
INTRODUCTION
Colorectal cancer (CRC) is a heterogeneous disease with progressive accumulation of genetic and epigenetic aberrations and defects in immune surveillance [1–3]. Activation of oncogenes and inactivation/loss of tumor suppressor genes (TSGs) are crucial for transformation of malignant cells [4, 5]. p53, as a stress-inducible transcription factor, regulates a diversity of biological functions, such as cell cycle arrest, apoptosis, senescence, and autophagy, via mediating its plethora of target genes [6–8]. In CRC, p53 mutation occurs in about 40–50% of cases [9–11]. Mutated p53 loses its tumor suppressive function, which is a critical event in the adenoma to carcinoma transition during colon carcinogenesis [12]. Scientists have been enthusiastic in developing different strategies to reactivate mutated p53 in cancer cells as an anti-cancer therapy [13, 14]. So far, a variety of compounds has been investigated to target mutant p53 [15, 16]. PRIMA-1 (p53-reactivation and induction of massive apoptosis-1), a low molecular weight compound (C9H15NO3), was discovered to restore the mutant p53 to the structure and function of wild-type p53, thus selectively killing cancer cells with mutant p53 [17]. PRIMA-1met (APR-246), a methylated and more potent analog of PRIMA-1, has already advanced to a phase I/II clinic trial in hematologic malignancies and prostate cancer [18, 19]. It has been reported that PRIMA-1/PRIMA-1met selectively restores the sequence-specific DNA binding region of mutated p53 via forming adducts with thiols and recovers its normal wild-type function to induce apoptosis in cancer cells [17, 20, 21]. However, some recent studies discovered that PRIMA-1/PRIMA-1met also has inhibitory effect on cancer cells without p53 mutation [20, 22–25].
In this study, we aim to screen the effect of PRIMA-1met on different CRC cell lines, in order to gain a deep and comprehensive understanding for the potential of PRIMA-1met as an anti-cancer agent especially for CRC patients. We demonstrated that PRIMA-1met inhibited CRC cells growth independently of p53 status. PRIMA-1met induced robust apoptosis preferably in p53 mutant CRC cell lines, which was mediated through upregulated expression of pro-apoptotic Noxa. In addition, PRIMA-1met also exerted effects on inhibition of proliferation, migration and colony formation in a p53-independent manner. PRIMA-1met impeded CRC tumor progression in xenograft mouse model.
RESULTS
PRIMA-1met inhibited CRC cell proliferation in a dose-dependent manner
We first investigated the effects of PRIMA-1met on the growth of 10 human CRC cell lines with different p53 status. The results from the CTG assay showed a gradual decrease in cell proliferation along with progressive increases in the concentrations of PRIMA-1met for 48 hours (Table 1 and Figure 1A and 1B). The IC50 and p53 status of these CRC cell lines were summarized in Table1. The inhibitory effect on cell growth induced by PRIMA-1met was in a dose-dependent manner in all CRC cell lines. However, HT29 cell line exhibited more resistance to PRIMA-1met than the others (Figure 1A). The means of IC50 in p53wt CRC cell lines, p53mt CRC cell lines, and p53ko CRC cell lines were 24.90 μM (ranging from 7.5 to 45.9 μM), 28.23 μM (ranging from 10.9 to 58.6 μM) and 23.70 μM, respectively. Surprisingly, we did not observe significant difference in the IC50 values among the 3 groups of cell lines with distinct p53 status (Figure 1B, p > 0.05).
