
INTRODUCTION
Primary malignant brain tumors make up one of the deadliest forms of cancer in both adults and children. Glioblastoma (GBM), the most common primary malignant brain tumor in adults, is a highly malignant diffuse astrocytoma that can present de novo or develop from the progression of a lower grade tumor. Children also present with GBM, although less commonly. For any age group, GBM share a similar histopathologic appearance between tumors, however GBM are highly heterogeneous with respect to their biologic and molecular characteristics. Using a combination of gene expression, genomic and proteomic data to identify patterns between GBMs, tumors can be stratified into potentially clinically relevant subtypes [1-8]. Future stratification of patients into subgroups based upon predicted responsiveness to specific therapies will likely lead to improved therapy. Despite these exciting advances, the current prognosis for patients with GBM is poor and median survival remains less than two years [9]. There is great need for improvement in our understanding of factors driving tumorigenesis within different tumor subtypes. This knowledge will propel the development of novel therapeutic strategies and advance our ability to target tumors and to predict response.
Aberrant activation of multiple receptor tyrosine kinase (RTK) signaling pathways is a unifying feature across GBM, promoting many aspects of tumorigenesis including tumor cell proliferation, survival, invasion, and induction of angiogenesis. Indeed the most common alterations in GBM include amplification of RTK receptors, such as EGFR and PDGFRA, and increased expression of ligands, such as PDGFB [5, 10-12]. Furthermore, overexpression of ligands such as PDGFB can drive tumorigenesis in murine brain tumor models [13-15]. Despite the role of RTKs in driving oncogenesis, small molecule inhibitors targeting single RTK pathways have been largely unsuccessful in improving overall survival [16]. A potential explanation for the limited efficacy of these targeted therapeutics is that GBM is driven by the summation of multiple signaling inputs [1, 17-19]. Simultaneous targeting of multiple abnormal signaling pathways will likely be required for the development of more effective therapies.
During normal development and tissue maintenance ligand-mediated signaling is exquisitely regulated, including the bioavailability of ligand in the extracellular environment. A prototypical example is the extracellular regulation of the Wnt family of secreted proteins. Once released from the cell Wnt ligands bind and are sequestered by proteins such as heparan sulfate proteoglycans (HSPGs) in the extracellular environment. Only after ligands are released from HSPGs can they bind to and activate their Frizzled receptors [20, 21]. While the mechanisms regulating ligand availability in the tumor microenvironment are just beginning to be elucidated in GBM, they likely play a role in driving oncogenic signaling pathways. Blocking these mechanisms has the potential to inhibit ligand-mediated activation of multiple oncogenic pathways in tumors.
Heparan sulfate proteoglycans and extracellular sulfatases in GBM.
HSPGs present on the cell surface as well as in the extracellular matrix, are a major component of the extracellular environment in normal brain and GBM [22, 23]. They regulate cellular signaling via their ability to bind diverse protein ligands including growth factors, chemokines, morphogens, matrix proteins, cell adhesion molecules, and proteases [24-28]. As illustrated in Figure 1A, HSPGs can bind and sequester ligands thereby preventing engagement with their cognate receptor, as discussed above with the Wnts for example. HSPG can also act as a co-receptor and promote receptor signaling, such as with FGF2 and VEGF [20, 29, 30]. HSPG-mediated signaling is also critical for normal brain development [31].
