has highlighted the critical roles of immune cells present in the tumor micro-environment [2, 3]. For instance, one way that tumor cells can modulate and escape immune destruction is by secretion of various factors such as pro-inflammatory eicosanoids, cytokines, chemokines and other soluble signaling molecules leading to the formation of an immunosuppressive tumor micro-environment [4].
Galectins are multifunctional proteins belonging to the animal lectin family. All galectins share similar binding affinities to β-galactosides and display high amino acid sequence homology among their carbohydrate-binding domains (CRDs) [5]. Fifteen different members have been identified and divided in three sub-groups according to their structure: prototype galectins containing one CRD (Gal-1, -2, -5, -7, -10, -13, -14 and -15), tandem-repeat galectins containing two-CRDs covalently linked (Gal-4, -6, -8, -9 and -12) and a chimera-type galectin containing multiple CRDs linked by their amino-terminal domain (Gal-3) [6]. While these proteins perform homeostatic functions inside normal cells, under pathological or stress conditions, cytosolic galectins are released either passively from dead cells or actively via non-classical secretion pathways [7]. Once in the extracellular milieu, they bind all glycosylated growth receptors on the surface of normal and cancer cells to set their signaling threshold [8, 9]. Such properties enable galectins to kill infiltrating immune cells while promoting growth of tumour cells [9]. Galectins are thus ideal targets for effective therapeutics, and new approaches are therefore being developed to modulate their activities [10]. These avenues have focused mainly on carbohydrate-based inhibitors disrupting extracellular galectins, which form multivalent complexes with cell surface glycoconjugates to deliver CRD-dependent intracellular signals that modulate cell activation and survival/apoptosis. Despite decades of research, however, the progression in this field has been very slow. In most cases, these inhibitors are high molecular weight, naturally occurring polysaccharides that are used to specifically block the binding of extracellular galectins to carbohydrate structures [11–14]. Unfortunately, such inhibitors often display low affinity, lack of selectivity for a given galectin due to highly conserved homology among galectin CRDs, and are not effective at targeting CRD-independent functions of galectins. Indeed, several studies have shown that several critical biological processes of galectins are mediated via CRD-independent interactions [15–18].
Sequencing of galectins isolated from amphibians, birds, fish, and mammals has revealed extensive sequence similarity [19, 20]. In addition to the presence of a CRD, all galectins harbor a highly conserved three-dimensional structure characterized by a jelly-roll topology composed of an 11- or 12-strand anti-parallel β-sandwich of approximately 135–140 amino acid residues [21]. One of the most common and important structural features associated with galectin function is their ability to form homodimers (Fig. 1B). This is particularly true for the prototype galectins, which consist of two ~14–15 kDa subunits that are non-covalently associated in a monomer-dimer equilibrium [22]. Studies of ancestral structures of fish galectins have indeed shown that galectins have gone through selective pressure for stabilizing this homodimer structure to increase their affinity for their ligand(s) [23]. Such multivalency is critical for galectins to trigger intracellular signaling following their binding to cell surface receptors [24–26]. In the present work, we report a novel peptide-based galectin inhibitor that was specifically designed to disrupt the formation of galectin-7 dimers and its pro-apoptotic function.
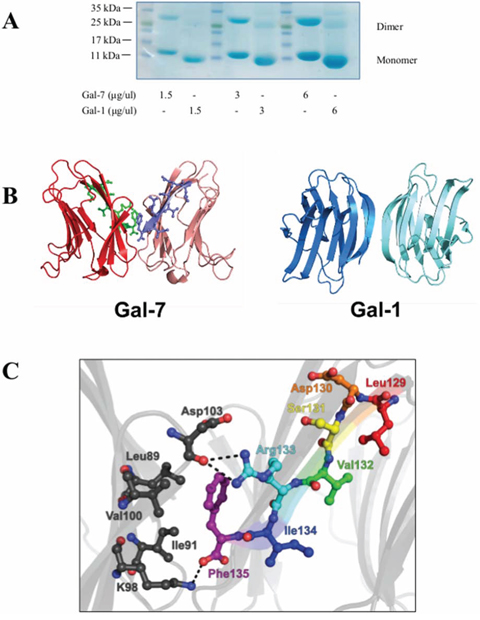
Figure 1: The dimeric structure of hGal-7. A. Dimer formation of recombinant hGal-7 and hGal-1 at increasing concentrations were compared by polyacrylamide gel electrophoresis in native conditions. B. Structural representation of the hGal-7 (PDB 1BKZ) and hGal-1 (PDB 3W58) dimers with residues 129–135 colored in green and magenta on the hGal-7 dimer interface. Dimer formation in hGal-7 proceeds through a “back-to-back” topology of the monomers while hGal-1 adopts a “side-by-side” structural arrangement, affording additional specificity for galectin inhibition. C. Molecular interactions implicated in the wild-type hGal-7 dimer interface between residues 129–135 of the first hGal-7 monomer (in various colors) and facing residues on the second hGal-7 monomer (in black) (PDB 1BKZ). Hydrogen bonding and electrostatic interactions are identified as dashed lines. The side chain of Phe135 is also involved in a number of van der Waals interactions [29]. The structures were prepared with PyMOL.