Table 1: The list of CRC cell lines with different p53 status and their IC50 values of PRIMA-1met
Cell lines |
p53 status |
IC50 (μM) |
|---|---|---|
HCT116 |
wild type |
7.5 |
RKO |
wild type |
21.3 |
LOVO |
wild type |
45.9 |
DLD-1 |
S241F, C > T |
14.3 |
SW480 |
R273H, G > A |
13.6 |
SW620 |
R273H, G > A |
10.9 |
Colo320 |
R248W, C > T |
13.8 |
Caco2 |
E204, G > T |
58.6 |
HT29 |
R273H, G > A |
58.2 |
HCT116KO |
knock out |
23.7 |
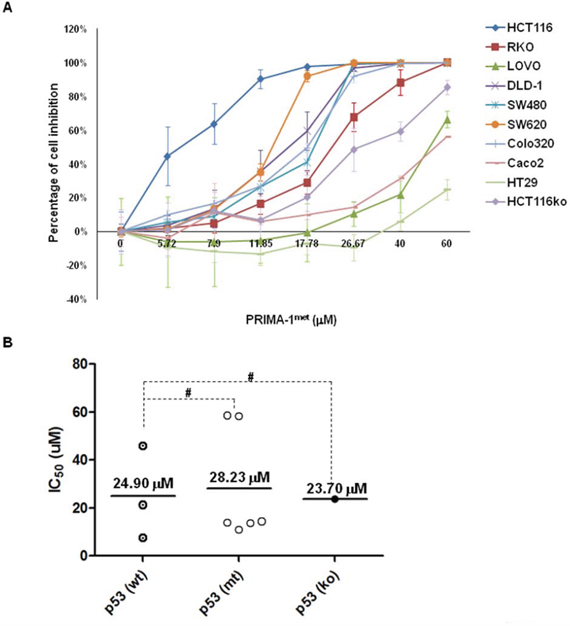
Figure 1: PRIMA-1met inhibited cell proliferation in CRC cell lines. A panel of CRC cell lines was treated with either DMSO control or PRIMA-1met at indicated dosages for 48 hours, followed by CTG assays. The percentage of cell growth inhibition was normalized with respective DMSO control. B. IC50 of PRIMA-1met in each cell line was calculated with CalcuSyn (Biosoft, Cambridge, UK) according to the fraction of cell inhibition. Dot plots were constructed and divided into 3 groups with wild type (wt) p53, mutant (mt) p53 and p53 knockout (ko) cell line, respectively. #p > 0.05. All the experiments were triplicated and error bars represented standard error of mean (SEM).
PRIMA-1met preferably induced robust apoptosis in CRC cells with mutant p53
To investigate whether PRIMA-1met induced apoptosis in CRC cell lines, we chose 2 cell lines with wild-type p53 (HCT116wt, RKO), 2 cell lines with mutant p53 (DLD-1, Caco2), and 1 cell line knocked out p53 (HCT116ko). As shown in Figure 2A, PRIMA-1met treatment induced apoptosis in two CRC cell lines carrying mutant p53, and showed a dose-dependent effect in DLD-1 cell line (Figure 2B). Interestingly, PRIMA-1met treatment also induced moderate apoptotic response in HCT116wt, in contrast, another p53 wild-type cell line RKO and p53-null HCT116 appeared to be resistant to PRIAM-1met treatment (Figure 2A). Western blotting analysis revealed an increase of PARP cleavage in DLD-1 and HCT116wt cell lines (Figure 2C). In agreement with the results from apoptosis assays by FACS analysis, an increase of PARP cleavage was not observed in p53-null HCT116 cells after PRIAM-1met treatment (Figure 2C). Apoptotic induction of PRIMA-1met was not only p53 dependent, but also related to p53 activation, because high expression of phospho-p53 (Ser15) could be detected after PRIMA-1met treatment (Figure 2D). These results suggest that, in general, PRIMA-1met induced-apoptosis is more prominent in CRC cells with mutant p53.
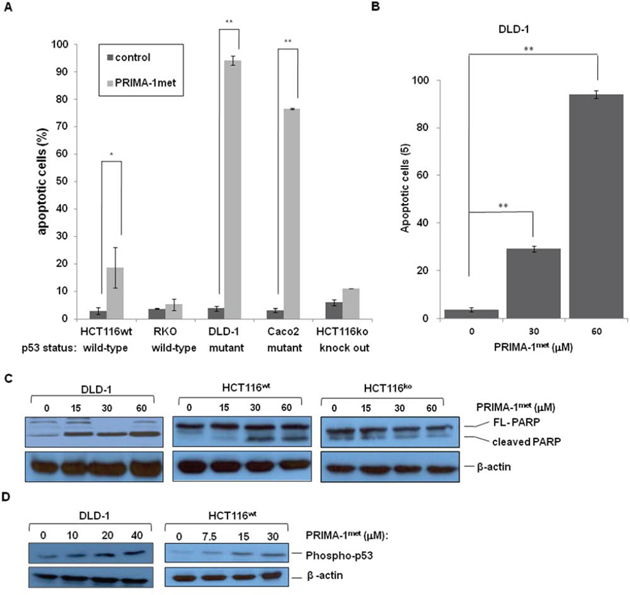
Figure 2: PRIMA-1met induced apoptosis in CRC cell lines. A. Apoptosis of different CRC cell lines was induced by PRIMA-1met. After treated with PRIMA-1met for 48 hours, cell apoptosis was estimated by flow cytometric analysis. B. DLD-1 cells were treated with PRIMA-1met at different concentrations, then followed by FACS analysis of apoptosis. In A. and B. N = 2, error bars show SEM. *p < 0.05; **p < 0.01. C. PARP cleavage was detected by western blotting analysis after treated with PRIMA-1met at different concentrations for 24 hours. D. Phosphorylation of p53 level was determined after PRIMA-1met treatment.