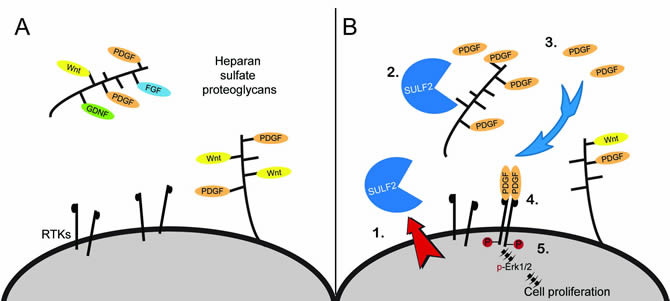
FIGURE 1: Heparan sulfate proteoglycan (HSPG) glycosaminoglycan side chains bind and sequester ligands in the extracellular environment. (A) Dependent on the HSPG core protein, HSPGs are found at the cell surface, in the extracellular matrix, or in secretory vesicles. HSPG function is critical for normal growth and development and includes regulation of ligand-mediated signaling, cell adhesion, and formation of the extracellular matrix for cell migration. (B) Model for SULF2 regulated RTK signaling in glioblastoma. SULF2 acts on HSPGs, present in the tumor microenvironment, to decrease 6O-sulfation, release sequestered ligands such as PDGF and increase activation of the RTK PDGFR-alpha and downstream signaling pathways in tumor cells. RTK, receptor tyrosine kinase.
In GBM, the expression levels of multiple HSPG core proteins and HSPG-modifying enzymes are significantly altered relative to normal brain (Figure 2). HSPGs consist of a protein core and heparan sulfate (HS) chains consisting of linear carbohydrate chains of repeating disaccharide units. Essential for their function in cell signaling, the HS chains undergo extensive post-translational modifications, including sulfation on the 6-O- position of glucosamine [32]. Indeed, 6-O-sulfation of HS is a critical determinant of growth factor binding and is essential for normal development [33-37]. While a number of intracellular enzymes regulate HSPG biosynthesis and sulfation, the recently discovered extracellular sulfatases, SULF1 and SULF2, reveal a novel mechanism for the regulation of HSPG-dependent signaling. By removing 6-O-sulfates on HS chains and mobilizing protein ligands from HSPG sequestration in the extracellular environment, the Sulfs can activate multiple key signaling pathways (e.g., Wnt, Shh, GDNF, and PDGF) [20, 29, 38-40].
Consistent with this ability, SULF transcripts are overexpressed in GBM and in many human cancers, including non-small cell lung cancer (NSCLC), hepatocellular carcinoma, breast cancer, head and neck cancer, pancreatic adenocarcinoma, multiple myeloma, and gastric carcinoma [40-42]. In GBM, we have found that SULF2 protein is expressed in adult and pediatric tumors (Figure 3) and, using knockdown and transgenic approaches, we have demonstrated ablation of SULF2 results in decreased activity of several RTKs, including PDGFR-alpha, decreased tumor cell proliferation, and prolonged survival in vivo[40]. Interestingly, SULF2 has also been directly implicated as a driver of carcinogenesis in NSCLC [43], pancreatic cancer [44], hepatocellular carcinoma [45], and a murine model for oligodendroglioma [46], further supporting its importance. The SULFs appear to regulate multiple signaling pathways important in cancer, likely upstream to the interaction of growth factors with RTKs and the activation of intracellular kinases (Figure 4). Defining the extent and timing of SULF2 function in tumorigenesis will be important.
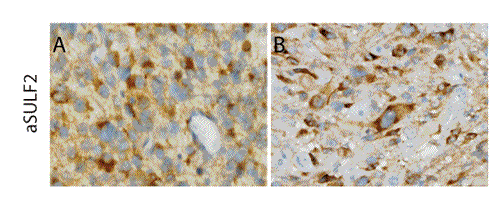
FIGURE 3: Expression of SULF2 protein in human GBM. Representative images of immunohistochemical staining for SULF2 in adult (A) and pediatric (B) GBM. Immunohistochemistry was performed as described previously [40]. Magnification 400x.
