
INTRODUCTION
miR-155 is an oncogenic pro-inflammatory microRNA (miRNA) that is up-regulated in a number of solid tumors and liquid malignancies [1-3]. High levels of miR-155 often correlate with a poor prognosis [4-5]. Targeted expression of miR-155 in B cells results in pre-B cell acute leukemia/high-grade lymphoma [6]. Furthermore, overexpression of miR-155 in lymphoid tissues results in disseminated lymphoma characterized by a clonal, transplantable pre-B-cell population of neoplastic lymphocytes “addicted” to miR-155-activity [7]. In hematopoietic cells, the expression of miR-155 is controlled by several immune signals [1-3]. Thus, LPS induces miR-155 expression in macrophage/monocytic cell lines of both mouse and human origin [8-9]. The oncogenic and pro-inflammatory effects of miR-155 have been attributed at least in part to its targeting of many transcripts encoding tumor suppressors and/or anti-inflammatory factors, especially Ship1 [10-12], Socs1 [13].
Quaking (QKI, KH domain containing, RNA binding) is a member of the signal transduction and activation of RNA (STAR) family of RNA-binding proteins. Three major QKI isoforms (QKI-5, QKI-6 and QKI-7), each with a specific carboxy-terminal end, are produced through alternative splicing both in mouse and human [14]. QKI-5 contains a nuclear localization signal, is predominantly localized in the nucleus, and is most likely to function in pre-mRNA splicing or RNA retention. In contrast, QKI-6 is localized in both the nucleus and the cytoplasm, while QKI-7, exclusively cytoplasmic, is pro-apoptotic [14-16]. QKI behaves as a tumor suppressor gene (TSG) in glioblastoma multiforme (GBM) [17]. QKI, under the direct control of p53 in GBM cells [17], is also downregulated in gastric and colon cancers [18-19]. In GBM cells, QKI can associate with and stabilize miR-20a, thus, increasing miR-20a down-regulatory effects on TGF-β receptor type II (TGF-βR2), whose activity is oncogenic in gliomagenesis [17].
As QKI is the first most probable target of miR-155 in apes and human (targetscan.org), and that the expression of miR-155 is upregulated in glioblastomas [20-21], the above-mentioned facts suggested that miR-155 might carry out its oncogenic and pro-inflammatory functions at least in part by targeting QKI. In the present study, we show that (i) QKI is indeed a target of miR-155 in B cells; (ii) the expression of QKI is lower in B-cells from CLL patients compare to B cells from healthy donors, and acts as TSG also in CLL; (iii) Qki is a target of LPS signaling, and its expression is downregulated following LPS challenge of macrophages; (iv) Qki modulates downstream LPS signaling in return; particularly p38 MAPK activation and IL-10 production, thus presenting with anti-inflammatory properties. We propose that miR-155 and QKI form a critical regulatory component downstream of TLR4 in target hematopoietic cells, and that miR-155 exerts its pro-inflammatory and oncogenic activities at least in part through the downregulation of QKI expression.
RESULTS
QKI is downregulated at the onset of the innate immune response to LPS
We have previously shown that the expression of miR-155 is upregulated in mouse RAW-264.7 macrophages treated with LPS [8]. As Qki is a potential target of miR-155, we monitored the expression of Qki, using a probe spanning exons 4 and 5 that recognizes all Qki isoforms, as well as of miR-155 and Tnf in mouse RAW-264.7 macrophages following LPS stimulation. Qki expression decreased 2-fold within 4 hours, while the expression of miR-155 as well as that of Tnf, both immediate downstream targets of LPS signaling, increased significantly (Figure 1A-1C). Beyond 12 hours, Qki transcripts progressively returned to their initial level at 2-days, while miR-155 level kept increasing. Of note, the expression of Qki in untreated cells subsequently increased above its initial level, an effect delayed by roughly 12 hours following LPS challenge (Figure 1A). This increase was probably related to the surge of cell density, as previously reported in HT29 colon cancer cells [22]. Semi-quantitative RT-PCR analysis using oligonucleotides designed to specifically recognize each of Qki isoforms, showed that both Qki-5 and Qki-6 are expressed in RAW-264.7 cells and their expression is downregulated at the beginning of LPS challenge (data not shown), further confirming our hypothesis. On Western blots, Qki levels also decreased significantly only for a short period of time (data not shown), suggesting that Qki might be a critical component of LPS signaling, potentially involved in preventing the initiation of an unnecessary innate immune response, assuring the robustness of the response at its onset, and resolving and terminating the response later on. Our pan-Qki antibody detected one Qki isoform only, and QKI-5 being the main isoform expressed in hematopoietic cells [23], this isoform is likely to represent Qki-5.
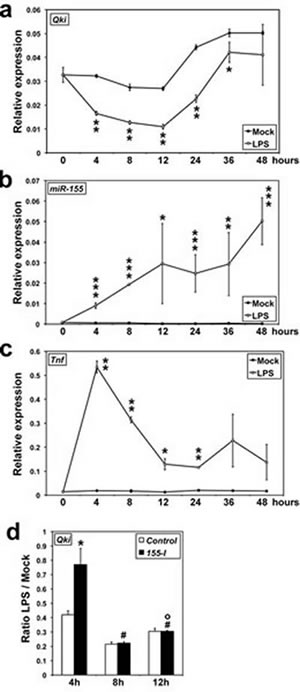
Figure 1: QKI expression is downregulated at the onset of LPS challenge. A.-C. Qki, miR-155 and Tnf levels in RAW-264.7 cells were measured by qRT-PCR (n = 3); A.: *P = 0.0636; **P < 0.000345. B.: *P = 0.06177; **P = 0.03106; ***P < 0.00168. C.: *P = 0.0124; **P < 0.000786. D. RAW-264.7 cells transfected with either a Control-RNA (Control) or an antisense miR-155 inhibitory RNA (155-I) were challenged with LPS 24 hours later. Qki transcripts levels were measured by qRT-PCR (n = 3). *miR-155-I different from Control, P < 0.037. # 8-hours different from 4-hours, P < 0.0052. o12-hours different from 8-hours, P < 0.0044.