PRIMA-1met increased expression of Noxa, but not PUMA
Under cellular stress, wild type (wt) p53 up-regulates its target proteins to promote apoptosis. Pro-apoptotic Bcl-2 (B-cell lymphoma-2) family members, such as Noxa and PUMA, are important target proteins [25, 31]. We performed western blotting analysis and qRT-PCR to evaluate expression of Noxa and PUMA. Cells were treated with PRIAM-1met that resulted in strong expression of Noxa in p53 mutant DLD-1 and SW620 cell lines and p53 wild-type HCT-116 cell line (Figure 3A and 3B). However, the highest fold increase of expression of Noxa mRNA in DLD-1 cell line occurred concomitantly with the largest percentage of apoptosis observed in DLD-1 cells after PRIMA-1met treatment (Figure 2A and Figure 3B). In contrast, PRIAM-1met treatment did not induce expression of another pro-apoptotic gene, PUMA, on either RNA level or protein level (Figure 3C and 3D). Taken together, these data indicated that Noxa played a pivotal role in the activity of PRIMA-1met.
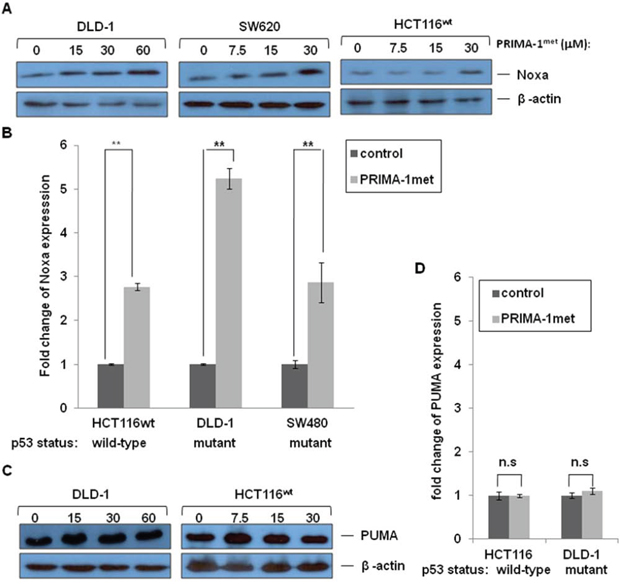
Figure 3: The effects of PRIMA-1met on pro-apoptotic molecules Noxa and PUMA. DLD-1, SW620 and HCT116wt cells were treated with different concentrations of PRIMA-1met for 24 hours. Cells then were harvested for analysis of Noxa protein levels A. and mRNA levels B. DLD-1 and HCT116wt cells were treated with different concentrations of PRIMA-1met for 24 hours. Cells then were harvested for analysis of PUMA protein levels C. and mRNA levels D. Expression of Noxa and PUMA genes was quantified using real-time RT-PCR on mRNA level. N = 2, error bars show SEM. *p < 0.05; **p < 0.01. n.s.: not significant, p > 0.05.
PRIMA-1met induced apoptosis in CRC cell lines with mutant p53 is mediated through Noxa
To further clarify the role of Noxa in PRIMA-1met induced-apoptosis, we knocked down Noxa gene in DLD-1, SW480 and HCT116wt cell lines. Western blotting analysis confirmed the reduced Noxa proten level upon Noxa specific siRNA treatment in these 3 cell lines (inserted pictures in Figure 4A, 4B and 4C). We observed that the inhibitory effect on two mutant p53 cell lines, DLD-1 and SW480, were significantly reduced after knocking down Noxa, whereas knocking down Noxa in wt p53 HCT1116 failed to produce any noticeable impact on the IC50 (Figure 4A, 4B, 4C and 4D).