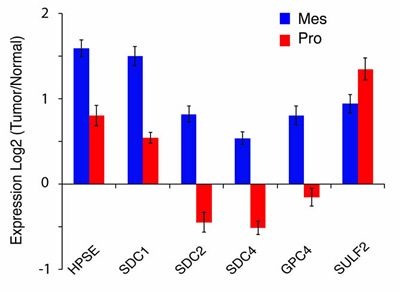
FIGURE 4: Subtype-specific alterations in the expression of HSPGs and HSPG modifying enzymes in GBM. The mean expression of HSPG-related genes are compared between the Mesenchymal (Mes) and Proneural (Pro) subtype of adult GBM. Bars represent the mean ratio of log2(Tumor/Normal) +/– SEM gene expression in each subgroup and a two-sided t-test was used to compare expression between the two groups. p<0.05, Mes n=56 and Pro n=53. Expression data from TCGA Data Portal [71]; http://cancergenome.nih.gov.
HSPGs are also the targets of heparanase (HPSE), an endoglycosidase, which generates biologically active fragments of HS chains. Heparanase is upregulated in many cancers, including GBM (Figure 2 and [47-49]). Increased expression of heparanase in tumors has been implicated in increased tumorigenesis, tumor angiogenesis, and invasiveness [47-49]. Together these studies suggest that tumors actively enzymatically modify components of the brain tumor microenvironment to help drive oncogenic signaling and invasion. Disruption of this partnership may be an important therapeutic strategy.
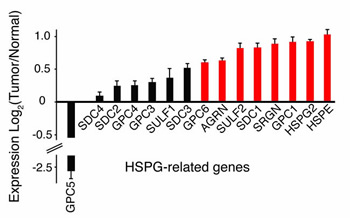
FIGURE 2: Altered HSPG-related gene expression in human GBM. The mean expression of a number of HSPG-related genes, including HSPG core proteins (GPCs, SDCs, AGRN, SRGN, and HSPG2) and modifying enzymes (HPSE and SULFs) are altered in GBM relative to normal controls. Bars represent the mean ratio of log2(Tumor/Normal) +/– SEM gene expression. Upregulated (red) and down regulated, log2 (Tumor/Normal) greater than or equal to 0.5 or less than or equal to -0.5, respectively. TCGA Data Portal [71]; http://cancergenome.nih.gov. (n=170 human tumors). GPC, glypican; SDC, syndecan; SULF, extracellular sulfatase; AGRN, agrin; SRGN, serglycin; HSPG2, perlecan; HSPE, heparanase.
In addition to enzymatic alterations in HSPGs, the identity and levels of the HSPG core proteins are also important determinants of cell signaling. The core protein specifies proteoglycan localization and can influence both extracellular and intracellular signaling. For example, the syndecans (SDCs), composed of four members, are membrane bound HSPGs and contain a cytoplasmic domain that binds cytoskeletal proteins and can serve as a substrate in cell signaling [50, 51]. SDCs play important roles in cell signaling, cell adhesion, and migration. Manipulation of syndecan-1 expression has been shown to alter HGF-Met signaling [52, 53] and Wnt signaling [54] in cancer. In contrast, the glypicans (GPCs) composed of six members, are GPI-linked to the cell membrane, and these are typically involved in growth factor and morphogen responses. Other HSPGs, such as perlecan (HSPG2) are found within the extracellular matrix. For those HSPGs associated with the cell membrane, the extracellular domain can also be shed, resulting in the release of biologically active proteoglycans. Indeed, shed syndecan-1 has been shown to mediate removal of CXC chemokines and facilitate resolution of inflammation [55]. In multiple myeloma high levels of shed syndecan-1 correlate with poor prognosis and have been associated with increased tumor growth in animal models [56, 57]. In GBM, both total and specific HSPGs core proteins are altered (Figure 2), including increased levels of syndecan-1 and glypican-1 as previously demonstrated [23, 58].