RESULTS
As depicted with G protein-coupled receptors, peptides derived from the dimeric interface were shown to disrupt GPCR dimers by interfering with critical interactions between amino acids located at the dimer interface [27, 28]. We hypothesized that the ability of hGal-7 to form homodimers is mediated by critical residues located at the homodimer interface located in a distant region of the CRD. Using a previously described dimeric crystal structure of hGal-7 [29], critical residues possibly involved in the formation of the dimer interface were identified based on their propensity to form hydrogen bonding, hydrophobic, or van der Waals interactions between both protomers of the complex (Fig. 1B). In addition, the design of our peptides was also based on the structural analysis of the hGal-7 dimeric interface, recently reported by Ermakova and colleagues [30]. Specifically, their analysis revealed that certain residues were likely to be involved in the dimerization of hGal-7, most notably residues R14, R20, R22, E87, L89, D94, D95, D103, A104, Q105, D130 and F135. These residues appear to symmetrically associate with their respective partner on the second monomer through H-bonding, electrostatic or hydrophobic interactions, or via the formation of disulfide bridges. As such, the peptides were designed as to rationally mimic and disrupt the hGal-7 segment between residues 13–25 and 129–135, since those residues appear to be directly involved in the stabilization of the dimeric structure (Fig. 1C) and consequently hindering the interaction of R14, R20, R22, D130 and F135 with their mirror partners. Accordingly, peptides corresponding to these regions were synthesized, i.e. hGal-7(13–25) (H-Ile-Arg-Pro-Gly-Thr-Val-Leu-Arg-Ile-Arg-Gly-Leu-Val-NH2) and hGal-7(129–135) (H-Leu-Asp-Ser-Val-Arg-Ile-Pro-NH2). To determine whether these peptides could inhibit the formation of the hGal-7 homodimer, we incubated 0.5 μM of recombinant hGal-7 with increasing concentrations of hGal-7(13–25) or hGal-7(129–135) and measured the formation of homodimers.
To measure the ability of the peptides to disrupt the formation of hGal-7 dimers, we used mild denaturing native gel electrophoresis, a commonly used approach to visualize monomer-dimer equilibrium (Fig. 1A and Supplementary Fig. S1) [31–35]. Our results showed a consistent decrease of hGal-7 homodimers starting at a 10 μM concentration of hGal-7(129–135), with a saturation dose of 100 μM (Fig. 2A). Similar results were obtained with hGal-7(13–25) but this compound appeared less potent than hGal-7(129–135) to disrupt hGal-7 homodimers and was consequently not used in subsequent experiments (data not shown). No such effect was observed using the control pituitary adenylate cyclase-activating polypeptide (PACAP) peptide, which was selected based on similarity in amino acid length and minimal toxicity on the cell line, PACAP(28–38) (Fig. 2A), or on a recombinant human gal-1 (hGal-1) or gal-2 (hGal-2) (Fig. 2B, 2C). hGal-1 and gal-2 were chosen as galectin selectivity controls since they are protype galectin that share the greatest sequence similarity to galectin-7 (38%) [29]. Moreover, the ability of the hGal-7(129–135) peptide to disrupt the formation of hGal-7 homodimers was not inhibited by the presence of lactose or LacNac (Fig. 2D, 2E). Further, the ability of hGal-7(129–135) to bind hGal-7 in a concentration-dependent and specific manner was further confirmed using a solid phase binding assay. In this assay, a biotinylated version of hGal-7(129–135) still capable of specifically inhibiting the formation of hGal-7 homodimers (Supplementary Fig. S2) was used to measure binding on immobilized recombinant hGal-7 (Fig. 3). Again, binding was shown to be specific since biotinylated hGal-7(129–135) could bind hGal-7 and not hGal-1 or hGal-2 (Supplementary Fig. S2). This specificity at disrupting hGal-7 dimer formation is provided by distinct three-dimensional arrangements between otherwise very similar galectin homologues. Indeed, while all monomeric galectins reveal identical topologies, dimer formation in hGal-7 proceeds through a “back-to-back” topology of the monomers while other galectins, including hGal-1 and hGal-2, adopt a “side-by-side” structural arrangement (Fig. 1B) [29]. This structural organization provides additional means to specifically target and disrupt galectin function.
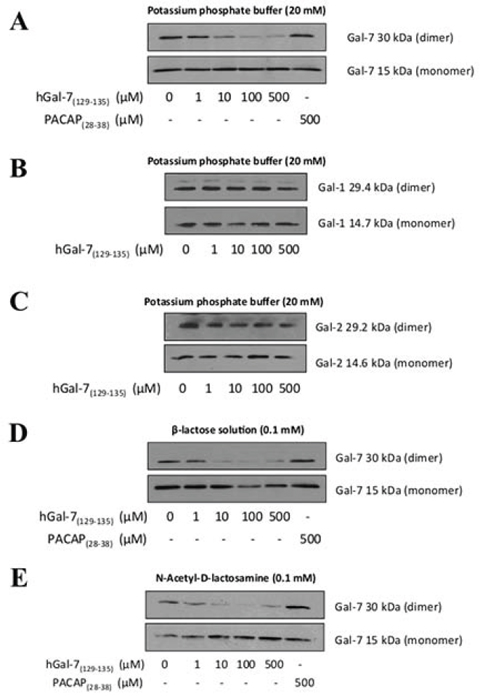
Figure 2: Disruption of the hGal-7 dimer due to increasing concentrations of hGal-7(129–135). A. The recombinant hGal-7 (0.5 μM) was incubated with increasing concentrations of hGal-7(129–135) in 20 mM potassium phosphate buffer (pH 7.1). Incubation of the recombinant B. hGal-1 and C. hGal-2 with hGal-7(129–135) was performed in the same potassium phosphate buffer. The effect of hGal-7(129–135) on the monomeric and dimeric forms of hGal-7/hGal-1/hGal-2 was assessed by Western blotting in native conditions with respective antibodies. The hGal-1/hGal-2 films were overexposed. PACAP28–38 was the control peptide used in order to ensure the specificity of hGal-7(129–135). Recombinant hGal-7 (0.5 μM) was also incubated with increasing concentrations of hGal-7(129–135) in 0.1 mM D. lactose or E. LacNac solutions. Results are representative of three independent experiments.
