
INTRODUCTION
The therapeutic options for malignant gliomas are still very limited, which is reflected by the low five-year survival of patients (<5%) and the low median patient survival (12–15 months) [1]. Therapy of malignant gliomas is based on the combined use of ionizing radiation and the DNA alkylating agent temozolomide (TMZ) [2]. TMZ exerts its effect via induction of O6-methylguanine (O6MeG), which represents a mutagenic and cytotoxic lesion [3]. DNA-crosslinking agents such as nimustine (ACNU) and lomustine (CCNU) are used as second-line chemotherapeutics. They chloroethylate the O6-position of guanine, thereby generating O6-chloroethylguanine (O6ClG) [4]. O6ClG undergoes intramolecular rearrangement to form N1-O6-ethenoguanine and finally N1-guanine-N3-cytosine inter-strand crosslinks (ICLs) [5, 6]. Besides high-grade gliomas, O6-chloroethylating agents are also used to treat malignant melanomas, gastrointestinal and pancreatic cancer, and Hodgkin's and non-Hodgkin's lymphomas [7].
The response to anticancer drug therapy largely depends on the DNA repair activity of the tumor and the activation of the DNA damage response (DDR). Exposure of tumor cells to DNA damaging anticancer drugs causes DNA replication arrest and DNA double-strand breaks (DSB), which are a direct trigger of the DDR. As a consequence, several important transcription factors like p53 and AP-1 become activated [8], which signal either cell survival or death pathways.
Activation of p53 results in cell cycle arrest, which occurs either in the G1 or the G2/M phase [9] and causes the induction of different DNA repair genes [8], thereby triggering resistance to anticancer drugs. On the other hand, p53 can also induce several important pro-apoptotic factors such as the Fas receptor (FAS) [10]. Therefore, the outcome of p53 activation largely depends on the anticancer drug and the cell system used. In malignant glioma cells, p53 provokes resistance to the topoisomerase I inhibitors topotecan and irinotecan [11–14] and the chloroethylating agents ACNU [15] and BCNU [16, 17]. In case of the chloroethylating nitrosoureas, this protective effect is caused by the induction of the nucleotide excision repair genes xpc and ddb2 [15] and the translesion polymerase eta [18]. Unlike chloroethylating agents, p53 stimulates apoptosis in U87MG glioma cells treated with TMZ [19]. Nevertheless, there are also opposite reports showing a protective function of p53 in glioma cells exposed to TMZ [16, 17, 20–22], indicating cell type-specific effects.
A second transcription factor that can be activated following anticancer drug treatment is AP-1, a dimeric transcription factor consisting of proteins belonging to the Fos, Jun or ATF family. AP-1 is activated via the MAPK (mitogene-activated protein kinase) pathway, involving JNK (c-Jun N-terminal kinase), p38K (p38 kinase) and ERK1/2 (extracellular signal-regulated kinases 1/2). Upon DNA damage, activation of AP-1 results in the induction of a plethora of AP-1 target genes, including DNA repair genes [8, 23–25] and pro-apoptotic genes [26–29]. Whereas for many genotoxins the activation of the MAPK cascade is experimentally well established [30], it is unclear whether DNA lesions induced by TMZ and CNUs are able to activate the MAPK/p38 kinase and whether this has an impact on therapy. Previously it was reported that JNK inhibition enhances senescence-associated β-galactosidase activity in TMZ-treated glioma cells with functional p53, whereas it induces mitotic catastrophe in p53 mutated cells [31]. Concerning p38K, it was reported that its inhibition sensitizes U87MG cells to TMZ due to abrogation of the G2 arrest [32, 33]. Regarding CNUs, it was reported that knockdown of the AP-1 component FRA1 sensitizes glioma cells towards ACNU via the attenuation of CHK1 phosphorylation and abrogation of the G2/M arrest [34], whereas carmustine (BCNU) induced ERK- and JNK-dependent cell death of neuronally-differentiated PC12 cells via generation of reactive oxygen species [35].
Here we show for the first time that the MAPK cascade triggered by JNK and its target c-Jun is involved in stimulating apoptosis upon TMZ and ACNU treatment of LN-229 and U87MG glioma cells. The cytotoxic effect results from AP-1 dependent induction of the BH3-only protein BIM, which reveals BIM as an important factor in TMZ and CNU-induced killing of glioma cells.
RESULTS
Induction of apoptosis following TMZ and ACNU treatment
Exploring the role of AP-1 for the sensitivity of malignant glioma cells to TMZ and ACNU, we first investigated the effectiveness of the anticancer drugs in the induction of apoptosis and the formation of DNA damage. Upon treatment of LN-229 cells with 100 μM TMZ or 50 μM ACNU, concentrations known to be reached in the serum of patients [36], a time-dependent induction of apoptosis was observed (Fig. 1A). Apoptosis started 96 h after TMZ treatment and earlier, after 72 h, in case of ACNU treatment, reaching 25% and 55%, respectively, 120 h after the onset of treatment. Parallel to the induction of cell death, cleavage of caspase-8 and -9 and the effector caspase-3 was observed (Fig. 1B). These events were preceded by phosphorylation of H2AX (γH2AX) (Fig. 1C), indicating activation of the DNA damage response pathway.
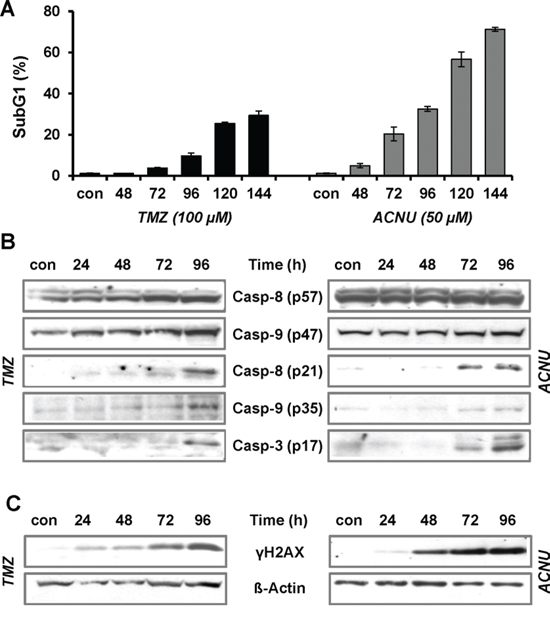
Figure 1: TMZ- and ACNU-induced apoptosis and DNA damage. A. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU. At different time points after exposure cells were stained with PI and the subG1 fraction was determined by flow cytometry. B/C. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU for indicated times. Protein extracts were prepared and subjected to western blot analysis. B. Expression of procaspase-8 (p57) and -9 (p47) as well as expression of the cleaved caspases-8 (p21), -9 (p35) and -3 (p17) was analyzed using specific antibodies. C. Expression of γH2AX was analyzed using specific antibodies. Detection of ß-Actin was used as loading control.
