
INTRODUCTION
Glioblastoma multiforme (GBM) is the most frequent, highly malignant primary tumor of the central nervous system [1], accounting for approximately 12–15% of all brain tumors [2]. The current standard of care is temozolomide plus radiotherapy [3]. However, almost all GBMs recur after first-line therapy and few second-line options have been identified that provide sustained clinical benefit [4]. Consequently, most patients die soon after tumor recurrence; median overall survival after disease progression is approximately 6–9 months [4, 5]. However, a small proportion of patients can survive for considerably longer [6].
As GBM is highly heterogeneous, optimal treatment tailored to the individual is difficult [5]. Nevertheless, several potential prognostic factors of survival have been identified and include the patient's age, and performance status, tumor location, and extent of surgical resection [7]. Furthermore, developments in genomic technology have facilitated the identification of molecular markers that could potentially drive treatment decisions. For example, methylation status of the gene encoding O6-methylguanine-DNA methyltransferase (MGMT) predicts response to temozolomide-based regimens in elderly patients [8]. Molecular studies have also identified markers that provide insights into the pathogenesis of GBM and potentially identify rationale for drug targets. For example, heritable genetic aberrations of the ErbB family of receptors, particularly the epidermal growth factor receptor (EGFR), has been implicated in GBM progression [9]. Overexpression of EGFR has been identified in 50–60% of GBM cases and is generally thought to confer poor prognosis [10–12], particularly in the presence of the EGFRvIII mutation [13].
To date, clinical trials with targeted agents in patients with GBM have been disappointing. For example, bevacizumab and cilengitide have been investigated in a first-line treatment setting, but these studies failed to show improvements in survival [14–16]. Furthermore, despite the likely role of EGFR in the pathogenesis of disease, reversible EGFR tyrosine kinase inhibitors (gefitinib and erlotinib) do not appear to be effective for recurrent GBM [17–19]. Recently, a phase I/II study assessed afatinib, a potent irreversible ErbB family blocker, with or without protracted temozolomide, in patients with recurrent GBM [20]. The rationale for this study was the observation that afatinib inhibits proliferation of cells with EGFR mutations that are commonly found in GBM, including EGFRvIII and R108K [21, 22]. Furthermore, unlike erlotinib and gefitinib, cytochrome P450 metabolism of afatinib is negligible [23], thus facilitating combination with chemotherapy or some anti-epileptic drugs. Also, clinical studies have shown afatinib to be effective in several tumor types, notably non-small cell lung cancer including patients with acquired resistance to gefitinib or erlotinib [24–28]. Unfortunately, the phase I/II study demonstrated that afatinib monotherapy, and afatinib plus temozolomide, had limited activity in unselected patients with recurrent GBM. However, certain selected patient populations (including patients with high levels of EGFR vIII immunoreactivity, EGFR amplification, or PTEN loss) appeared to have promising response and durable progression-free survival.
Here, we present a patient with multifocal recurrent GBM who demonstrated a remarkable response to treatment with afatinib plus temozolomide. We undertook broad molecular analysis on this patient's tumor to assess possible mechanistic explanations for the sustained clinical benefit that was observed. Informed consent has been obtained.
CASE PRESENTATION
This right-handed, 58-year-old, previously healthy female patient presented in October 2009 with constant right frontal headache, mild weakness of the left side, gait disturbance, and behavioral changes (anger, forgetfulness).
Radiological analysis
Three lesions were identified from radiological assessment (Figure 1). These included a right frontal lesion (4.0 × 3.7 × 4.9 cm) with surrounding edema and mass effect on the adjacent right frontal horn (9 mm to the left of midline); a subcortical left basal frontal area lesion (maximum diameter 1.3 cm) with thick marginal enhancement and central cystic/necrotic appearance, and a left inferior frontal gyrus lesion evident in T2 and Flair images.