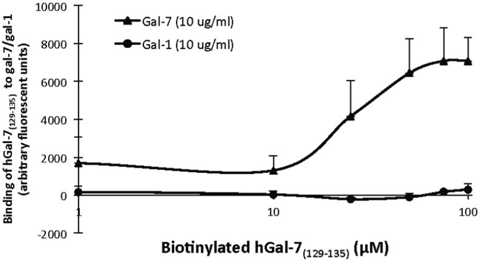
Figure 3: Biotin-labeled hGal-7(129–135) is capable of binding to recombinant hGal-7. Binding curve showing a dose-dependent interaction between biotin-labeled hGal-7(129–135) and hGal-7 or hGal-1. Recombinant hGal-7 or hGal-1 (10 μg/ml) were coated on 96-well plates overnight and then incubated 60 min with unlabeled hGal-7(129–135) (1 mM) to eliminate non-specific binding. Incubation with increasing concentrations of biotin-labeled hGal-7(129–135) was performed for 120 min. Results are representative of three independent experiments. Error bars represent standard deviation.
Galectins are well known for their ability to bind glycosylated cell surface receptors, most notably on Jurkat T cells, on which galectin binding induces apoptosis [36–42]. We thus tested whether hGal-7(129–135) could modulate the binding of hGal-7 on Jurkat T cells, a cell model that is routinely used to test the pro-apoptotic activity of galectins [43–45]. For this purpose, recombinant hGal-7 was labeled with fluorescein isothiocyante (FITC) and its binding on the surface of Jurkat T cells was measured by flow cytometry in absence or presence of increasing concentrations of hGal-7(129–135). Our results showed that hGal-7(129–135) increased the fluorescent intensity of Jurkat T cells in a concentration-dependent manner following incubation with equal amounts (0.1 μM) of FITC-labeled hGal-7, as compared to fluorescence measured in absence of peptide (Fig. 4A). No such effect was observed in presence of a high concentration of the control peptide (PACAP(28–38)). The effect of hGal-7(129–135) was specific, statistically significant (Fig. 4B), and consistent with the increased number of monomers, which bind to surface glycosylated receptors through their CRDs [38]. The hGal-7(129–135) peptide also inhibited the ability of hGal-7 to induce apoptosis in Jurkat T cells, as measured by poly [ADP-ribose] polymerase 1 (PARP-1) cleavage (Fig. 5A). This inhibitory effect was dose-dependent and also observed using human peripheral blood monocytes (Supplementary Fig. S3). No such effect was observed with the control peptide. This effect on apoptosis was confirmed by flow cytomtery using Annexin V/propidium iodide (PI) staining (Fig. 5B and 5C). Incubation of hGal-7(129–135) (or PACAP(28–38)) alone did not induce apoptosis in Jurkat T cells (Supplementary Fig. S4).
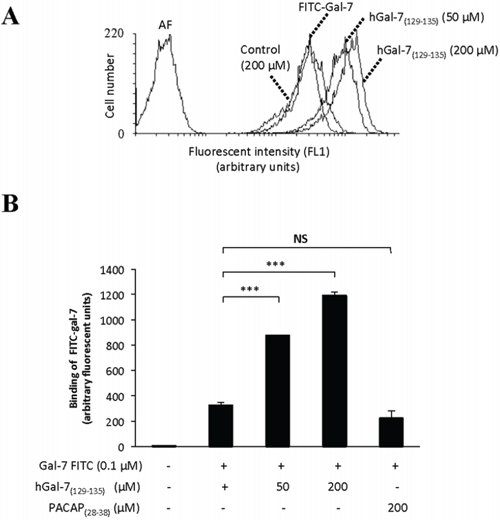
Figure 4: Increased binding of hGal-7 on Jurkat T cells due to increasing concentrations of hGal-7(129–135). A. Histogram showing the mean fluorescence intensities (MFI) of cells following binding of fluorescein isothiocyanate (FITC)-labeled hGal-7 on the surface of Jurkat T cells. B. Binding of FITC-labeled hGal-7 (0.1 μM) on Jurkat T cells following pre-incubation with increasing concentrations of hGal-7(129–135). The PACAP(28–38) peptide was used as a control. Results are representative of three independent experiments. ***p ≤ 0.05. Error bars represent standard deviations.