Impact of p53 signaling on TMZ and ACNU-induced cell death and DNA repair in glioma cells
An important transcription factor associated with survival and death is p53. In order to analyze its impact on TMZ and ACNU-induced cytotoxicity, LN-229 glioblastoma cells, wild-type (wt) for p53 (Suppl. Fig. 1A) were pre-treated with the p53 inhibitor pifithrin α (Pthα). This drug enhanced the cytotoxic potential of ACNU, but at the same time slightly protected LN-229 cells against TMZ (Suppl. Fig. 1B, left panel). Moreover we utilized LN-229 pSUPERp53 knockdown cells and LN-229 pSUPER control transfectants [37], which showed similar results (Suppl. Fig. 1B, right panel). Interestingly, despite the different biological outcomes, the expression of proapoptotic (Fas) and prosurvival factors (p21, XPC and DDB2) was similar in p53 wt cells following TMZ and ACNU treatment on mRNA (Suppl. Fig. 1C) and protein level (Suppl. Fig. 1D). The data support the notion that for ACNU the p53-induced activation of NER (induction of XPC and DDB2) is most important for cellular protection [38]. Upon TMZ exposure, activation of NER also occurs. However, it does not contribute to the repair of TMZ-induced DNA damage since O6-MeG is repaired via MGMT and not by NER [3]. In case of TMZ, enhanced expression of Fas seems to be the predominant p53 regulated pro-apoptotic trait.
Activation of MAPK signaling upon TMZ and ACNU in glioma cells
In addition to p53, the MAPK cascade also becomes activated upon treatment of glioma cells with TMZ and ACNU. Thus, TMZ induced a long-lasting phosphorylation of the p38K and the JNK starting 24 h and the reaching its maximum 72–120 h after the begin of treatment (Fig. 2A, left panel). Both kinases became strongly phosphorylated 24–72 h after ACNU treatment, thereafter the phosphorylation of JNK declined (Fig. 2A, right panel). ACNU, but not TMZ, enhanced the phosphorylation of the ERK kinase (ERK1/2). In effect of this, downstream transcription factors were activated as shown by increased AP-1 binding activity (Fig. 2B) and phosphorylation of its main component c-Jun (Fig. 2C).
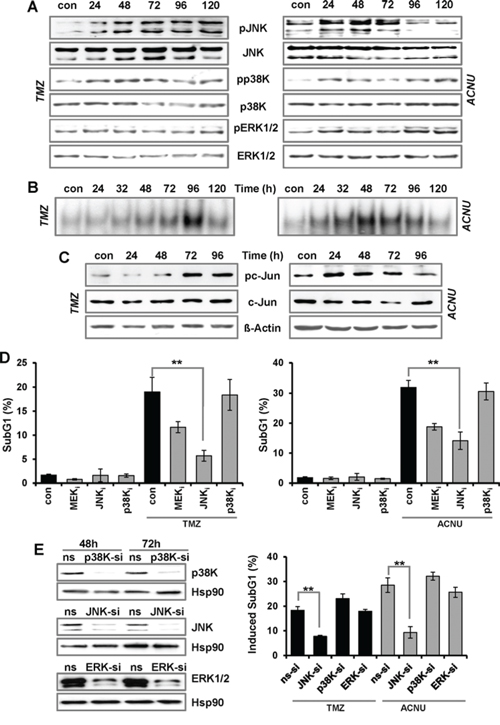
Figure 2: TMZ- and ACNU-induced activation of AP-1 and impact on sensitivity. A/B. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU for indicated times. Protein extracts were prepared and subjected to western blot analysis. A. Expression of JNK, p38K and ERK1/2, as well as expression of the phosphorylated forms (pJNK, pp38K and pERK1/2) was analyzed using specific antibodies. B. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU for indicated times. Nuclear extracts were isolated and incubated with radioactively labeled oligonucleotides containing the AP-1 binding site of the collagenase promoter (mmp1). The binding of AP-1 was visualized by EMSA. C. Expression of c-Jun as well as expression of the phosphorylated form (pc-Jun) was analyzed using specific antibodies. Detection of ß-Actin was used as loading control. D. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125), p38K (SB203580) and MEK1/2 (U0126) thereafter not exposed or exposed to 100 μM TMZ for 120 h or to 50 μM ACNU for 96 h. Cells were stained with PI and the subG1 fraction was determined by flow cytometry. E. LN-229 cells were transfected with siRNA against JNK, p38K and ERK1/2 or a non-silencing RNA (ns). Expression of JNK, p38K and ERK1/2 was analyzed 48 and 72 h later using specific antibodies. Detection of Hsp90 was used as loading control (left panel). LN-229 cells were exposed or not exposed to 100 μM TMZ or 50 μM ACNU. 24 h after exposure, cells were transfected with 50 nM siRNA against JNK, p38K and ERK1/2 or a non-silencing RNA (ns). Apoptosis was measured 120 h upon TMZ and 96 h after the onset of ACNU treatment via measurement of the subG1 fraction (right panel). Apoptotic frequency in relation to the untreated (siRNA transfection) control is shown.
Impact of the MAPK cascade on TMZ and ACNU-induced cell death in glioma cells
To analyze whether activation of the MAPK cascade has an impact on the sensitivity to TMZ and ACNU, the kinases were inhibited and the drug sensitivity was analyzed by determining the apoptosis frequency. Inhibition of JNK by a JNK1/2/3 inhibitor (SP600125) clearly protected LN-229 cells against TMZ and ACNU (Fig. 2D). Inhibition of p38K (SB203580) had no effect on cell death whereas inhibition of ERK1/2 by U0126 (an inhibitor of the upstream MEK1/2 kinase) slightly protected the cells (Fig. 2D). To further analyze the impact of the MAPK cascade on the sensitivity to TMZ and ACNU, we repressed JNK, p38K and ERK1/2 with specific siRNAs (Fig. 2E, left panel). Also in this case, knockdown of JNK strongly protected the cells against TMZ and ACNU (Fig. 2E, right panel). On the other hand, knockdown of ERK1/2 and p38K had no effect on TMZ/ACNU-induced apoptosis.