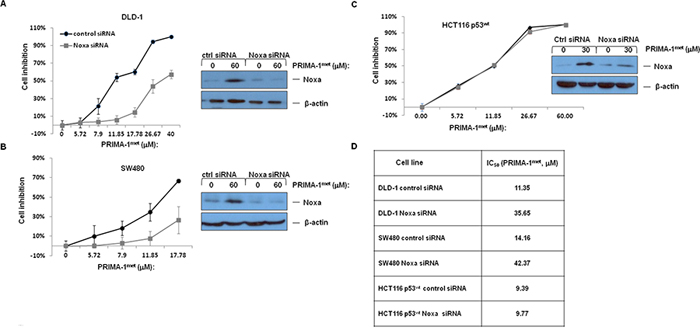
Figure 4: The impact of PRIMA-1met on cell proliferation after treatment of si-Noxa in DLD-1. A. SW480 B. and HCT116wt C. N = 3, error bars show SEM. Insert pictures at the left showing alterations in Noxa protein after transfection with Control siRNA or siRNA specifically targeting Noxa (si-Noxa) for 24 hours with or without PRIMA-1met. D. IC50 values of PRIMA-1met in CRC cell lines transfected with either control siRNA or Noxa specific siRNA.
PRIMA-1met inhibited cell migration, invasion and colony formation in CRC cells
To explore the effect of PRIMA-1met on the migration and invasion of CRC cells, a wound healing assay was employed to compare CRC cells treated with either DMSO or PRIMA-1met. As shown in Figure 5, relative to DMSO treated controls, wound healing was significantly impaired in HCT116wt and DLD-1 cells treated with PRIMA-1met. These findings strongly suggest that PRIMA-1met inhibits motility signalling in CRC cells. Morphological transformation of cell colonies in vitro has a high correlation with carcinogenesis in vivo. We conducted soft agar assay to assess the impact of PRIMA-1met on colony formation of CRC cells. After treated with PRIMA-1met for 48 hours, cells were seeded in soft agar and incubated for 21 days. We found that PRIMA-1met greatly reduced the number and size of HCT116wt and DLD-1 cell colonies (Figure 5B). Altogether, the in vitro migration and soft agar assay indicated that the ability of PRIMA-1met to inhibition cell migration and colony formation of CRC cells also contributed to its anti-cancer activity, independent of p53 status.
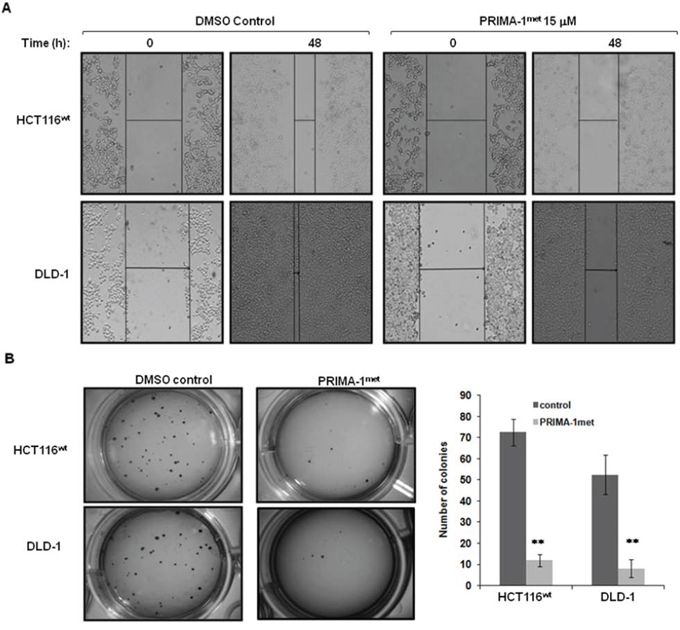
Figure 5: PRIMA-1met suppressed CRC cell migration and colony formation. A. DLD-1 and HCT116wt cells were seeded into a six-well tissue culture dish and allowed to grow to 90% confluency in complete medium. Cell monolayers were wounded by a plastic tip (1 mm) that touched the plate and put back into the incubator for 48 hours. Cells were monitored under a microscope equipped with a camera (Olympus, Japan). Each experiment was repeated three times and representative images were shown. B. To assay colony formation, 2 × 103 DLD-1 and HCT116wt cells were plated in triplicates into 12-well cell culture plates with DMSO or PRIMA-1met in the soft agar. After 21 days, the number of foci on each dish was counted. Colonies with more than 20 cells were scored as positive. Each experiment was repeated three times. The bar figure represented the data of mean ± S.E. of three determinations. **p < 0.001.