Interestingly, alterations in HSPGs vary across tumor subtypes suggesting there may be subtype-specific HSPG functions in GBM, Figure 4. The proneural GBM subtype, characterized by alterations in PDGFR signaling, has high SULF2 expression [4]. In contrast, the mesenchymal GBM subtype exhibits increased expression of multiple other HSPG-related genes. This latter subtype has increased expression of genes involved in interactions with the extracellular environment, cell signaling, and the immune response, and, consistent with gene expression data, the number of tumor-associated microglia/macrophages is significantly greater in the mesenchymal compared to the proneural subtype (Mann-Whitney, p=0.042; J. Engler and J. Phillips, unpublished observation). While the mechanisms driving the increased inflammatory response in the mesenchymal subtype are not fully elucidated, proteoglycans have the potential to influence the immune response in cancer [59]. Indeed, tumor-derived versican has been shown to activate macrophages and increase metastatic tumor growth in a model for lung carcinoma [60]. Furthermore, targeting of HSPGs with a heparan sulfate (HS) mimetic normalized myeloid-derived suppressor cell levels in a murine mammary carcinoma model [61]. Understanding the function of HSPGs and the enzymes that modify them in a subtype-specific context will be important for the optimization of future therapeutic strategies. This has recently been illustrated by the report of an immunogenic therapy in which patients with a GBM of the mesenchymal subtype had a more robust immune response than patients with a tumor of the proneural subtype [62].
HSPGs and the enzymes that regulate them as potential therapeutic targets in GBM.
Accumulating data suggest the extracellular HSPGs, and the enzymes that modify them, may regulate ligand-mediated signaling pathways in GBM. Given their role in disease combined with their accessibility in the extracellular environment, they represent clinically relevant, druggable therapeutic targets. In malignant astrocytoma, we recently determined that knockdown of SULF2 resulted in decreased activity of multiple RTK signaling pathways including PDGFR-alpha, IGF1R-beta and EPHA2 [40], three pathways known to be involved in astrocytoma growth and invasion [10, 13, 14, 63-65]. Furthermore, ablation of SULF2, in a relevant murine model for astrocytoma, resulted in decreased activation of PDGFR-alpha, decreased tumor cell proliferation, and prolonged survival [40]. These data, combined with the high expression of SULF2 in a significant number of human GBMs, suggest SULF2 may be considered an upstream therapeutic target in the treatment of GBM and other cancers in which it is overexpressed.
Since HSPGs regulate multiple upstream signaling pathways, and some of these same pathways are critical in malignancy, it is of great interest that a recent class of compounds has been developed to inhibit some of these oncogenic functions. Heparan sulfate mimetics are highly sulfated oligosaccharides that inhibit heparanase, sequester HSPG-binding factors, and inhibit SULF2 [66-68]. In preclinical studies, HS mimetics have effectively targeted multiple HSPG-dependent phenotypes and have resulted in decreased in vivo tumor growth, tumor invasion, tumor metastasis, and angiogenesis [61, 69]. Furthermore, a human Phase II clinical trial demonstrated safety and preliminary efficacy for a HS mimetic in recurrent hepatocellular carcinoma [70], and a recent preclinical study of a new rationally engineered HS mimetic, M402, suggests additional potential as a therapeutic agent [61]. While HS mimetics have not yet been tested in GBM, they are known to inhibit SULF2 activity [67] and represent a promising strategy.
Currently the prognosis for patients with GBM is challenging. With recent advances in imaging, genomic sequencing and proteomics, there is great hope that we are entering into a new era for detection and treatment of GBM. Stratification of patients into therapeutically relevant subgroups will likely be an essential component for treatment. Large-scale analyses of bulk tumors have revealed significant differences in expression of genes involved in tumor-microenvironment interactions between tumor subgroups, including proteoglycans and immune response-related gene. Targeting HSPGs and related components of the tumor microenvironment has the potential to simultaneously inhibit multiple oncogenic signaling pathways in tumor cells and to disrupt critical tumor-microenvironment interactions. Future efforts will be aimed at identifying the relevant tumor-microenvironment interactions that help drive GBM and how to effectively target them therapeutically.