Next, we analyzed which kinase is responsible for the activation of AP-1 after treatment with TMZ and ACNU. Therefore, phosphorylation of the main AP-1 component c-Jun was analyzed in cells pre-treated with the kinase inhibitors. The results indicate that JNK, but not p38K or ERK1/2 activation, is responsible for c-Jun phosphorylation upon TMZ and ACNU (Fig. 3A). To analyze whether the pro-apoptotic effect of JNK activation is caused by c-Jun, siRNA-mediated knockdown was used to abrogate the expression of c-Jun or another component of the AP-1 complex, c-Fos (Fig. 3B). Knockdown of c-Jun, but not c-Fos, provoked an effect comparable to the JNK inhibitor, namely protection against TMZ and ACNU-induced cytotoxicity (Fig. 3C), indicating that the pro-apoptotic effect triggered by the MAPK cascade activation is caused by c-Jun. In contrast, knockdown of c-Fos did not protect against TMZ-induced apoptosis and even slightly sensitized cells to ACNU (Fig. 3C).
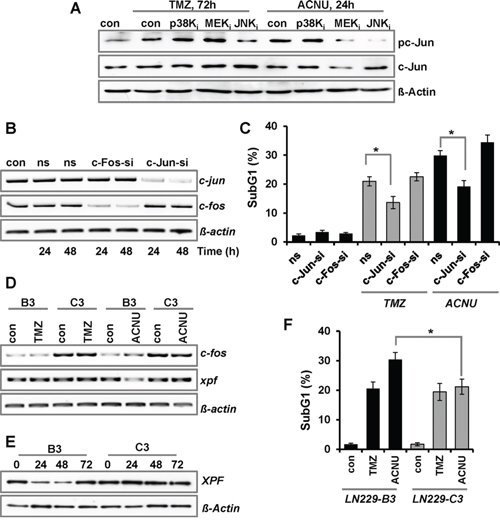
Figure 3: Impact of c-Jun and c-Fos on TMZ- and ACNU-induced cell death. A. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125), p38K (SB203580) or MEK1/2 (U0126) and thereafter not exposed or exposed to 100 μM TMZ for 72 h or to 50 μM ACNU for 24 h. Expression of c-Jun and its phosphorylated form (pc-Jun) was analyzed using specific antibodies. Detection of ß-Actin was used as loading control. B/C. LN-229 cells were transfected with 20 nM siRNA directed against c-Jun (si-cJun) and c-Fos (si-cFos) or a non-silencing RNA (ns). B. 24 and 48 h later RNA was isolated and endpoint RT-PCR was performed using c-fos, c-jun or, as loading control, gapdh specific primers. C. 24 h later cells were not exposed or exposed to 100 μM TMZ for 120 h or 50 μM ACNU for 96 h, stained with PI and the sub-G1 fraction was determined by flow cytometry. D–F. LN-229 cells stably overexpressing c-Fos (C3) and LN-229 mock-transfected cells (B3) were exposed to 100 μM TMZ or 50 μM ACNU. D. 24 h later RNA was isolated and endpoint RT-PCR was performed using c-fos, xpf or, as loading control, gapdh specific primers. E. At different time points upon ACNU exposure, expression of XPF was analyzed. F. 96 h after exposure to ACNU cells were stained with PI and the subG1 fraction was determined by flow cytometry.
This sensitization effect deserves to be considered in more detail. Previously, a protective function of c-Fos was reported in human fibroblasts treated with UV-light, which was associated with the induction and re-synthesis of the NER-endonuclease XPF [24, 25]. In LN-229 cells, 24 h after exposure to ACNU, both c-fos and xpf were slightly reduced; however, at later times c-fos became induced and the basal expression of xpf was restored (Suppl. Fig. 2A). To analyze whether c-Fos is responsible for the re-synthesis of xpf, we utilized LN-229 cells overexpressing c-Fos (Suppl. Fig. 2B, clone C3). Indeed c-Fos overexpressing cells showed no reduction of xpf mRNA following ACNU treatment, whereas reduction still occurred in the mock-transfected clone B3 (Fig. 3D). An accumulation of XPF was also observed at protein level (Fig. 3E), which was associated with a reduced sensitivity to ACNU (Fig. 3F). These data corroborate a protective function of c-Fos against ACNU-induced cell death by stimulation of NER. Overall, the data indicate that in LN-229 cells treated with TMZ or ACNU, MAPK activation leads to enhanced cell death which is regulated by JNK and c-Jun, but not c-Fos. For ACNU, this process appears to be counteracted by c-Fos triggered stimulation of XPF dependent repair of chloroethylated DNA adducts.
MAPK cascade and the expression of proapoptotic factors following TMZ and ACNU exposure
To analyze whether activation of JNK/c-Jun negatively influences DNA damage signaling and repair, the TMZ/ACNU-induced phosphorylation of H2AX (γH2AX) was analyzed upon inhibition of JNK (Fig. 4A). Furthermore, in the case of ACNU, the formation and repair of interstrand crosslinks was analyzed (Fig. 4B). In none of the experiments, inhibition of JNK had an effect, indicating that JNK has no negative impact on the cell's DNA damage signaling and repair capacity. Therefore, we posit that the JNK/c-Jun axis enhances glioma cell sensitivity via stimulation of the apoptotic pathway. An important AP-1 induced proapoptotic gene encodes the Fas ligand, a factor of the extrinsic apoptotic pathway [28, 39]. Indeed, treatment of LN-229 cells with TMZ and ACNU led to an enhanced expression of fasL mRNA (Fig. 4C). In the case of TMZ, fasL induction was slightly reduced by inhibition of JNK and p38K, and in the case of ACNU, JNK inhibition completely abrogated fasL induction whereas p38K inhibition had no effect (Fig. 4D). This indicates that in TMZ- and ACNU-treated glioma cells the fasL gene is a target of AP-1. However, in contrast to fasL mRNA, only a weak induction of the FasL protein (Fig. 4E) and no translocation of it to the outer cell membrane were observed (Fig. 4F). Thus, we conclude that the fasL is most likely not responsible for the pro-apoptotic effect of JNK/c-Jun.