PRIMA-1met suppressed tumor growth in xenograft mouse CRC model
To validate the anti-tumorigenic potential of PRIMA-1met in vivo, we utilised a xenograft CRC mouse model by subcutaneously injecting DLD-1 cells into female NOD/SCID mice. After the mice developed palpable tumor masses, we began PRIMA-1met treatment (intraperitoneal injection, 50 mg/kg) on the mice daily for 8 days. Tumor growth during the treatment was monitored by caliper measurement every other day. At day 8 of post treatment, the mice were euthanized, and the tumors were removed, and photographed. We observed a substantial reduction in tumor growth in the PRIMA-1met treated group in comparison with the vehicle control treated group (Figure 6A). The PRIMA-1met treatment group presented with tumor sizes substantially smaller than those of the controls (Figure 6B), indicating the efficacy of the PRIMA-1met in CRC regression. It is important to note that during the treatment, the mice showed tolerance to PRIMA-1met injections and maintained normal activities and no loss of weight were recorded (data not shown).
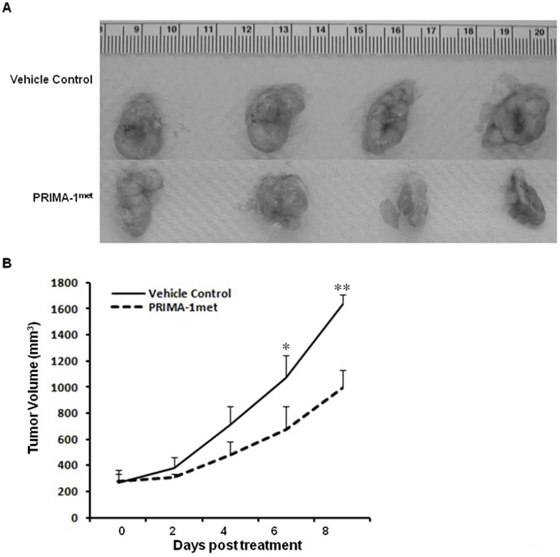
Figure 6: PRIMA-1met significantly suppressed CRC xenograft growth. A. The images of DLD-1 tumors from mice treated with vehicle control versus PRIMA-1met (n = 4 for each group). B. PRIMA-1met significantly suppressed tumor growth. *p < 0.05; **p < 0.01.
DISCUSSION
In this study, we shed new light on the effect of PRIMA-1met on CRC. Our results showed that PRIMA-1met inhibited CRC cell growth independent of p53 status in a dose-dependent manner, but preferably induced robust apoptosis in mutant p53 CRC cells. We further uncovered the molecular mechanism underlying PRIMA-1met induced-apoptosis through up-regulation of pro-apoptotic protein Noxa, leading to activation of cleavage of PARP. These results were consistent with previous studies that demonstrated the ability of PRIMA-1met to restore the mutant p53 to wild-type p53 properties in other cancers [15, 17, 20, 21, 25]. Reduction of Noxa expression by siRNA accentuated the inhibitive effect of PRIMA-1met and increased IC50 of PRIMA-1met in DLD-1 and SW480 cell lines. These results suggest that upregulation of Noxa plays an influential role in the mediation of cell proliferation and apoptosis in CRC cells treated with PRIMA-1met. The importance of Noxa also has been noted in PRIMA-1met treated multiple myeloma cells [25].
Most studies reported that PRIMA-1met targets mutant p53 proteins [17, 32, 33]. Interestingly, in this study, CRC cell lines with wild-type p53 or without p53 responded to PRIMA-1met treatment. Some IC50 values of these cell lines with wild-type p53 were even less than those carrying mutant p53. However, different mechanisms might contribute to the effectiveness of PRIMA-1met observed in these two types of CRC cells. Unlike in p53 mutant CRC cells, PRIMA-1met mainly induced cytostasis, but to less extent apoptosis, in CRC cells with wt p53 or p53 null. The percentage of apoptotic cells did not increased significantly after treated with PRIMA-1met in cell lines RKO (p53 wt) and HCT116ko. In addition, knocking down Noxa gene reduced PRIMA-1met sensitivity in mutant p53 CRC cells, but it didn’t significantly decrease PRIMA-1met sensitivity in HCT116wt cell line. Taken together, these results support a model where although PRIMA-1met inhibits proliferation of CRC cells independently on p53 status. Nevertheless, PRIMA-1met mainly induces cytostasis in CRC cells with wt p53 or p53 null, whereas PRIMA-1met promotes apoptosis in CRC cells with mutant p53. More importantly, PRIMA-1met treatment significantly reduced tumor growth in our mouse model and showed no noticeable toxicity to the mice.