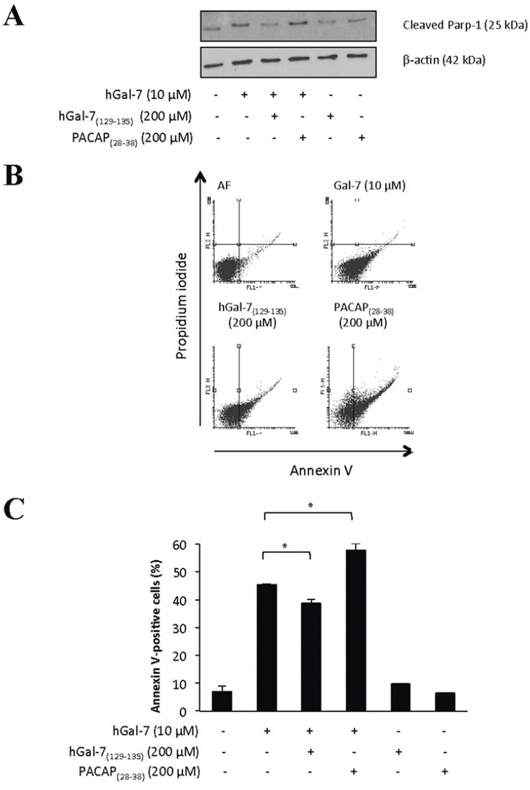
Figure 5: Apoptotic levels of Jurkat T cells induced by hGal-7 were decreased due to the presence of hGal-7 (129–135). A. Recombinant hGal-7 was pre-incubated with the respective peptide concentrations prior to its addition to Jurkat T cells for 4 h a 37°C in serum-free RPMI. Apoptosis was measured by Western blot analysis of PARP-1 cleavage. The PACAP28–38 was used as a control. β-actin was used as a loading control. B. Flow cytometric analysis of Jurkat T cells with stained Annexin V (FL1) and propidium iodide-PI (FL3) following binding of recombinant hGal-7 with or without hGal-7(129–135). Cells in the lower right quadrant are representative of annexin V-positive/PI-negative, or early apoptotic cells. Cells in the upper right quadrant indicate annexin V-positive/PI-positive, or late apoptotic cells. C. Histogram showing the percentages of annexin V-positive Jurkat T cells obtained by adding the percentages of cells found in the lower and upper right quadrants. Results are representative of three independent experiments. Error bars represent standard deviation.
DISCUSSION
In the present work, we report a new peptidic inhibitor that specifically binds hGal-7, disrupts the formation of homodimers, and inhibits the pro-apoptotic activity of hGal-7 on Jurkat T cells. These results show that the dimer interface of the prototype galectins is a viable option for the inhibition of galectins and a potential therapeutic target for the treatment of many diseases in which galectins are involved. To our knowledge, there have been no published reports of dimerization-disrupting inhibitors of galectins. Such inhibitors provide a new avenue for blocking hGal-7 activity and represent attractive agents against the prototype galectins because they target a second distant site from the active CRD. They are also a complementary alternative to soluble glycans or other small molecular weight allosteric inhibitors that are currently being developed for the targeting of prototype galectins. This peptidic inhibitor also represents an additional means to achieve specificity when targeting galectins. Although soluble glycans have shown physiological responses in in vivo assays for various pathologies where galectins are involved, there is yet little evidence of their specificity. This may not be surprising since galectins display high homology among their CRDs and often show redundancy when binding to glycosylated moieties [29, 46]. Hence, their specific targeting remains ambiguous and challenging, and the use of peptides could improve the current library of selective galectin inhibitors.
We attempted to synthesize other peptides covering other potential sites at the dimeric interface of hGal-7. This strategy was unsuccessful since these peptides were either toxic to Jurkat T cells, showed poor solubility, or were not as effective at disrupting the dimeric interface as hGal-7(129–135) (data not shown). Although we did find a direct interaction between hGal-7(129–135) and hGal-7 through a classical solid-phase binding assay, further tests are required to determine how hGal-7(129–135) binds to the critical residues located at the interface. Interestingly, our preliminary data using an alanine scan strategy has already shown that the substitution of the Asp130 residue by an Ala moiety completely abrogates the ability of the peptide to disrupt hGal-7 homodimers (Supplementary Table 1 and Supplementary Fig. S5). It is clear, however, that hGal-7(129–135) was effective at relatively high concentrations and future modifications will be needed to improve its binding affinity. These alanine scans nevertheless suggest that binding of hGal-7(129–135) to monomeric hGal-7 proceeds in a non-native fashion relative to wild-type hGal-7 dimer formation. Indeed, it is currently impossible to structurally rationalize some of the molecular effects of the alanine scan results in the context of wild-type hGal-7 dimer formation. For instance, while Asp130 does not participate in the formation or stabilization of the wild-type hGal-7 dimer interface (Fig. 1C), its replacement to alanine clearly shows significant alteration of the hGal-7(129–135) potency Supplementary Fig. S5. These results suggest a distinct binding mode between hGal-7(129–135) and monomeric hGal-7, requiring further structural analyses.
Moreover, the modulation of hGal-7 binding on the surface of Jurkat T cells and an apoptotic response were observed in the presence of the hGal-7(129–135) peptide. The increase in fluorescence was the manifestation of the increased hGal-7 binding on cell surface rather than its accumulation inside the cell, since the binding assays were performed at 0°C and in the presence of sodium azide, which would limit protein internalization [47, 48]. Whether this reflects increased binding of monomeric forms of Gal-7, however, is currently unclear and will require more experiments. Nevertheless, the increase of hGal-7 cell surface binding, seen in the presence of hGal-7(129–135), is specific since the control peptide, PACAP(28–38), did not display such effects. Interestingly, even though an increase of cell surface hGal-7 binding was observed, a reduction in the ability of the protein to induce apoptosis of T cells was observed. This supports the idea that the increase in hGal-7 binding on cell surface is due to the increased access of the monomer's CRDs binding glycosylated residues on cell surface receptors while lacking intracellular signaling, highlighting that effective crosslinking of cell surface receptors is critical for inducing apoptosis [25, 49]. Such peptide may thus be a valable tool to study the ability of Gal-7 to modulate T cells functions in vitro or in vivo. We and others have indeed shown that Gal-7 has a strong cytotoxic and inhibitory effects on T cell functions and survival [38, 50], consistent with the ability of galectins to induce to a local (and systemic ?) immunosuppression when produced by cancer cells. Intriguingly, a slight increase in apoptosis is observed with the control peptide, even though the PACAP(28–38) peptide did not display any toxicity on Jurkat T cells alone. PACAP(28–38) is a positively charged peptide that binds cell surface phospholipids but does not translocate within the cytoplasm [51]. Even if the peptide alone does not induce apoptosis, the perturbation exerted by PACAP(28–38) at the cell surface might slightly increase hGal-7-induced necrosis.