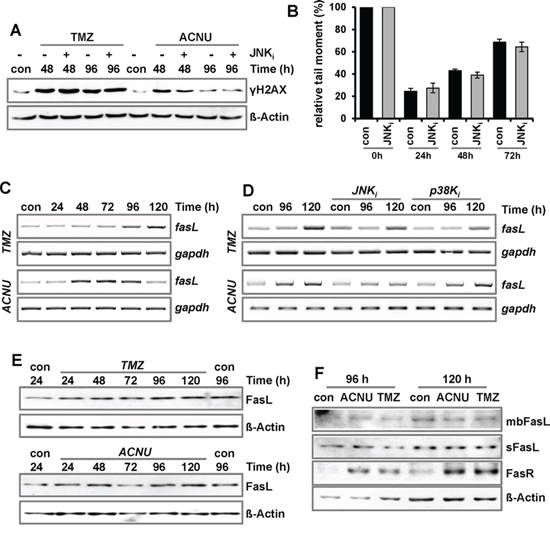
Figure 4: Impact of MAPK-signaling on DNA repair and expression of FasL. A/B. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125) and thereafter non-exposed or exposed to 100 μM TMZ or 50 μM ACNU for 24 h. A. At indicated time points expression of γH2AX was analyzed using specific antibodies. Detection of ß-Actin was used as loading control. B. Cells were non-exposed or exposed to 50 μM ACNU. At indicated time points, cells were harvested and subsequently irradiated with 8 Gy. Thereafter, the alkaline comet assay was performed. C. LN-229 cells were exposed to 100 μM TMZ (upper panel) or 50 μM ACNU (lower panel). D. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125) or p38K (SB203580) and thereafter non-exposed or exposed to 100 μM TMZ (upper panel) or 50 μM ACNU (lower panel). C/D. At indicated time points RNA was isolated and endpoint RT-PCR was performed using fasL or, as loading control, gapdh specific primers. E/F. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU. E. At indicated time points expression of the FasL was analyzed. F. 96 and 120 h later the expression of the soluble FasL (sFasL), the membrane bound form of the FasL (mbFasL) and the FasR was analyzed.
AP-1 was also shown to induce cyclinA, cyclinD1 and p16INK4 [40–43], which are involved in cell cycle regulation, and the proapoptotic genes bak and bim [26, 27]. Upon TMZ and ACNU we observed in LN-229 cells only the induction of bim (Fig. 5A, Suppl. Fig. 2C and data not shown). For BIM, three proapoptotic isoforms (bimEL, bimL, and bimS) were described. In our experiments, induction of bimEL was observed (Suppl. Fig. 2D). Similar to fasL, induction of bimEL in TMZ- and ACNU-treated cells was abrogated by pharmacological inhibition of JNK, but not p38K (Fig. 5B). The importance of JNK for the transcriptional activation of BIM was further shown by using JNK siRNA, leading to abrogation of BIM induction (Fig. 5C). Most importantly, BIM was also induced on protein level (Fig. 5D).
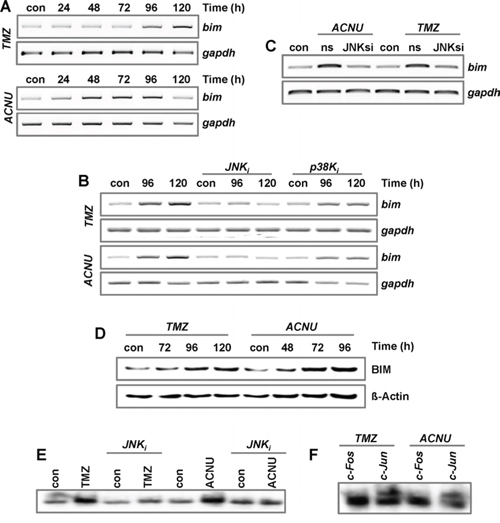
Figure 5: Impact of MAPK-signaling on the expression of BIM. A. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU. B. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125) or p38K (SB203580) and thereafter non-exposed or exposed to 100 μM TMZ (upper panel) or 50 μM ACNU (lower panel). C. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU. 24 h later, cells were transfected with 50 nM siRNA against JNK or a non-silencing control RNA (ns). A-C. At indicated time points RNA was isolated and endpoint RT-PCR was performed using bim or, as loading control, gapdh specific primers. D. LN-229 cells were exposed to 100 μM TMZ or 50 μM ACNU and expression of BIM was analyzed at indicated time points. E. LN-229 cells were pre-incubated for 1 h with a specific inhibitor for JNK1/2/3 (SP600125) or p38K (SB203580) and thereafter non-exposed or exposed to 100 μM TMZ (for 120 h) or 50 μM ACNU (for 96 h). Nuclear extracts were isolated and incubated with radioactively labeled oligonucleotides containing the AP-1 binding site of the bim promoter. The binding of AP-1 was visualized by EMSA. F. EMSA supershift assay: The binding of c-Jun and c-Fos to oligonucleotides containing the AP-1 binding site of the bim promoter was analyzed by the addition of specific antibodies to the EMSA reaction utilizing proteins from TMZ- and ACNU-exposed cells.
To further verify that BIM is an AP-1 target in glioma cells, EMSA assays were performed using radioactively labeled oligonucleotides harboring the potential AP-1 binding site of the BIM promoter (Fig. 5E). These experiments, together with supershift experiments, demonstrate the binding of c-Jun, but not of c-Fos, to this AP-1 binding site in cell extracts of TMZ- and ACNU-treated LN-229 cells (Fig. 5F).
BIM stimulates TMZ- and ACNU-induced death in glioma cells
To analyze whether BIM has an impact on the resistance of glioblastoma cells to TMZ and ACNU, BIM was transiently down-regulated by shRNAs in LN-229 and U87MG cells. Expression of BIM was determined by PCR, showing a strongly reduced expression 48 to 96 h after transfection (Fig. 6A, the effect on all three isoforms is shown). The response of mock-transfected and BIM-downregulated glioma cells to TMZ and ACNU was determined by flow cytometry. Apoptosis occurred at a significantly lower frequency in cells transfected with BIM-shRNA plasmid than in mock-transfected cells (Fig. 6B). Furthermore, cleavage of caspase-9 was significantly reduced upon knockdown of BIM (Fig. 6C). The data demonstrate a direct involvement of BIM in the induction of the caspase-9 driven apoptotic pathway in glioma cells following TMZ and ACNU treatment.