In conclusion, in our study PRIMA-1met inhibits CRC cells growth, colonies formation, proliferation and migration in a p53-independent manner. However, the ability of to induce apoptosis appears specific in CRC cells with mutant p53. Our in vitro and in vivo results suggest that PRIMA-1met is an attractive candidate for further investigation as a potential anti-cancer agent in clinical trials for patients with CRC. However, we also acknowledge that the work is limited by studying CRC lines. A deeper understanding of the molecular mechanisms of anti-tumorigenic activity of PRIMA-1met will allow for precise treatment regimens and the possibility of co-treatment with other therapeutics to greatly improve the outcome of CRC patients.
MATERIALS AND METHODS
Cell lines and drugs
A panel of colorectal cancer cell lines with different p53 status was used in this study. The details of these cell lines were: LOVO (wild-type p53), RKO (wild-type p53), SW480 (mutant p53-R237H), SW620 (mutant p53-R237H), HT29 (mutant p53-R237H), Colo320 (mutant p53-R248W) and Caco2 (mutant p53-E204X). These cells were provided by Dr. Motomi Osato (Cancer Science Institute of Singapore, National University of Singapore). HCT116 (wild-type p53+/+, HCT116wt) and HCT116 null (p53-/-, HCT116ko) cell lines were gifts from Dr. Jimmy Chao (Bioprocessing Technology Institute, A*Star, Singapore). DLD-1 (mutant p53-S241F) cell line was obtained in house. Cells were grown in DMEM (Dulbecco's Modified Eagle's Medium) (Invitrogen, Carlsbad, CA) with 10% inactivated fetal bovine serum (FBS, JRH Bioscience Inc, Lenexa, KS) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA), in a humid incubator with 5% CO2 at 37°C. PRIMA-1met (APR-246, Santa Cruz Biotechnology, Santa Cruz, CA) was dissolved in DMSO (Dimethyl sulfoxide) at a concentration of 50 mM and stored at −20°C. Working solutions were then diluted to appropriate concentrations in cell culture medium. Same amount of DMSO as control reagent was added in each experiment.
Cell proliferation assay
After 24 hours of seeding at 5000 cells/well in 100 μl medium in a 96-well plate, cells were treated with PRIMA-1met at concentrations from 5.72 μM to 60 μM, and incubated for 48 hours. CellTiter-Glo (CTG) Luminescent Cell Viability Assay (Promega, Madsion, WI) was used to measure the cell proliferation by quantitating the cell's ATP as previously reported [26]. The reading of DMSO control samples were set as 100% and the reading of other samples were calculated as relative proliferation to the DMSO control samples. Each experiment was in triplicate. The results were presented in percentage changes relative to their respective DMSO controls.
Cell apoptosis assay
Cells were seeded on 6-well plates at a density of 0.2 × 106 cells per well and treated with 30 μM, 60 μM PRIMA-1met, respectively, for 48 hours. Apoptosis assay was conducted with flow cytometric analysis of Annexin V-FITC (Invitrogen) and Sytox Blue (BD Biosciences, San Jose, CA) staining cells [27]. After suspending using trypsin and washing twice with cold PBS, cells were counted and adjusted at 0.5 - 1.0 × 106 for each sample, and resuspended in 100 μl 1 × binding buffer. Each sample was added 1.5 μl Annexin V-FITC, and incubated on ice in dark for 20 minutes. Then after added 1 μl Sytox Blue and incubated at room temperature for 5 minutes in dark, each sample was mixed with 400 μl of 1 × binding buffer, filtered to a 5 ml BD Falcon tube (BD Biosciences), then to analyze by flow cytometry within 1 hour.
Western blotting analysis
We plated cells at 2 × 106 per 10cm dish. After 24 hours, cells were treated with DMSO control and different dose of PRIMA-1met as indicated. After 24 hours, cells were lysed and analyzed by western blotting as described previously [28]. Primary antibodies used in this study were against p53 (sc-126, Santa Cruz Biotechnology), Actin (sc-1616, Santa Cruz Biotechnology), cleaved PARP (D214), Phospho-p53 (Ser15) and PUMA from Cell Signaling Technology (Danvers, MA), Noxa (EMD chemicals, Gibbstown, NJ). Secondary antibodies were goat-anti-rabbit IgG HRP (sc-2030, Santa Cruz Biotechnology) and goat-anti-mouse IgG HRP (sc-2031, Santa Cruz Biotechnology).