MATERIALS AND METHODS
Cell lines and reagents
The Jurkat cell line was maintained in RPMI 1640 medium. The culture medium was supplemented with 10% [v/v] fetal bovine serum, 2 mmol/L L-glutamine, 10 mM HEPES buffer, and 1 mM sodium pyruvate. All cell culture products were purchased from Life Technologies (Burlington, ON, Canada). Isolation of peripheral mononuclear cell (PBMCs) was performed as previously described [17].
Peptide synthesis
The hGal-7(129–135) peptide and its Ala-substituted derivatives ([Ala130]hGal-7(129–135), [Ala131]hGal-7(129–135), [Ala133]hGal-7(129–135) and [Ala135]hGal-7(129–135)), as well as the control peptide PACAP28–38 (an inactive fragment of the human Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP)), were synthesized using standard Fmoc chemistry, as previously described [52]. Briefly, all peptides were assembled using a semi-automatic multi-reactor system. The Rink-amide resin was used as the solid support, and the amino acids of the peptide sequences were introduced under their Fmoc-N-protected form, i.e. 3 eq based on the original substitution of the resin (0.7 mmol.g-1). Couplings of the protected amino acids were mediated by (Benzotriazol-1-yloxy)tris(dimethylamino) phosphonium hexafluorophosphate (BOP, 3 eq) and N, N-Diisopropylethylamine (DIPEA, 5 eq) in DMF for 1 h. Coupling efficiency was monitored with the qualitative ninhydrin test. Fmoc removal was achieved with 20% piperidine in DMF for 20 min. In order to obtain a biotin-conjugated peptide, an ε-amino acid (Fmoc-Ahx-OH) linker was first coupled, as described above, to peptidyl resins and then, following the removal of the Fmoc protecting group, a Biotin-NHS derivative (6 eq, Aapptec) was attached to the peptidyl resins with triethylamine (TEA, 6 eq) in dimethylformamide. Peptides were then deprotected and removed from the resin via an acidolytic treatment with trifluoroacetic acid (TFA) containing 1,2-ethanedithiol (2.5%), phenol (3%) and water (2.5%) for 2 h at room temperature. The diethyl ether-precipitated crude peptides were purified by preparative RP-HPLC performed on a Waters PrepLC 4000 System with a Waters 2487 detector set at 220 nm and an XTerra Prep MS C18 column. A linear gradient from eluent A to B with 1% B per 2-min increments (Eluent A = H2O, 0.1% TFA, Eluent B = 60% CH3CN/40% H2O, 0.1% TFA) was used for each purification. Collected fractions were then analyzed by matrix-assisted laser desorption/ionization – time-of-flight (MALDI-TOF) mass spectrometry (Voyager DE System from Applied Biosystems) in linear mode using the α-cyano-4-hydroxycinnamic acid matrix (Carlsbad, CA, USA) and analytical RP-HPLC with a Phenomenex Jupiter C18 column to ensure their homogeneity. Fractions corresponding to the desired product with purity greater than 98% were pooled and lyophilized.
Production of recombinant hGal-7 and hGal-1
Human Gal-7 cDNA was cloned into pET-22b(+) using NdeI and HindIII restriction enzymes. Human pET-Gal-1 vector was generously donated by Dr. S. Sato (McGill University, QC, Canada). The proteins were produced in E. coli BL21 (DE3) cells at 37°C. Isopropyl β-D-1-thiogalactopyranoside (IPTG) (1 mM) was added to the bacterial culture at OD600nm = 0.6–0.7 and then incubated for 4 h at 37°C to allow protein production. Bacterial pellets were resuspended in lysis buffer (0.7 mg/mL lysozyme, 10 mM Tris pH 8, 100 mM NaCl, 1 mM EDTA, 1 mM DTT and a protease inhibitor cocktail) and then incubated for 1 h at 37°C prior to centrifugation for 30 min at 15,000 × g (4°C). The supernatant was then filtered with 500 mL bottle top filter (22 μm) (Corning, New York, NY, USA) and then ran through a lactose-agarose column (Sigma, St. Louis, MO, USA). The protein was eluted in 1 mL fractions with 150 mM lactose solution. Purified fractions were analyzed by SDS-PAGE. The hGal-7 was then concentrated and purified using Centrifugal filter units (Amicon Ultra-15, 10K) (EMD, Millipore, Etobicoke, ON) in 20 mM potassium phosphate at pH 7.1. All subsequent experiments with the recombinant proteins were performed in the same buffer solution unless mentioned otherwise. Brilliant Coomassie blue was purchased from BioRad (Bio-Rad Laboratories, Mississauga, ON, Canada). The recombinant protein hGal-2 was purchased from MyBiosource (San Diego, CA, USA).