ACKNOWLEDGEMENTS
This work was supported by funds from the National Institutes of Health (K08 NS063456) and the Pediatric Brain Tumor Foundation Institute Award at UCSF. Zena Werb, Steven D. Rosen, and Joseph T.C. Shieh provided helpful discussions and Aaron E. Robinson provided assistance with graphics.
Reference
1. Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A and Holland E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009; 4(11):e7752.
2. Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, Popoff S, Nutt CL, Louis DN, Cairncross JG, Gilbert MR, Phillips HS, Mehta MP, Chakravarti A, Pelloski CE, Bhat K, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010; 12(1):49-57.
3. Mischel PS, Shai R, Shi T, Horvath S, Lu KV, Choe G, Seligson D, Kremen TJ, Palotie A, Liau LM, Cloughesy TF and Nelson SF. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003; 22(15):2361-2373.
4. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG and Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9(3):157-173.
5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17(1):98-110.
6. Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, James JD, Gumin J, Diefes KL, Kim SH, Turski A, Azodi Y, Yang Y, Doucette T, Colman H, Sulman EP, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011; 25(24):2594-2609.
7. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010; 28(18):3061-3068.
8. Duarte CW, Willey CD, Zhi D, Cui X, Harris JJ, Vaughan LK, Mehta T, McCubrey RO, Khodarev NN, Weichselbaum RR and Gillespie GY. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PloS One. 2012; 7(1):e29653.
9. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114(2):97-109.
10. Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B and Nister M. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992; 52(11):3213-3219.
11. Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, Huse JT, Pedraza A, Utsuki S, Yasui Y, Tandon A, Fomchenko EI, Oka H, Levine RL, Fujii K, Ladanyi M, et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010; 24(19):2205-2218.
12. Guha A, Dashner K, Black PM, Wagner JA and Stiles CD. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer. 1995; 60(2):168-173.
13. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN and Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001; 15(15):1913-1925.
14. Uhrbom L, Hesselager G, Nister M and Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998; 58(23):5275-5279.
15. Liu KW, Feng H, Bachoo R, Kazlauskas A, Smith EM, Symes K, Hamilton RL, Nagane M, Nishikawa R, Hu B and Cheng SY. SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest. 2011; 121(3):905-917.
16. Fan QW and Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Cur Top Microbiol Immunol. 2010; 347:279-296.
17. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L and DePinho RA. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007; 318(5848):287-290.
18. Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch A, Heppner FL, Yonekawa Y, Merlo A, Frei K, Mariani L and Hofer S. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther. 2011; 10(6):1102-1112.
19. Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN and Iafrate AJ. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011; 20(6):810-817.
20. Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM and Emerson CP, Jr. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001; 293(5535):1663-1666.
21. Logan CY and Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004; 20:781-810.
22. Steck PA, Moser RP, Bruner JM, Liang L, Freidman AN, Hwang TL and Yung WK. Altered expression and distribution of heparan sulfate proteoglycans in human gliomas. Cancer Res. 1989; 49(8):2096-2103.
23. Watanabe A, Mabuchi T, Satoh E, Furuya K, Zhang L, Maeda S and Naganuma H. Expression of syndecans, a heparan sulfate proteoglycan, in malignant gliomas: participation of nuclear factor-kappaB in upregulation of syndecan-1 expression. J Neurooncol. 2006; 77(1):25-32.
24. Smith EM, Mitsi M, Nugent MA and Symes K. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. Proc Natl Acad Sci U S A. 2009; 106(51):21683-21688.
25. Feyzi E, Lustig F, Fager G, Spillmann D, Lindahl U and Salmivirta M. Characterization of heparin and heparan sulfate domains binding to the long splice variant of platelet-derived growth factor A chain. J Biol Chem. 1997; 272(9):5518-5524.
26. Ono K, Hattori H, Takeshita S, Kurita A and Ishihara M. Structural features in heparin that interact with VEGF165 and modulate its biological activity. Glycobiology. 1999; 9(7):705-711.
27. Kreuger J, Salmivirta M, Sturiale L, Gimenez-Gallego G and Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001; 276(33):30744-30752.
28. Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH and Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004; 279(13):12346-12354.
29. Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U and Emerson CP, Jr. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol. 2003; 162(2):341-351.
30. Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest. 2001; 108(3):357-361.
31. Sarrazin S, Lamanna WC and Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011; 3(7).
32. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J and Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999; 68:729-777.
33. Bink RJ, Habuchi H, Lele Z, Dolk E, Joore J, Rauch GJ, Geisler R, Wilson SW, den Hertog J, Kimata K and Zivkovic D. Heparan sulfate 6-o-sulfotransferase is essential for muscle development in zebrafish. J Biol Chem. 2003; 278(33):31118-31127.
34. Bulow HE and Hobert O. Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron. 2004; 41(5):723-736.
35. Kamimura K, Fujise M, Villa F, Izumi S, Habuchi H, Kimata K and Nakato H. Drosophila heparan sulfate 6-O-sulfotransferase (dHS6ST) gene. Structure, expression, and function in the formation of the tracheal system. J Biol Chem. 2001; 276(20):17014-17021.
36. Esko JD and Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001; 108(2):169-173.
37. Habuchi H, Habuchi O and Kimata K. Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconj J. 2004; 21(1-2):47-52.
38. Danesin C, Agius E, Escalas N, Ai X, Emerson C, Cochard P and Soula C. Ventral neural progenitors switch toward an oligodendroglial fate in response to increased Sonic hedgehog (Shh) activity: involvement of Sulfatase 1 in modulating Shh signaling in the ventral spinal cord. J Neurosci. 2006; 26(19):5037-5048.
39. Ai X, Kitazawa T, Do AT, Kusche-Gullberg M, Labosky PA and Emerson CP, Jr. SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007; 134(18):3327-3338.
40. Phillips JJ, Huillard E, Robinson AE, Ward A, Lum DH, Polley MY, Rosen SD, Rowitch DH and Werb Z. Heparan sulfate sulfatase SULF2 regulates PDGFR-alpha signaling and growth in human and mouse malignant glioma. J Clin Invest. 2012; 122(3):911-922.
41. Bret C, Moreaux J, Schved JF, Hose D and Klein B. SULFs in human neoplasia: implication as progression and prognosis factors. J Transl Med. 2011; 9:72.
42. Rosen SD and Lemjabbar-Alaoui H. Sulf-2: an extracellular modulator of cell signaling and a cancer target candidate. Expert Opin Ther Targets. 2010; 14(9):935-949.
43. Lemjabbar-Alaoui H, van Zante A, Singer MS, Xue Q, Wang YQ, Tsay D, He B, Jablons DM and Rosen SD. Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene. 2009; 29(5):635-646.
44. Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M and Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS One. 2007; 2(4):e392.
45. Lai JP, Oseini AM, Moser CD, Yu C, Elsawa SF, Hu C, Nakamura I, Han T, Aderca I, Isomoto H, Garrity-Park MM, Shire AM, Li J, Sanderson SO, Adjei AA, Fernandez-Zapico ME, et al. The oncogenic effect of sulfatase 2 in human hepatocellular carcinoma is mediated in part by glypican 3-dependent Wnt activation. Hepatology. 2010; 52(5):1680-1689.
46. Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, Uhrbom L and Westermark B. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A. 2004; 101(31):11334-11337.
47. Hong X, Nelson KK, deCarvalho AC and Kalkanis SN. Heparanase expression of glioma in human and animal models. J Neurosurg. 2010; 113(2):261-269.
48. Ridgway LD, Wetzel MD and Marchetti D. Heparanase Modulates Shh and Wnt3a Signaling in Human Medulloblastoma Cells. Exp Ther Med. 2011; 2(2):229-238.
49. Kurokawa H, Katsube K, Podyma KA, Ikuta M, Iseki H, Nakajima M, Akashi T, Omura K, Takagi M and Yanagishita M. Heparanase and tumor invasion patterns in human oral squamous cell carcinoma xenografts. Cancer Sci. 2003; 94(3):277-285.