Real-time quantitative reverse transcriptase-PCR (qRT-PCR)
All CRC cells were seeded in a 6-well plate at a density of 0.4 × 106 cells/well to grow overnight. RKO, Colo320, DLD-1 and SW480 cells were treated with 60 μM PRIMA-1met for 24 hours. HCT116wt cells with 30 μM PRIMA-1met and HCT116ko cells with 120 μM PRIMA-1met were incubated for 24 hours. RNA was isolated from cells using RNeasy mini kit (Qiagen, Valencia, CA). Typically, 1 μg of total RNA was used to generate cDNA by using SuperScript® III RT (Invitrogen) with oligo-dT primer. qRT-PCR was performed using the Power SYBR® Green PCR Master Mix as recommended by the manufacturer (Applied Biosystems, Carlsbad, CA). GAPDH was used as the internal control. SDS 2.2.1 software (Applied Biosystems) was used to perform relative quantification of target genes using the comparative CT (ΔΔCT) method. The primer sequences were described as the following: Noxa (Forward: 5′-GCTGGAAGTCGAGTGTGCTA-3′, Reverse: 5′-CCTGAGCAGAAGAGTTTGGA-3′) and PUMA (Forward: 5′-ACGACCTCAACGCACAGTACG-3′, Reverse: 5′-TCCCATGA TGAGATTGTACAGGAC-3′) (Integrated DNA Technologies, Singapore).
siRNA transfection
HCT116wt, DLD-1 and SW480 cells were seeded in 6-well plates 1 day for transfection at a density of 3 × 106 cells/well. Mixture containing 110 pmoles of Noxa siRNA (sc-37305, Santa Cruz Biotechnology) or control siRNA (sc-36869, Santa Cruz Biotechnology), 200 μl of jetPRIME® buffer (Polyplus-Transfection, Illkich, Franc) and 4 μl of jetPRIME® in vitro siRNA transfection reagent (Polyplus-Transfection) was vortexed 10 seconds, spun down and incubated 10 minutes at room temperature. This transfection mixture was added to each well. After incubated for 24 hours, cells were harvested for further analysis.
Wound healing assay
HCT116wt and DLD-1 cells were seeded to four wells in 6-well plates at 1 × 106 cells/well for duplication. After 3 days, cells were cultured to near confluence and scratched a wound through the centre of the well. Rinsed with PBS three times gently, two of these wells were replaced with 2 ml of media containing DMSO as control. The others were added with PRIMA-1met at concentration of 15 μM. After taken pictures under microscope (10 × magnification), plates were transferred to humidified incubator at 37°C and incubated for 48 hours. Pictures were taken again.
Soft agar assay
After seeded in 6-well plate at a density of 2 × 105 cells/well overnight, HCT116wt and were harvested and 2 × 103 each cell line to mix with 3 ml top agar (containing PRIMA-1met 10 μM, 20 μM, respectively) gently. The mixture was added to each of the duplicated wells in a 12-well plate with base agar (containing 0.75 ml 1 × DMEM+10% FCS and 0.75 ml 1% agar in each well) prepared before. The plate was incubated in humidified incubator at 37°C for 21 days, and then to stained with 0.5 ml of 0.005% crystal violet (Sigma-Aldrich, St. Louis, MO) for 1 hour. Cell colonies formation was observed under microscope (200× magnification).
Xenograft mouse model
Female Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice (17–20 g, 4–6 weeks old) were purchased from InVivos (Singapore). The preparation and injection of DLD-1 cells were followed the reported methods [29, 30]. In briefly, exponentially growing DLD-1 cells (4 × 106) were subcutaneously injected into loose skin between the shoulder blades and left front leg of recipient mice. All treatments were started 14 days after the injection, when the mice had palpable tumors of an average size of approximately 200 mm3. PRIMA-1met was dissolved in 1x PBS and administrated at 50 mg/kg/day by in intraperitoneal injection (I.P.) daily. Mice in vehicle control group were treated with 1 X PBS daily. Treatments lasted for 14 days. Each group was comprised of 10 mice.
The length (L) and width (W) of the tumor were measured with callipers, and tumor volume (TV) was calculated as TV = (L × W2)/2. The protocol was reviewed and approved by Institutional Animal Care and Use Committee in compliance to the guidelines on the care and use of animals for scientific purpose.