Western blotting
For the apoptosis tests, whole-cell extracts were homogenized and resuspended in RIPA buffer (Thermo Fisher Scientific, Rockford, IL, USA) containing protease inhibitors (Roche, Laval, QC, Canada). Equal amounts of whole-cell extracts (25 μg) were separated on SDS-PAGE and transferred onto nitrocellulose membranes (Bio-Rad Laboratories). The membranes were first blocked with 5% milk [w/v] in TBS/0.5% Tween 20 [v/v] for 1 h at room temperature and subsequently blotted overnight in a solution of TBS containing 3% BSA [w/v] and 0.5% Tween 20 [v/v]. The following antibodies were used: a rabbit anti-poly-(ADP-ribose) polymerase (PARP)-1 (p25) polyclonal antibody (1:5000; Epitomics, Burlingame, CA, USA) and a mouse anti-β-actin (1:10000; Sigma-Aldrich, St. Louis, MO, USA). Secondary antibodies consisted of horseradish peroxidase-conjugated donkey anti-rabbit (GE Healthcare, Buckinghamshire, England) and sheep anti-mouse (GE Healthcare) IgG. Detection was performed using the enhanced chemiluminescence method (GE Healthcare). For the recombinant protein tests, each peptide was dissolved and maintained in 20 mM potassium phosphate at pH 7.1. The β-lactose and N-Acetyl-D-lactosamine (LacNAc) powders were purchased from Sigma-Aldrich (St. Louis, MO, USA). The recombinant proteins and peptide dilutions were pre-incubated for 1 h at 4°C prior to gel migration. The native polyacrylamide gel was made without SDS to allow molecular weight differentiation between the dimer and monomer. The following antibodies were used: a goat anti-Gal-7 antibody (1:10000; R&D Systems, Minneapolis, MN, USA), a mouse anti-Gal-1 antibody (1:1000; Proteintech, Chicago, IL, USA) and a rabbit anti-Gal-2 antibody (1:1000; Proteintech, Chicago, IL, USA). Secondary antibodies consisted of donkey anti-goat (R&D Systems) or sheep anti-mouse (GE Healthcare) IgG. Detection was performed as mentioned above.
Fluorescent binding assay
Recombinant hGal-7 or hGal-1 (10 μg/mL) was coated overnight at 4°C on black, flat bottom, 96-well polystyrene microplates (Ultident, Montreal, QC, Canada). Thereafter, the plate was blocked with Reagent diluent (PBS-BSA 1%) for 1 h, then incubated with unlabeled hGal-7(129–135) (cold) for 60 min and finally incubated with biotin-labeled [Ahx0]hGal-7(129–135) for 2 h. Lastly, Streptavidin R-phycoerythrin (1/500, Jackson Immunoresearch, West Grove, PA, USA) was applied to samples for 30 min. All incubations were performed at room temperature and the washes between incubations were done with 20 mM potassium phosphate buffer pH 7.1. The plate was read by a Tecan Infinite M1000 PRO microplate reader at excitation and emission wavelengths of 488 nm and 670 nm, respectively.
FITC conjugation and hGal-7 binding assay
To assess hGal-7 binding onto Jurkat T cells, 5 μL of a 1.25 mg/mL fluorescein isothiocyanate (FITC)/DMSO solution was added to 300 μL of 1.7 μg/μL recombinant hGal-7 in a 0.1 M NaHCO3 pH 9.2 solution and incubated for 1 h at room temperature on a roller. FITC-labeled hGal-7 was then purified using a PD-10 Sepharose column (GE healthcare) and eluted with PBS containing 0.01% [v/v] sodium azide. FITC-labeled hGal-7 (0.1 μM) was then pre-incubated with hGal-7(129–135) (or related peptides) in 20 mM potassium phosphate buffer pH 7.1 for 1 h at 4°C. Jurkat cells (5 × 105 cells per sample) were harvested in PBS-0.01% [v/v] sodium azide and incubated for 30 min on ice with the FITC-labeled hGal-7 with and without peptides. Cells were then washed with PBS-0.01% [v/v] sodium azide and resuspended in 400 μL of the same buffer and analyzed on a FACSCalibur (BD Biosciences).
Apoptosis assays with Annexin V/PI staining
Apoptosis was measured by flow cytometry using FITC-labeled Annexin V (Biolegend, San Diego, CA, USA) and propidium iodide (PI). Briefly, the corresponding dilutions of recombinant hGal-7 and hGal-7(129–135) peptides were pre-incubated for 1 h at 4°C in serum free RPMI 1640 medium. 2.5 × 105 Jurkat cells were then harvested in the same medium and incubated with their corresponding dilutions at 37°C for 4 h. Cells were washed once in PBS and once in binding buffer (0.01 M HEPES, 0.14 M NaCl, 2.5 mM CaCl2, pH 7.4). Cells were then incubated for 15 min with Annexin V in the dark at room temperature. A total of 400 μL of binding buffer containing 0.25 μg/mL propidium iodide was added to the cells before analysis by flow cytometry.
Statistical analysis
Statistical significance of the experiments was evaluated using the unpaired Student's t-test or the Fisher's exact test. Results were considered statistically significant at P ≤ 0.05.