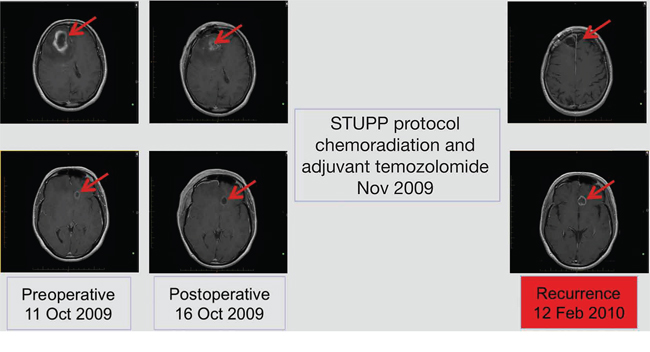
Figure 1: Radiological disease assessments relating to first-line treatment (surgical resection and STUPP protocol).
Surgical resection
Subtotal resection of the right frontal lesion was performed in October 2009. Postoperative reduction in tumor size was evident upon radiological assessment (Figure 1). The patient was diagnosed with multifocal glioma and pathological findings were consistent with GBM World Health Organization (WHO) grade IV. Tumor MGMT promoter and isocitrate dehydrogenase 1 (IDH1) status were determined to be methylated and wild-type, respectively. Two weeks following surgery, the patient developed a deep vein thrombosis (DVT) in the right lower extremity and a pulmonary embolism. Consequently, the patient was treated with heparin followed by low molecular weight heparin. Within 10 days, she had fully recovered from the DVT.
Treatments administered
First-line treatment, initiated in November 2009, comprised the STUPP protocol: radiotherapy (60 Gy over 6 weeks; intensity modulated radiotherapy [IMRT]) with concurrent oral temozolomide (75 mg/m2 daily for 42 days), followed by adjuvant temozolomide 150–200 mg/m2 every 5/28 days [29]. The patient tolerated the concurrent treatment well. However, in February 2010, disease progression was observed by magnetic resonance imaging (MRI) after 3 cycles of adjuvant temozolomide (increase in left frontal lesion with mass effect; Figure 1).
Second-line treatment, initiated in April 2010, was determined according to participation in a clinical trial of afatinib with or without daily temozolomide. The patient was randomized to the combined afatinib (20–40 mg daily for 28 days) and temozolomide (50 mg/m2 orally [p.o.] every 21/28 days) arm. After 1 cycle of second-line treatment, MRI revealed minimal decrease in lesion size. After 5 cycles, significant disease regression was observed and maintained in subsequent assessments to 54 months (Figure 2). A full spine MRI performed in November 2014 showed no evidence of metastases in the cervical, thoracic or lumbar regions and the most recent MRI (January 2015) showed stable disease. At the time of writing (April 2015), the patient had completed 63 cycles and treatment was ongoing.
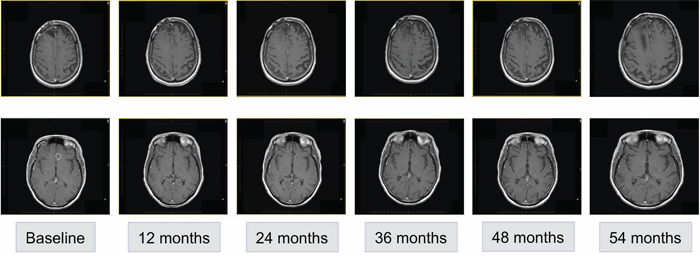
Figure 2: Radiological disease assessments relating to second-line treatment (afatinib and temozolomide).
Adverse events
In total, the patient experienced four incidences of grade III adverse events that were considered to be at least probably related to second-line treatment: one incidence of maculopapular rash and three incidences of paronychia. The maculopapular rash occurred during the third cycle and was managed with a 2-week interruption of treatment followed by a dose reduction (afatinib from 40 to 30 mg p.o. daily; temozolomide from 140 to 90 mg p.o. daily). Grade III paronychia occurred during cycle 16, cycle 20 (leading to a dose reduction of afatinib from 30 to 20 mg p.o. daily), and cycle 36 (leading to a treatment interruption of 7 weeks). Dose reductions led to marked improvement in symptoms.