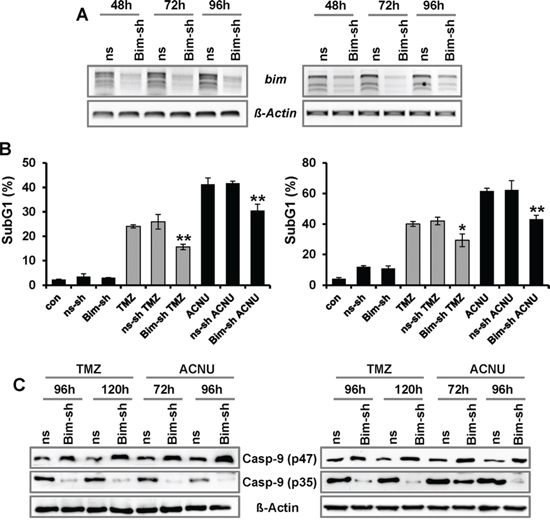
Figure 6: Impact of BIM on TMZ- and ACNU-induced cell death. A–C. LN-229 (left panel) and U87MG (right panel) glioblastoma cells were transiently transfected with plasmids expressing two different shRNA constructs specific for BIM (BIM-sh) or plasmids expressing a non-silencing shRNA (ns-sh). A. At indicated time points RNA was isolated and endpoint RT-PCR was performed using bim or, as loading control, ß-actin specific primers. B. 24 h later cells were non-exposed or exposed to 100 μM TMZ (for 120 h) or 50 μM ACNU (for 96 h). Cells were stained with PI and the sub-G1 fraction was determined by flow cytometry. C. 24 h later cells were exposed to 100 μM TMZ or 50 μM ACNU and 120 or 96 later the expression of procaspase-9 (p47) as well as the expression of cleaved caspases-9 (p35) was analyzed using specific antibodies.
DISCUSSION
Here, we analyzed the impact of the MAPK cascade and its target, the transcription factor AP-1, on the sensitivity of glioma cells to TMZ and ACNU, which are important alkylating drugs used in glioma therapy.
TMZ and ACNU induce apoptosis at late times in glioma cells (this data and [15]). The activation of the MAPK cascade precedes the induction of apoptosis, as shown by phosphorylation of p38K and JNK, increased phosphorylation of c-Jun and enhanced AP-1 binding activity. Inhibition and siRNA mediated knockdown of JNK protected LN-229 cells against TMZ- and ACNU-induced apoptosis. The same result was obtained upon siRNA-mediated silencing of the AP-1 component c-Jun, but not of c-Fos, indicating that the proapoptotic effect of JNK activation is caused by c-Jun. Interestingly, knockdown of c-Fos slightly sensitized LN-229 cells to ACNU, which was explained by lack of upregulation of the NER protein XPF.
We are aware that our data reporting JNK inhibition to block apoptosis in TMZ-treated cells are in contrast to Ohba et al., showing that inhibition of JNK enhances the TMZ-induced cytotoxicity in U87MG cells [31]. In these experiments cell death was analyzed by the MTT assay 72 h after TMZ exposure. In our hands, apoptosis is not yet executed in LN-229 or U87MG cells at this time following treatment (Suppl. Fig. 3A). Using the MTT assay, we also observed a reduced metabolic activity in U87MG cells upon JNK inhibition, whereas in LN-229 cells this effect was only marginal (Suppl. Fig. 3B). Further, Ohba et al. reported that the increased sensitivity following JNK inhibition is associated with enhanced senescence while the effect on apoptosis was not analyzed. Taken together, it is reasonable to conclude that JNK signals different events, depending on the time period following exposure. In line with this, we previously showed that glioma cells undergo autophagy, senescence and apoptosis in a specific time-dependent manner after TMZ treatment [44]. In this case, autophagy, which is the earliest event upon TMZ treatment, stimulates cells to undergo senescence rather than apoptosis, showing that the different endpoints are interrelated. Furthermore, inhibition of autophagy and thereby senescence leads to an increase in apoptosis upon TMZ [44], explaining why inhibition of JNK at the same time can enhance senescence [31] and reduce apoptosis (this paper). In another study, sensitization of glioma cells to TMZ by JNK inhibitors was observed in MGMT expressing cells due to an impact of JNK on the basal expression of this DNA repair gene [45]. Both cell lines used in our study (LN-229 and U87MG) are MGMT deficient (due to promoter methylation) and, therefore, a possible repression of MGMT following JNK inhibition cannot explain the data reported here.
To investigate the molecular mechanism underlying JNK/c-Jun-induced apoptosis in TMZ- and ACNU-treated glioma cells, we analyzed whether JNK/c-Jun is directly involved in the activation of the extrinsic apoptotic pathway. The extrinsic pathway is activated via binding of FasL to the Fas receptor (Fas) [46, 47]. While the FasR is induced via p53, the FasL is a prominent target of AP-1 [28, 29, 39]. Previously, we showed that apoptosis induced by UV-light and cisplatin is triggered by JNK/c-Jun mediated induction of FasL [48, 49]. Here, we demonstrate that LN-229 cells treated with TMZ and ACNU respond with an enhanced expression of fasL mRNA, which was abrogated by inhibition of JNK. However, only weak induction on the protein level and no translocation to the cell membrane was observed, indicating that the transcriptional induction of fasL alone is not responsible for the proapoptotic activity of JNK/c-Jun following TMZ and ACNU exposure.
A search for additional AP-1 induced apoptotic targets revealed the induction of the proapoptotic BIM gene upon TMZ/ACNU exposure. BIM was originally identified as a Bcl-2-interacting protein [50]. Multiple isoforms were characterized, however only for BimEL, BimL, and BIMS the pro-apoptotic function has been confirmed, BIMS being the most potent. This is partly explained via the sequestration of BIMEL and BIML by the cytoskeleton-associated motor complex and binding to dynein light chain LC8, from which they are released upon various apoptotic stimuli [51]. In our experiments, we observed strong induction of bimEL (Suppl. Fig. 2C). BIM induces apoptosis by causing mitochondrial permeabilization and the release of apoptogenic factors (e.g. cytochrome c, Smac/DIABLO). In this process, BIM can bind BAX and BAK and induces cytochrome c release from mitochondria [52]. In addition, it was reported that BIM preferentially activates BAX whereas BAK is mostly activated by BID [53]. Upon formation, these factors can activate caspases, including caspase-9, and thereby induce apoptosis [54].