CONCLUSIONS
We have identified a new class of galectin inhibitors that specifically target the dimer interface of hGal-7. Such inhibitor provides an interesting alternative to more conventional galectin inhibitors that target the CRD with soluble glycans. Given the critical role of hGal-7 in cancer, studies are underway to determine whether both types of inhibitors could be used in combination as anti-cancer agents.
ACKNOWLEDGMENTS
The authors acknowledge the support from the FRQNT Strategic Cluster ‘‘Regroupement Québécois de Recherche sur la Fonction, la Structure et l’Ingénierie des Protéines’’ (PROTEO) and the FRQS Strategic Cluster ‘‘Groupe de Recherche Axé sur la Structure des Protéines’’ (GRASP).
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
GRANT SUPPORT
Supported by a team grant to Y.S.P.-N.D.-D.C. from the Fonds de recherche du Québec-Nature et Technologies (FRQ-NT) and by Natural Sciences and Engineering Research Council Discovery grants (RGPIN 402623-2011 to N.D and RGPIN 298215-2013). A.A.G. and M.L. are supported by studentships from Fonds de Recherche du Québec-Santé (FRQS) and N.D. by a Research Scholar Career Award (Junior 1) from the FRQS.
REFERENCES
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
2. Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015; 25:198–213.
3. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19:1423–1437.
4. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010; 10:181–193.
5. Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, et al. Galectins: a family of animal β-galactoside-binding lectins. Cell. 1994; 76:597–598.
6. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005; 5:29–41.
7. Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. 2009; 7:424–438.
8. Wang W, Guo H, Geng J, Zheng X, Wei H, Sun R, Tian Z. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J Biol Chem. 2014; 289:33311–33319.
9. Stannard KA, Collins PM, Ito K, Sullivan EM, Scott SA, Gabutero E, Darren Grice I, Low P, Nilsson UJ, Leffler H, Blanchard H, Ralph SJ. Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett. 2010; 299:95–110.
10. Yang RY, Rabinovich GA, Liu FT. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med. 2008; 10:e17.
11. Inohara H, Raz A. Effects of natural complex carbohydrate (citrus pectin) on murine melanoma cell properties related to galectin-3 functions. Glycoconj J. 1994; 11:527–532.
12. Pienta KJ, Naik H, Akhtar A, Yamazaki K, Replogle TS, Lehr J, Donat TL, Tait L, Hogan V, Raz A. Inhibition of spontaneous metastasis in a rat prostate cancer model by oral administration of modified citrus pectin. J Natl Cancer Inst. 1995; 87:348–353.
13. Nangia-Makker P, Hogan V, Honjo Y, Baccarini S, Tait L, Bresalier R, Raz A. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J Natl Cancer Inst. 2002; 94:1854–1862.
14. Dings RP, Miller MC, Nesmelova I, Astorgues-Xerri L, Kumar N, Serova M, Chen X, Raymond E, Hoye TR, Mayo KH. Antitumor agent calixarene 0118 targets human galectin-1 as an allosteric inhibitor of carbohydrate binding. J Med Chem. 2012; 55:5121–5129.
15. Villeneuve C, Baricault L, Canelle L, Barboule N, Racca C, Monsarrat B, Magnaldo T, Larminat F. Mitochondrial proteomic approach reveals galectin-7 as a novel BCL-2 binding protein in human cells. Mol Biol Cell. 2011; 22:999–1013.
16. Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001; 20:7486–7493.
17. Akahani S, Nangia-Makker P, Inohara H, Kim HR, Raz A. Galectin-3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 1997; 57:5272–5276.
18. Shimura T, Takenaka Y, Fukumori T, Tsutsumi S, Okada K, Hogan V, Kikuchi A, Kuwano H, Raz A. Implication of galectin-3 in Wnt signaling. Cancer Res. 2005; 65:3535–3537.
19. Cooper DN. Galectinomics: finding themes in complexity. Biochim Biophys Acta. 2002; 1572:209–231.
20. Fink NE. Soluble β-galactoside-binding lectins [Article in Spanish]. Acta Physiol Pharmacol Ther Latinoam. 1996; 46:1–10.
21. Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins. Structure and function of a large family of animal lectins. J Biol Chem. 1994; 269:20807–20810.
22. Giudicelli V, Lutomski D, Levi-Strauss M, Bladier D, Joubert-Caron R, Caron M. Is human galectin-1 activity modulated by monomer/dimer equilibrium? Glycobiology. 1997; 7:viii–x.
23. Levi G, Teichberg VI. Isolation and physicochemical characterization of electrolectin, a β-D-galactoside binding lectin from the electric organ of Electrophorus electricus. J Biol Chem. 1981; 256:5735–5740.
24. Fred Brewer C. Binding and cross-linking properties of galectins. Biochim Biophys Acta. 2002; 1572:255–262.
25. Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol. 2007; 17:513–520.
26. Garner OB, Baum LG. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans. 2008; 36:1472–1477.
27. Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996; 271:16384–16392.
28. George SR, Lee SP, Varghese G, Zeman PR, Seeman P, Ng GY, O’Dowd BF. A transmembrane domain-derived peptide inhibits D1 dopamine receptor function without affecting receptor oligomerization. J Biol Chem. 1998; 273:30244–30248.
29. Leonidas DD, Vatzaki EH, Vorum H, Celis JE, Madsen P, Acharya KR. Structural basis for the recognition of carbohydrates by human galectin-7. Biochemistry. 1998; 37:13930–13940.