50. Rapraeger AC. Syndecan-regulated receptor signaling. J Cell Biol. 2000; 149(5):995-998.
51. Whiteford JR, Xian X, Chaussade C, Vanhaesebroeck B, Nourshargh S and Couchman JR. Syndecan-2 is a novel ligand for the protein tyrosine phosphatase receptor CD148. Mol Biol Cell. 2011; 22(19):3609-3624.
52. Derksen PW, Keehnen RM, Evers LM, van Oers MH, Spaargaren M and Pals ST. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood. 2002; 99(4):1405-1410.
53. Ramani VC, Yang Y, Ren Y, Nan L and Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011; 286(8):6490-6499.
54. Alexander CM, Reichsman F, Hinkes MT, Lincecum J, Becker KA, Cumberledge S and Bernfield M. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet. 2000; 25(3):329-332.
55. Hayashida K, Parks WC and Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood. 2009; 114(14):3033-3043.
56. Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, Waage A and Borset M. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000; 95(2):388-392.
57. Yang Y, Yaccoby S, Liu W, Langford JK, Pumphrey CY, Theus A, Epstein J and Sanderson RD. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. 2002; 100(2):610-617.
58. Su G, Meyer K, Nandini CD, Qiao D, Salamat S and Friedl A. Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am J Pathol. 2006; 168(6):2014-2026.
59. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006; 6(9):633-643.
60. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL and Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009; 457(7225):102-106.
61. Zhou H, Roy S, Cochran E, Zouaoui R, Chu CL, Duffner J, Zhao G, Smith S, Galcheva-Gargova Z, Karlgren J, Dussault N, Kwan RY, Moy E, Barnes M, Long A, Honan C, et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PloS One. 2011; 6(6):e21106.
62. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF and Liau LM. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011; 17(6):1603-1615.
63. Nister M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin CH, Schlessinger J and Westermark B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988; 48(14):3910-3918.
64. Carapancea M, Cosaceanu D, Budiu R, Kwiecinska A, Tataranu L, Ciubotaru V, Alexandru O, Banita M, Pisoschi C, Backlund ML, Lewensohn R and Dricu A. Dual targeting of IGF-1R and PDGFR inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in JNK-1 expressing cells. J Neurooncol. 2007; 85(3):245-254.
65. Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, Jackson D, de Groot J and Yung WK. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007; 6(4):1357-1367.
66. Dredge K, Hammond E, Handley P, Gonda TJ, Smith MT, Vincent C, Brandt R, Ferro V and Bytheway I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br J Cancer. 2011; 104(4):635-642.
67. Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, van Kuppevelt TH, Jenniskens GJ, Kimata K, Rosen SD and Uchimura K. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology. 2010; 20(2):175-186.
68. Johnstone KD, Karoli T, Liu L, Dredge K, Copeman E, Li CP, Davis K, Hammond E, Bytheway I, Kostewicz E, Chiu FC, Shackleford DM, Charman SA, Charman WN, Harenberg J, Gonda TJ, et al. Synthesis and biological evaluation of polysulfated oligosaccharide glycosides as inhibitors of angiogenesis and tumor growth. J Med Chem. 2010; 53(4):1686-1699.
69. Joyce JA, Freeman C, Meyer-Morse N, Parish CR and Hanahan D. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene. 2005; 24(25):4037-4051.
70. Liu CJ, Lee PH, Lin DY, Wu CC, Jeng LB, Lin PW, Mok KT, Lee WC, Yeh HZ, Ho MC, Yang SS, Lee CC, Yu MC, Hu RH, Peng CY, Lai KL, et al. Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: a randomized phase II trial for safety and optimal dosage. J Hepatol. 2009; 50(5):958-968.
71. The Cancer Genome Atlas. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455(7216):1061-1068.