The impact of JNK/c-Jun on the upregulation of BIM was shown by the finding that BIM induction following TMZ and ACNU was abrogated by pharmacological inhibition and siRNA based silencing of JNK. Also, binding of AP-1/c-Jun to the AP-1 binding site of the BIM promoter was demonstrated by EMSA experiments. The data are in line with reports showing AP-1 dependent induction of BIM following exposure to nitric acid and upon nerve growth factor deprivation [26, 27]. Moreover, BIM was shown to be upregulated in U87MG cells treated with lovastatin [55]. Interestingly, BIM induction was also observed in head and neck squamous cell carcinoma (HNSCC) cell lines upon replication stress provoked by the ribonucleotide reductase inhibitor hydroxyurea (HU) [56]. Since ACNU and TMZ induce DNA replication blocking lesions and DSBs in the 1st and 2nd post-exposure cell cycle, respectively, we suppose that these lesions represent the initial trigger for BIM induction [57, 58].
Besides transcriptional induction, BIM activity can also be regulated via post-translational modifications. Thus ERK1/2-dependent phosphorylation of BimEL promotes its dissociation from Mcl-1 and Bcl-x and causes its proteasomal degradation [59, 60]. Therefore we analyzed the expression of BIM upon siRNA mediated knockdown of ERK1/2. The results show that knockdown of ERK1/2 leads to an increased expression of BIM, supporting a role of ERK1/2 in controlling BIM stability in glioma cells (Suppl. Fig. 3C). However, ERK1/2 induced degradation of BIM seems not to contribute to TMZ/ACNU sensitivity, as indicated by ERK1/2 knockdown experiments (Fig. 2E). We should note that pharmacological inhibition of MEK1/2, but not siRNA based repression of ERK1/2, slightly protected glioma cells against TMZ and ACNU. This may indicate that the effect of MEK1/2 inhibition may be caused either by ERK1/2-independent signals activated by MEK1/2 or by off-target effects of the inhibitor.
To address the question of whether BIM is involved in TMZ/ACNU-induced apoptosis in glioma cells, experiments were performed using shRNA-mediated silencing of BIM. These experiments showed that knockdown of BIM reduced the cleavage of caspase-9 and the level of apoptosis in LN-229 and U87MG cells treated with TMZ or ACNU. Our findings are in line with a report showing that the JNK activator anisomycin can sensitize glioma cells to apoptosis in a mechanism requiring both JNK activation and BIM upregulation [61]. Furthermore it was shown that the sphingosine analogue FTY720, which also induces upregulation of BIM, synergistically induces apoptosis in combination with TMZ in brain tumor stem cells [62]. A synergistic effect of combined anisomycin/TMZ treatment was also observed in our study (Suppl. Fig. 3D). Therefore, it is reasonable to conclude that targeting BIM represents a novel concept in anticancer therapy [63].
In summary, we show that TMZ and ACNU induce the activation of JNK and subsequently of c-Jun/AP-1 in glioma cells. Pharmacological inhibition and siRNA mediated repression of JNK and c-Jun reduced the level of apoptosis in these cells treated with TMZ and ACNU. Apoptosis was a result of AP-1 triggered induction of the proapoptotic protein BIM, indicating that BIM represents an important factor that contributes to the outcome of TMZ- and ACNU-based therapy of malignant gliomas.
MATERIALS AND METHODS
Cell lines and anticancer drug treatment
The glioma cell lines LN-229 and U87MG were described previously [37]. The glioma cell lines LN229-Super and LN229-p53sh were kindly provided by Prof. Weller (Laboratory of Molecular Neuro-Oncology, University Hospital and University of Zurich, Zurich, Switzerland). The cells were grown in Dulbecco's minimal essential medium (DMEM) containing 10% fetal bovine serum (FBS), in 7% CO2 at 37°C. The JNK1/2/3 inhibitor SP600125 and the p38K inhibitor SB203580 were purchased from Sigma-Aldrich (Hamburg, Germany); the MEK1/2 inhibitor U0126 was from Promega (Mannheim, Germany). The inhibitors were added to the medium at a concentration of 10 μM 1 h prior to ACNU or TMZ treatment and were not removed until harvest.
Knockdown experiments
Knockdown of c-Fos and c-Jun was performed using 20 nM siRNAs from Santa Cruz (sc29221 and sc29223). Knockdown of JNK, p38K and ERK1/2 was performed using 50 nM SignalSilence® SAPK/JNK siRNA I #6233, SignalSilence® p38 MAPK siRNA I #6564, and SignalSilence® p44/42 MAPK (Erk1/2) siRNA #6560. For transfection the Lipofectamine RNAiMAX Transfection Kit (Invitrogen) was used. Down-regulation of BIM was achieved by the transfection with shRNA vectors [56].
Preparation of cell extracts and western blot analysis
Whole cell extracts and nuclear cell extracts were prepared as described [25]. Proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham) by semi-dry blotting. For immuno-detection, the antibodies against ß-Actin (C4, Santa Cruz), BIM (B7929, Sigma), FasL (G247-4, BD Pharmingen), ERK2 (sc-154, Santa Cruz), γH2AX (#05-164, Upstate), XPF (Ab-5, NeoMarkers), c-Fos (sc-52, Santa Cruz) were diluted 1:1000 in 5% non-fat dry milk/Tween-TBS. For western blot analysis with phospho-specific antibodies, cells were directly lyzed in 1 x SDS-PAGE sample buffer (Roti®-Load 1, Carl Roth GmbH) and subsequently sonified. Rabbit phospho-specific antibodies (anti-p-cJun: #3270, anti-p-JNK: #4668P; anti-p-p38K: #4511P; anti-p-ERK1/2: #4370P) as well as non-phospho-specific antibodies (anti-cJun: #9165; anti-JNK: #9258P; anti-p38K: #9212P and anti-ERK1/2: #4695P) were purchased from Cell Signaling Technology (Boston, MA, USA) and diluted 1:1000 in 5% BSA/Tween-TBS. The secondary anti-rabbit or anti-mouse antibodies (Rockland) were diluted 1:2000 in 5% non-fat dry milk/Tween-TBS.
Preparation of RNA and endpoint PCR
Total RNA was isolated using the RNA II Isolation Kit (Machery and Nagel, Düren, Germany) and cDNA synthesis was performed using the Verso cDNA Kit (Thermo Scientific, Bonn, Germany). Endpoint PCR was performed using Red-Taq Ready Mix (Sigma-Aldrich). The specific primers are shown in Suppl. Table 1.