Other drug-related adverse events included grade I nausea and vomiting (cycle 1; managed with anti-emetics), grade II diarrhea (cycle 1; managed with loperamide), grade I rash (cycles 1 and 2; managed with hydrocortisone cream), weight loss, and fatigue.
The patient's neurologic and physical status were stable (Karnofsky Performance Status > 90) throughout second-line treatment.
Molecular analysis
DNA was isolated from a brain tumor specimen from the primary resection and subjected to FoundationOne™ next-generation sequencing [30]. The entire coding sequence of 236 cancer-related genes, plus 47 introns from 19 genes often rearranged or altered in cancer, was assessed. Fifteen cancer-related gene alterations were evident.
A number of EGFR aberrations were observed (Table 1). The patient was positive for EGFR amplification (estimated copy number of 60) and was likely positive for the EGFRvIII mutation (the extent of the EGFRvIII variant is difficult to quantify with next-generation sequencing). It is not unusual to observe amplification of both wild-type EGFR and EGFRvIII in the same tumor; EGFRvIII is rarely observed in isolation [31]. Two subclonal somatic mutations were observed (2% of reads), P596L and G598V. Both of these mutations have previously been identified in patients with GBM [21]. Interestingly, an additional unprecedented variant of unknown significance was observed in the extracellular domain of EGFR (D247Y; 89% of reads). This could potentially be a rare single nucleotide polymorphism (SNP), but could also be a somatically acquired allele that may possibly be linked to the observed clinical response to afatinib.
Table 1: Details of gene alterations detected in 15 genes
Number |
Gene |
Description |
|---|---|---|
1 |
EGFR |
Amplification of full gene, estimated gene copy number = 60 |
2 |
EGFR |
Known somatic mutation P596L (c.1787C > T) |
3 |
EGFR |
Known somatic mutation G598V (c.1793G > T) |
4 |
EGFR |
Known somatic mutation EGFRvIII |
5 |
EGFR |
Variant of unknown significance D247Y (c.739G > T) |
6 |
PTEN |
Known somatic mutation R130* (c.388C > T) |
7 |
CDKN2A (p16) |
Homozygous deletion of full gene |
8 |
CDKN2B (p15) |
Homozygous deletion of full gene |
9 |
BAP1 |
Variant of unknown significance V447I (c.1339G > A) |
10 |
BCORL1 |
Variant of unknown significance T1111M (c.3332C > T) |
11 |
C17orf39 |
Variant of unknown significance N285Y (c.853A > T) |
12 |
CDH1 |
Variant of unknown significance P30T (c.88C > A) |
13 |
EPHA5 |
Variant of unknown significance M987T (c.2960T > C) |
14 |
ESR1 |
Variant of unknown significance H6Y (c.16C > T) |
15 |
GRIN2A |
Variant of unknown significance C800 (c.2400C > A) |
16 |
MAP3K1 |
Variant of unknown significance S939C (c.2816C > G) |
17 |
NOTCH3 |
Variant of unknown significance R1669H (c.5006G > A) |
18 |
STAG2 |
Variant of unknown significance splice (c.2026–1G > C) |
19 |
IKZF1 |
Amplification of full gene, estimated gene copy number = 51 |
A: adenine; C: cytosine; EGFR: epidermal growth factor receptor; G: guanine; GBM: glioblastoma; SNP: single nucleotide polymorphism; T: thymine.
Other genetic aberrations of interest included a null mutation in PTEN and deletion of CDKN2A and B, all of which are common genetic features of GBM. A number of other genes were found to have single base-pair changes, including BAP1, BCORL1, C170rf39, CDH1, EPHA5, ESR1, GRIN2A, and MAP3K1. It is likely that all of these aberrations represent rare heterozygous SNPs without any functional relevance (Table 1). Of note, several other common molecular genetic features of GBM, such as mutations in TP53, NF1, and IDH1, were absent in this patient. In independent analysis, the MGMT promoter was found to be methylated.