Statistical analysis
The statistical analysis was performed by SPSS version 13.0. Values were expressed as mean ± standard error of the mean (SEM). Comparison between groups of data was made using one-way analysis of variance (ANOVA). Tumour volume reduction of the treatment groups was compared to the vehicle control group by Student's t-test. p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
We gratefully acknowledge Dr Teoh Phaik Ju for her critical advice on this study.
CONFLICTS OF INTEREST
None.
FINANCIAL SUPPORT
This research is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative and NMRC Clinician-Scientist IRG Grant CNIG11nov38 (Zhou J). Chng WJ is also supported by NMRC Clinician Scientist Investigator award.
Authors’ contributions
Zhou J, Li XL, Chen ZR and Chng WJ designed the experiments; Zhou J and Li XL wrote the manuscript; Chng WJ and Chen ZR revised the manuscript; Li XL, Zhou J, Chan ZL, and Chooi JY performed the experiments. All authors approved the final version of the manuscript.
REFERENCES
1. Grady WM, Pritchard CC. Molecular alterations and biomarkers in colorectal cancer. Toxicologic pathology. 2014; 42:124–39.
2. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nature reviews Gastroenterology & hepatology. 2011; 8:686–700.
3. Patai AV, Molnar B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World journal of gastroenterology: WJG. 2013; 19:607–15.
4. Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 2011; 6:479–507.
5. Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nature reviews Cancer. 2003; 3:695–701.
6. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Current opinion in cell biology. 2001; 13:332–7.
7. Cario E. The human TLR4 variant D299G mediates inflammation-associated cancer progression in the intestinal epithelium. Oncoimmunology. 2013; 2:e24890.
8. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews Cancer. 2014; 14:359–70.
9. Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. Journal of gastroenterology. 2006; 41:185–92.
10. Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N, et al. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005; 23:7518–28.
11. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World journal of gastroenterology: WJG. 2015; 21:84–93.
12. Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011; 60:116–29.
13. Bouchet BP, Caron de Fromentel C, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Critical reviews in oncology/hematology. 2006; 58:190–207.
14. Stegh AH. Targeting the p53 signaling pathway in cancer therapy - the promises, challenges and perils. Expert opinion on therapeutic targets. 2012; 16:67–83.
15. Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS letters. 2014; 588:2622–7.
16. Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007; 26:2226–42.
17. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature medicine. 2002; 8:282–8.
18. Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012; 30:3633–9.
19. Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nature reviews Clinical oncology. 2011; 8:25–37.
20. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer cell. 2009; 15:376–88.
21. Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010; 29:1329–38.
22. Ali D, Jonsson-Videsater K, Deneberg S, Bengtzen S, Nahi H, Paul C, et al. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. European journal of haematology. 2011; 86:206–15.
23. Rieber M, Strasberg-Rieber M. Hypoxia, Mn-SOD and H(2)O(2) regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochemical pharmacology. 2012; 84:1563–70.
24. Russo D, Ottaggio L, Foggetti G, Masini M, Masiello P, Fronza G, et al. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochimica et biophysica acta. 2013; 1833:1904–13.
25. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Molecular cancer therapeutics. 2013; 12:2331–41.
26. Chong PS, Zhou J, Cheong LL, Liu SC, Qian J, Guo T, et al. LEO1 is regulated by PRL-3 and mediates its oncogenic properties in acute myelogenous leukemia. Cancer research. 2014; 74:3043–53.
27. Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009; 113:4052–62.
28. Zhou J, Chong PS, Lu X, Cheong LL, Bi C, Liu SC, et al. Phosphatase of regenerating liver-3 is regulated by signal transducer and activator of transcription 3 in acute myeloid leukemia. Experimental hematology. 2014; 42:1041–52. e1–2.
29. Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011; 118:2830–9.
30. Zhou J, Cheong LL, Liu SC, Chong PS, Mahara S, Bi C, et al. The pro-metastasis tyrosine phosphatase, PRL-3 (PTP4A3), is a novel mediator of oncogenic function of BCR-ABL in human chronic myeloid leukemia. Molecular cancer. 2012; 11:72.
31. Saha MN, Qiu L, Chang H. Targeting p53 by small molecules in hematological malignancies. Journal of hematology & oncology. 2013; 6:23.
32. Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010; 29:4245–52.
33. Zache N, Lambert JM, Wiman KG, Bykov VJ. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cellular oncology: the official journal of the International Society for Cellular Oncology. 2008; 30:411–8.