Determination of apoptosis
For monitoring drug-induced apoptosis and cell cycle distribution, ethanol-fixed cells were treated with DNAse free RNAse and stained with propidium iodide (PI). The Sub-G1 fraction was determined by flow cytometry. The experiments were repeated at least three times. Mean values ± SD are shown.
Preparation of nuclear extracts and EMSA
Nuclear cell extracts were prepared and subjected to electromobility shift assay (EMSA) or Western blot analysis as described [64]. The sequence of the oligonucleotides specific for the AP-1 binding site of the mmp1 promoter was 5′-AGTGGTGACTCATCACT-3′. The sequence of the oligonucleotides specific for the AP-1 binding site of the BIM promoter was 5′-GTTAGCGGTGACTCACATTCCCAG-3′. For supershift analysis, 2 μg antibodies specific for c-Jun (sc45, Santa Cruz) or c-Fos (sc-52, Santa Cruz) were incubated with 8 μg protein extract for 20 min at room temperature in the reaction buffer before addition of the labeled oligonucleotides.
ICL repair measured by single cell gel electrophoresis (SCGE, comet assay)
For the modified comet assay [65] cells were treated with 50 μM ACNU and, after the indicated time periods, cells were trypsinized and washed with ice cold PBS. Thereafter, all cells were irradiated by 8 Gy. Immediately after irradiation, alkaline cell lysis and electrophoresis was performed as described [66]. The results are expressed as relative tail moment (%) = fluorescence intensity tail (%) x distance head center to tail end.
ACKNOWLEDGMENTS
We greatly appreciate the technical assistance of Ms. Birgit Rasenberger. The work was supported by Mildred Scheel Stiftung (Deutsche Krebshilfe) and Wilhelm Sander Stiftung.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Wen PY, Kesari S. Malignant gliomas in adults. The New England Journal of Medicine. 2008; 359:492–507.
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine. 2005; 352:987–96.
3. Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair. 2007; 6:1079–99.
4. Ludlum DB. The chloroethylnitrosoureas: sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Investigation. 1997; 15:588–98.
5. Tong WP, Kirk MC, Ludlum DB. Formation of the cross-link 1-[N3-deoxycytidyl),2[N1-deoxyguanosinyl]-ethane in DNA treated with N,N′-bis(2-chloroethyl)-N-nitrosourea. Cancer Research. 1982; 42:3102–5.
6. Fischhaber PL, Gall AS, Duncan JA, Hopkins PB. Direct demonstration in synthetic oligonucleotides that N,N′-bis (2-chloroethyl)-nitrosourea cross links N1 of deoxyguanosine to N3 of deoxycytidine on opposite strands of duplex DNA. Cancer Research. 1999; 59:4363–8.
7. Brulikova L, Hlavac J, Hradil P. DNA interstrand cross-linking agents and their chemotherapeutic potential. Current Medicinal Chemistry. 2012; 19:364–85.
8. Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Research. 2013; 41:8403–20.
9. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991; 51:6304–11.
10. Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998; 188:2033–45.
11. Tomicic MT, Christmann M, Kaina B. Topotecan-triggered degradation of topoisomerase I is p53-dependent and impacts cell survival. Cancer Research. 2005; 65:8920–6.
12. Tomicic MT, Christmann M, Kaina B. Topotecan triggers apoptosis in p53-deficient cells by forcing degradation of XIAP and survivin thereby activating caspase-3-mediated Bid cleavage. J Pharmacol Exp Ther. 2010; 332:316–25.
13. Tomicic MT, Kaina B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochimica et Biophysica Acta. 2013; 1835:11–27.
14. Wang Y, Zhu S, Cloughesy TF, Liau LM, Mischel PS. p53 disruption profoundly alters the response of human glioblastoma cells to DNA topoisomerase I inhibition. Oncogene. 2004; 23:1283–90.
15. Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Research. 2007; 67:11886–95.
16. Xu GW, Mymryk JS, Cairncross JG. Inactivation of p53 sensitizes astrocytic glioma cells to BCNU and temozolomide, but not cisplatin. Journal of Neuro-oncology. 2005; 74:141–9.
17. Xu GW, Mymryk JS, Cairncross JG. Pharmaceutical-mediated inactivation of p53 sensitizes U87MG glioma cells to BCNU and temozolomide. International Journal of Cancer. 2005; 116:187–92.
18. Tomicic MT, Aasland D, Naumann SC, Meise R, Barckhausen C, Kaina B, Christmann M. Translesion polymerase eta is upregulated by cancer therapeutics and confers anticancer drug resistance. Cancer Research. 2014; 74:5585–96.
19. Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2007; 26:186–97.
20. Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Research. 2001; 61:1957–63.
21. Dinca EB, Lu KV, Sarkaria JN, Pieper RO, Prados MD, Haas-Kogan DA, Vandenberg SR, Berger MS, James CD. p53 Small-molecule inhibitor enhances temozolomide cytotoxic activity against intracranial glioblastoma xenografts. Cancer Research. 2008; 68:10034–9.
22. Blough MD, Beauchamp DC, Westgate MR, Kelly JJ, Cairncross JG. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. Journal of Neuro-oncology. 2011; 102:1–7.
23. Christmann M, Tomicic MT, Aasland D, Berdelle N, Kaina B. Three prime exonuclease I (TREX1) is Fos/AP-1 regulated by genotoxic stress and protects against ultraviolet light and benzo(a)pyrene-induced DNA damage. Nucleic Acids Research. 2010; 38:6418–32.
24. Christmann M, Tomicic MT, Origer J, Aasland D, Kaina B. c-Fos is required for excision repair of UV-light induced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Research. 2006; 34:6530–9.
25. Tomicic MT, Reischmann P, Rasenberger B, Meise R, Kaina B, Christmann M. Delayed c-Fos activation in human cells triggers XPF induction and an adaptive response to UVC-induced DNA damage and cytotoxicity. Cellular and Molecular Life Sciences : CMLS. 2011; 68:1785–98.
26. Jin HO, Park IC, An S, Lee HC, Woo SH, Hong YJ, Lee SJ, Park MJ, Yoo DH, Rhee CH, Hong SI. Up-regulation of Bak and Bim via JNK downstream pathway in the response to nitric oxide in human glioblastoma cells. Journal of Cellular Physiology. 2006; 206:477–86.