AFATINIB IN MOUSE XENOGRAFT MODELS OF GBM
The anti-tumor activity of afatinib has been evaluated in two different glioblastoma models derived from adult patients. These experiments were performed before trial initiation with the aim of illustrating that afatinib could have single-agent activity in glioblastoma models. Of note, both models have some degree of genetic similarity with the tumor profile of the patient described in this report (likely positive for EGFRvIII mutation and positive for EGFR amplification). The GB218 glioblastoma model is characterized by the presence of EGFRvIII mutation. In this model, monotherapy treatment (44 days) with afatinib (10, 7.5 or 5mg/kg/day) or erlotinib (50mg/kg/day) resulted in tumor growth inhibition (TGI) of 60.0, 69.3, 46.9, and 52.2%, respectively (Figure 3A). These doses are below the usual maximum tolerated doses in mice. GB218 also displayed sensitivity to temozolomide (50 or 25 mg/kg/day) resulting in complete suppression of tumor growth (TGI = 103.4%). The GB138 glioblastoma model is characterized by EGFR gene copy number gain (EGFR amplification). In this model, monotherapy treatment (28 days) with afatinib (10 or 7.5 mg/kg/day) or erlotinib (40mg/kg/day) resulted in TGI of 91.5, 81.8 and 72.7%, respectively (Figure 3B). Temozolomide (25 mg/kg/5 days on - 2 days off) resulted in complete suppression of tumor growth (TGI = 110.5%).
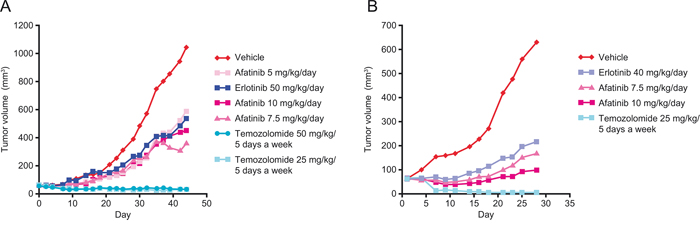
Figure 3: Effect of afatinib, erlotinib and temozolomide in two different human patient-derived xenograft models of glioblastoma. A. GB218 tumor growth kinetics. Groups of GB218 tumor-bearing mice (n = 7/group) were treated orally in weekly schedules either with afatinib 10, 7.5 or 5 mg/kg/day, erlotinib 50 mg/kg/day, temozolomide 25 or 50 mg/kg/5 days, or with the vehicle only. Median tumor volumes are plotted over time. Day 1 was the first and Day 44 the final day of treatment. B. GB138 tumor growth kinetics. Groups of GB138 tumor-bearing mice (n = 7/group) were treated orally in weekly schedules either with afatinib 10 or 7.5 mg/kg/day, erlotinib 40 mg/kg/day, temozolomide 25 mg/kg/5 days, or with the vehicle only. Median tumor volumes are plotted over time. Day 1 was the first and Day 28 the final day of treatment.
DISCUSSION
This case report describes a sustained clinical and radiographic response in a patient with recurrent multifocal GBM. Since progression on first-line therapy, the patient has survived for 60 months, approximately 6 times longer than the median overall survival generally observed in patients with recurrent GBM. It is not possible to say with certainty that these striking observations are solely attributable to treatment with afatinib and temozolomide. Moreover, pseudo-progression cannot be definitively excluded because the tumor is MGMT-methylated and disease progression was observed only 4 months after chemoradiation treatment.