30. Ermakova E, Miller MC, Nesmelova IV, Lopez-Merino L, Berbis MA, Nesmelov Y, Tkachev YV, Lagartera L, Daragan VA, Andre S, Canada FJ, Jimenez-Barbero J, Solis D, Gabius HJ, Mayo KH. Lactose binding to human galectin-7 (p53-induced gene 1) induces long-range effects through the protein resulting in increased dimer stability and evidence for positive cooperativity. Glycobiology. 2013; 23:508–523.
31. Jahnel R, Dreger M, Gillen C, Bender O, Kurreck J, Hucho F. Biochemical characterization of the vanilloid receptor 1 expressed in a dorsal root ganglia derived cell line. Eur J Biochem. 2001; 268:5489–5496.
32. Klodmann J, Lewejohann D, Braun HP. Low-SDS Blue native PAGE. Proteomics. 2011; 11:1834–1839.
33. Lin CL, Huang YT, Richter JD. Transient CPEB dimerization and translational control. RNA. 2012; 18:1050–1061.
34. Hoang T, Smith MD, Jelokhani-Niaraki M. Expression, folding, and proton transport activity of human uncoupling protein-1 (UCP1) in lipid membranes: evidence for associated functional forms. J Biol Chem. 2013; 288:36244–36258.
35. Giudici AM, Molina ML, Ayala JL, Montoya E, Renart ML, Fernandez AM, Encinar JA, Ferrer-Montiel AV, Poveda JA, Gonzalez-Ros JM. Detergent-labile, supramolecular assemblies of KcsA: relative abundance and interactions involved. Biochim Biophys Acta. 2013; 1828:193–200.
36. Ito K, Stannard K, Gabutero E, Clark AM, Neo SY, Onturk S, Blanchard H, Ralph SJ. Galectin-1 as a potent target for cancer therapy: role in the tumor microenvironment. Cancer Metastasis Rev. 2012; 31:763–778.
37. Radosavljevic G, Volarevic V, Jovanovic I, Milovanovic M, Pejnovic N, Arsenijevic N, Hsu DK, Lukic ML. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol Res. 2012; 52:100–110.
38. Labrie M, Vladoiu MC, Grosset AA, Gaboury L, St-Pierre Y. Expression and functions of galectin-7 in ovarian cancer. Oncotarget. 2014; 5:7705–7721.
39. Norambuena A, Metz C, Vicuna L, Silva A, Pardo E, Oyanadel C, Massardo L, Gonzalez A, Soza A. Galectin-8 induces apoptosis in Jurkat T cells by phosphatidic acid-mediated ERK1/2 activation supported by protein kinase A down-regulation. J Biol Chem. 2009; 284:12670–12679.
40. Petelenz T, Knosala P, Korzeniowska B, Flak Z, Skrzypek-Wanha J. Pol Tyg Lek. 1991; 46:39–42.
41. Tribulatti MV, Mucci J, Cattaneo V, Aguero F, Gilmartin T, Head SR, Campetella O. Galectin-8 induces apoptosis in the CD4(high)CD8(high) thymocyte subpopulation. Glycobiology. 2007; 17:1404–1412.
42. Fukata Y, Itoh A, Nonaka Y, Ogawa T, Nakamura T, Matsushita O, Nishi N. Direct cytocidal effect of galectin-9 localized on collagen matrices on human immune cell lines. Biochim Biophys Acta. 2014; 1840:1892–1901.
43. Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995; 378:736–739.
44. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, Baum LG. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006; 176:778–789.
45. Xue J, Gao X, Fu C, Cong Z, Jiang H, Wang W, Chen T, Wei Q, Qin C. Regulation of galectin-3-induced apoptosis of Jurkat cells by both O-glycans and N-glycans on CD45. FEBS Lett. 2013; 587:3986–3994.
46. Henrick K, Bawumia S, Barboni EA, Mehul B, Hughes RC. Evidence for subsites in the galectins involved in sugar binding at the nonreducing end of the central galactose of oligosaccharide ligands: sequence analysis, homology modeling and mutagenesis studies of hamster galectin-3. Glycobiology. 1998; 8:45–57.
47. Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP. Azide-resistant mutants of Escherichia coli alter the SecA protein, an azide-sensitive component of the protein export machinery. Proc Natl Acad Sci U S A. 1990; 87:8227–8231.
48. Kubak BM, Yotis WW. Staphylococcus aureus adenosine triphosphatase: inhibitor sensitivity and release from membrane. J Bacteriol. 1981; 146:385–390.
49. Boscher C, Dennis JW, Nabi IR. Glycosylation, galectins and cellular signaling. Curr Opin Cell Biol. 2011; 23:383–392.
50. Yamaguchi T, Hiromasa K, Kabashima-Kubo R, Yoshioka M, Nakamura M. Galectin-7, induced by cis-urocanic acid and ultraviolet B irradiation, down-modulates cytokine production by T lymphocytes. Exp Dermatol. 2013; 22:840–842.
51. Doan ND, Letourneau M, Vaudry D, Doucet N, Folch B, Vaudry H, Fournier A, Chatenet D. Design and characterization of novel cell-penetrating peptides from pituitary adenylate cyclase-activating polypeptide. J Control Release. 2012; 163:256–265.
52. Brkovic A, Hattenberger A, Kostenis E, Klabunde T, Flohr S, Kurz M, Bourgault S, Fournier A. Functional and binding characterizations of urotensin II-related peptides in human and rat urotensin II-receptor assay. J Pharmacol Exp Ther. 2003; 306:1200–1209.