27. Biswas SC, Shi Y, Sproul A, Greene LA. Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. The Journal of Biological Chemistry. 2007; 282:29368–74.
28. Faris M, Latinis KM, Kempiak SJ, Koretzky GA, Nel A. Stress-induced Fas ligand expression in T cells is mediated through a MEK kinase 1-regulated response element in the Fas ligand promoter. Molecular and Cellular Biology. 1998; 18:5414–24.
29. Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa, B, and AP-1. Molecular Cell. 1998; 1:543–51.
30. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer. 2009; 9:537–49.
31. Ohba S, Hirose Y, Kawase T, Sano H. Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced cytotoxicity in human glioma cells. Journal of Neuro-oncology. 2009; 95:307–16.
32. Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Molecular and Cellular Biology. 2003; 23:8306–15.
33. Hirose Y, Katayama M, Berger MS, Pieper RO. Cooperative function of Chk1 and p38 pathways in activating G2 arrest following exposure to temozolomide. Journal of Neurosurgery. 2004; 100:1060–5.
34. Meise R, Tomicic MT, Kaina B, Christmann M. The chloroethylating anticancer drug ACNU induces FRA1 that is involved in drug resistance of glioma cells. Biochimica et Biophysica Acta. 2012; 1823:1199–207.
35. An JM, Kim SS, Rhie JH, Shin DM, Seo SR, Seo JT. Carmustine induces ERK- and JNK-dependent cell death of neuronally-differentiated PC12 cells via generation of reactive oxygen species. Toxicology in vitro. 2011; 25:1359–65.
36. Hammond LA, Eckardt JR, Kuhn JG, Gerson SL, Johnson T, Smith L, Drengler RL, Campbell E, Weiss GR, Von Hoff DD, Rowinsky EK. A randomized phase I and pharmacological trial of sequences of 1,3-bis (2-chloroethyl)-1-nitrosourea and temozolomide in patients with advanced solid neoplasms. Clinical Cancer Research. 2004; 10:1645–56.
37. Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene. 2003; 22:8233–45.
38. Barckhausen C, Roos WP, Naumann SC, Kaina B. Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene. 2014; 33:1964–74.
39. Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA, Nel A. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J Immunol. 1998; 160:134–44.
40. Katabami M, Donninger H, Hommura F, Leaner VD, Kinoshita I, Chick JF, Birrer MJ. Cyclin A is a c-Jun target gene and is necessary for c-Jun-induced anchorage-independent growth in RAT1a cells. The Journal of Biological Chemistry. 2005; 280:16728–38.
41. Andrecht S, Kolbus A, Hartenstein B, Angel P, Schorpp-Kistner M. Cell cycle promoting activity of JunB through cyclin A activation. The Journal of Biological Chemistry. 2002; 277:35961–8.
42. Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. The EMBO Journal. 1999; 18:188–97.
43. Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. The EMBO Journal. 2000; 19:2056–68.
44. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, Kaina B. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PloS One. 2013; 8:e55665.
45. Okada M, Sato A, Shibuya K, Watanabe E, Seino S, Suzuki S, Seino M, Narita Y, Shibui S, Kayama T, Kitanaka C. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. International Journal of Oncology. 2014; 44:591–9.
46. Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995; 181:71–7.
47. Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995; 373:441–4.
48. Christmann M, Tomicic MT, Aasland D, Kaina B. A role for UV-light-induced c-Fos: Stimulation of nucleotide excision repair and protection against sustained JNK activation and apoptosis. Carcinogenesis. 2007; 28:183–90.
49. Brozovic A, Fritz G, Christmann M, Zisowsky J, Jaehde U, Osmak M, Kaina B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. International Journal of Cancer. 2004; 112:974–85.
50. O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. The EMBO Journal. 1998; 17:384–95.
51. Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Molecular Cell. 1999; 3:287–96.
52. Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular Cell. 2005; 17:525–35.
53. Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Molecular Cell. 2013; 51:751–65.
54. Fesik SW, Shi Y. Structural biology. Controlling the caspases. Science. 2001; 294:1477–8.
55. Jiang Z, Zheng X, Lytle RA, Higashikubo R, Rich KM. Lovastatin-induced up-regulation of the BH3-only protein, Bim, and cell death in glioblastoma cells. Journal of Neuro-Chemistry. 2004; 89:168–78.
56. Stauber RH, Knauer SK, Habtemichael N, Bier C, Unruhe B, Weisheit S, Spange S, Nonnenmacher F, Fetz V, Ginter T, Reichardt S, Liebmann C, Schneider G, et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget. 2012; 3:31–43.
57. Nikolova T, Hennekes F, Bhatti A, Kaina B. Chloroethylnitrosourea-induced cell death and genotoxicity: cell cycle dependence and the role of DNA double-strand breaks, HR and NHEJ. Cell Cycle. 2012; 11:2606–19.
58. Quiros S, Roos WP, Kaina B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle. 2010; 9:168–78.
59. Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003; 22:6785–93.
60. Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. The Journal of Biological Chemistry. 2003; 278:18811–6.
61. Xia S, Li Y, Rosen EM, Laterra J. Ribotoxic stress sensitizes glioblastoma cells to death receptor induced apoptosis: requirements for c-Jun NH2-terminal kinase and Bim. Molecular Cancer Research. 2007; 5:783–92.
62. Estrada-Bernal A, Palanichamy K, Ray Chaudhury A, Van Brocklyn JR. Induction of brain tumor stem cell apoptosis by FTY720: a potential therapeutic agent for glioblastoma. Neuro-oncology. 2012; 14:405–15.
63. Harada H, Grant S. Targeting the regulatory machinery of BIM for cancer therapy. Critical Reviews in Eukaryotic Gene Expression. 2012; 22:117–29.
64. Christmann M, Kaina B. Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. The Journal of Biological Chemistry. 2000; 275:36256–62.
65. Spanswick VJ, Hartley JM, Ward TH, Hartley JA. Measurement of drug-induced DNA interstrand crosslinking using the single-cell gel electrophoresis (comet) assay. Methods Mol Med. 1999; 28:143–54.
66. Tomicic MT, Thust R, Sobol RW, Kaina B. DNA polymerase beta mediates protection of mammalian cells against ganciclovir-induced cytotoxicity and DNA breakage. Cancer Research. 2001; 61:7399–403.