This patient was participating in a clinical trial of afatinib with or without temozolomide for recurrent GBM (NCT00727506) [20]. The main finding of this study was that afatinib has limited effect on survival in unselected patients. Overall, median progression-free survival in patients treated with afatinib plus temozolomide (n = 39) was 1.5 months versus 1.9 months in patients treated with temozolomide only (n = 39). Nevertheless, subanalysis suggested that patients with certain molecular characteristics may benefit from combination therapy versus temozolomide monotherapy. For example, median progression-free survival was 2.7 versus 1.0 months in patients with EGFR amplification, and 2.7 versus 1.9 months in patients with PTEN loss. The present patient exhibited characteristics associated with a stronger afatinib response: EGFR amplification, EGFRvIII positivity, and a null mutation in PTEN. Our preclinical data in mouse xenograft models demonstrate that tumors with these EGFR aberrations are sensitive to tyrosine kinase inhibitors.
We have undertaken broad molecular analysis of the patient to try to more clearly define the molecular aberrations that could potentially identify patients who might achieve significant clinical benefit from afatinib plus temozolomide. The patient's overall molecular pathology was largely consistent with the ‘classical GBM’ subtype proposed by Verhaak, et al. [32], which is characterized by EGFR amplification and a lack of abnormalities in TP53, NF1, PDGFRα, and IDH1. It is possible that EGFR amplification and EGFRvIII positivity could underlie the encouraging response to afatinib plus temozolomide. However, 3 additional EGFR aberrations were also identified. Two of these aberrations, P596L and G598V, were clonally rare within the tumor and are therefore unlikely candidates for driving response to combination therapy. However, the third aberration, D247Y, appeared to be clonally amplified in the tumor and could therefore conceivably contribute to the observed response to afatinib plus temozolomide. Interestingly, this aberration affects the extracellular domain of EGFR. A previous study has shown that variants in the extracellular domain are relatively common in patients with GBM (13.6%) and confer sensitivity to EGFR tyrosine kinase inhibitors [21]. Based on these observations, we hypothesize that the complex EGFR genotype comprising extracellular aberrations in concert with focal amplification and the EGFRvIII mutation may underlie the observed sensitivity to afatinib and temozolomide. This hypothesis may be tested further in a suitable in vitro model. Moreover, it would be interesting to assess patients who have been treated with the combination in the phase I/II trial for EGFR extracellular aberrations. It is important to note that the tumor specimen sequenced was from initial resection, prior to any treatment; as such, the profile may have evolved upon exposure to subsequent therapies, but we cannot speculate on the nature of any treatment-driven evolution.
Adverse events in this case were manageable and in line with previous studies of the safety profiles of afatinib and temozolomide, including the GBM trial [20, 24, 26, 33]. In relation to safety, the GBM study also reported a lack of pharmacokinetic interactions between afatinib and temozolomide [20], supporting a strategy of co-administration of these drugs.
In conclusion, we describe a novel extracellular EGFR mutation in a recurrent GBM patient who demonstrated uncharacteristically prolonged survival following afatinib plus daily temozolomide. Studies to confirm the criteria for predicting afatinib response, and to determine the optimal dosing regimen, are awaited with interest.
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors’ institutional review board.
ACKNOWLEDGMENTS AND FUNDING
The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Lynn Pritchard, of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this article. This study was supported by Boehringer Ingelheim.
CONFLICTS OF INTEREST
David A. Reardon has received consultancy fees for Merck, Roche/Genentech, Novartis, Stemline and Amgen, and speaking fees for Merck and Roche/Genentech. Neil Gibson, Flavio Solca, Agnieszka Cseh are employees of Boehringer Ingelheim. For the remaining authors none were declared.
REFERENCES
1. Lim SK, Llaguno SR, McKay RM, Parada LF. Glioblastoma multiforme: a perspective on recent findings in human cancer and mouse models. BMB Rep. 2011; 44:158–164.
2. Kleihues P, Burger P, Collins V. Glioblastoma. In: Kleihues P, Cavenee W, editors. Pathology and genetics of tumours of the nervous system. (Lyon: IARC Press)(2000)29–39.
3. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, Mahajan A, Schultz CJ, Erridge S, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013; 31:4085–4091.
4. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro Oncol. 2013; 15:4–27.
5. Chaudhry NS, Shah AH, Ferraro N, Snelling BM, Bregy A, Madhavan K, Komotar RJ. Predictors of long-term survival in patients with glioblastoma multiforme: advancements from the last quarter century. Cancer Invest. 2013; 31:287–308.
6. Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G. Long-term survival with glioblastoma multiforme. Brain. 2007; 130:2596–2606.
7. Mazaris P, Hong X, Altshuler D, Schultz L, Poisson LM, Jain R, Mikkelsen T, Rosenblum M, Kalkanis S. Key determinants of short-term and long-term glioblastoma survival: a 14-year retrospective study of patients from the Hermelin Brain Tumor Center at Henry Ford Hospital. Clin Neurol Neurosurg. 2014; 120:103–112.
8. Thon N, Kreth S, Kreth FW. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 2013; 6:1363–1372.
9. Andersson U, Schwartzbaum J, Wiklund F, Sjostrom S, Liu Y, Tsavachidis S, Ahlbom A, Auvinen A, Collatz-Laier H, Feychting M, Johansen C, Kiuru A, Lonn S, et al. A comprehensive study of the association between the EGFR and ERBB2 genes and glioma risk. Acta Oncol. 2010; 49:767–775.
10. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000; 60:1383–1387.
11. Etienne MC, Formento JL, Lebrun-Frenay C, Gioanni J, Chatel M, Paquis P, Bernard C, Courdi A, Bensadoun RJ, Pignol JP, Francoual M, Grellier P, Frenay M, et al. Epidermal growth factor receptor and labeling index are independent prognostic factors in glial tumor outcome. Clin Cancer Res. 1998; 4:2383–2390.
12. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, Bhat K, McDonald JM, Yung WK, Colman H, Woo SY, Heimberger AB, Suki D, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007; 25:2288–2294.
13. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003; 63:6962–6970.
14. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:709–722.
15. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:699–708.
16. Stupp R, Hegi ME, Gorlia T, Erridge S, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Weyerbrock A, Hau P, Taphoorn MJB, Nabors LB, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study. J Clin Oncol. 2013; 31. LBA2009.
17. Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crino L, Bres AA. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer. 2007; 96:1047–1051.
18. Norden AD, Raizer JJ, Abrey LE, Lamborn KR, Lassman AB, Chang SM, Yung WK, Gilbert MR, Fine HA, Mehta M, Deangelis LM, Cloughesy TF, Robins HI, et al. Phase II trials of erlotinib or gefitinib in patients with recurrent meningioma. J Neurooncol. 2010; 96:211–217.
19. Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010; 96:219–230.
20. Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, Mathieu D, Phuphanich S, Cseh A, Fu Y, Cong J, Wind S, Eisenstat DD. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol. 2014; 17:430–439.
21. Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, Yoshimoto K, King JC, Nghiemphu P, Yuza Y, Xu Q, Greulich H, Thomas RK, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006; 3:e485.
22. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008; 27:4702–4711.
23. Stopfer P, Marzin K, Narjes H, Gansser D, Shahidi M, Uttereuther-Fischer M, Ebner T. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol. 2012; 69:1051–1061.
24. Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, Heo DS, Crino L, Tan EH, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012; 13:528–538.
25. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014; 15:213–222.
26. Yang JC, Shih JY, Su WC, Hsia TC, Tsai CM, Ou SH, Yu CJ, Chang GC, Ho CL, Sequist LV, Dudek AZ, Shahidi M, Cong XJ, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012; 13:539–548.
27. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327–3334.
28. Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, Nishio K, Seki Y, Ebisawa R, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013; 31:3335–3341.
29. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl.J Med. 2005; 352:987–996.
30. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023–1031.
31. Zadeh G, Bhat KP, Aldape K. EGFR and EGFRvIII in glioblastoma: partners in crime. Cancer Cell. 2013; 24:403–404.
32. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98–110.
33. Brandes AA, Tosoni A, Cavallo G, Bertorelle R, Gioia V, Franceschi E, Biscuola M, Blatt V, Crino L, Ermani M. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: phase II study from Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br J Cancer. 2006; 95:1155–1160.