Accordingly, an antisense miR-155 inhibitory RNA (155-I) protected Qki transcript from downregulation only during the first 4 hours of LPS challenge, while remaining without effects later on (Figure 1D). Altogether, the above results suggest that, in RAW-264.7 macrophages, Qki transcripts are potentially targets of miR-155 at the onset of LPS signaling only.
QKI is a direct target of miR-155 in U937 cells
Human QKI-3’-UTR contains three putative miR-155 target sites, the first one being conserved among eutherian mammals but absent in muridae and in some human isoforms, the second highly conserved across vertebrates, and the third one primate-specific (targetscan.org). To determine whether miR-155 could directly target QKI transcripts, we prepared three Luciferase reporter constructs from human QKI-3’-UTR, each containing one miR-155 target site (Figure 2A). A miR-155 mimic co-transfected in U937 cells with each of these constructs decreased the Luciferase activity produced from the constructs containing a WT, but not a mutant, miR-155 site (Figure 2B). Finally, the Luciferase activity also decreased when U937 cells were first transfected with constructs containing the WT, but not the mutant, miR-155 site and then challenged with LPS (Figure 2C), giving a further evidence that QKI downregulation at the onset of the innate immune response to LPS results from its direct targeting by miR-155. Intriguingly, mutating the miR-155 target site of QKI-UTR-1 increased the Luciferase activity produced following miR-155 transfection beyond that of the Control (Figure 2B, 2C). This may possibly result from miR-155 targeting immune factor(s) that bind to this particular region of Qki-3’-UTR, given that the stability of several immune transcripts is regulated by the fixation of RNA binding proteins on their 3’-UTR, and that we observed a similar phenomenon in RAW264.7 macrophages, but not in HEK-293 kidney cells (not shown).
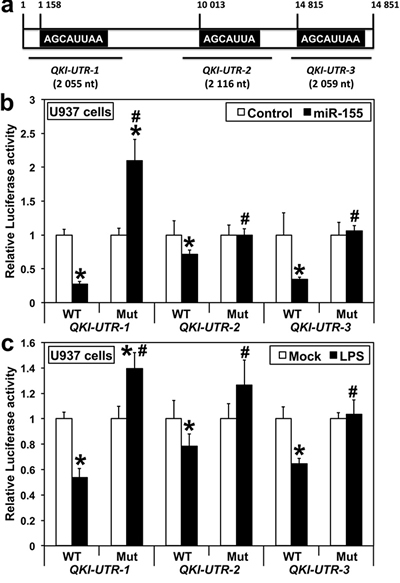
Figure 2: QKI is a direct target of miR-155 in human U937 monocytes. A. Schematic (not-to-scale) representation of human QKI-3’-UTR. The sequences of miR-155 consensus target sites (highlighted) present in the three QKI-3’-UTR Luciferase reporter constructs are shown. B. Cells were co-transfected with either QKI-UTR-1, QKI-UTR-2 or QKI-UTR-3, each containing either a wild type (WT) or a mutated (Mut) miR-155 target site, along with either pre-miR-Control (Control) or pre-miR-155 (n = 12). C. Cells were transfected with either WT/Mut QKI-UTR-1, QKI-UTR-2 or QKI-UTR-3 24 hours before LPS challenge (n = 12). Values for pre-miR-Control B. and Mock C. were arbitrarily set to 1. Assays were performed three times in quadruplicates 48 hours after transfection. B. *miR-155 different from Control, P < 0.00052. C. *LPS different from Mock C., P < 0.0022. B., C. #Mutant different from WT, P < 1x10-6.
Qki regulates cytokine production and MAPK phosphorylation
Qki being a target of LPS signaling suggested that Qki itself might be a modulator of the innate immune response. We thus transfected RAW-246.7 cells with a construct expressing QKI-5 (hereafter referred to as QKI), chosen because: (i) it is the only QKI isoform that contains a nuclear localization signal and functions in pre-miRNA splicing and/or RNA retention [15], thus being the most likely isoform to impact the stability and processing of immune transcripts; and (ii) QKI-5 is the main isoform expressed in hematopoietic progenitors and differentiated cells [23], and our experiments in Figure 1 showed that it is well expressed in RAW-264.7 cells. Indeed, the over-expression of QKI-5 doubled the level of anti-inflammatory IL-10 in cell supernatant (Figure 3A), 24 hours after LPS challenge. In contrast, targeting Qki transcripts with small interfering RNAs (siQKI), lead to higher levels of inflammatory IL-1α, IL-1β, IL-6 and GM-CSF (Figure 3B). The fact that QKI enhanced IL-10 production, suggests that its downregulation during the first hours of LPS challenge is a prerequisite for the immune response to reach the optimal threshold level of activation. As LPS binding to TLR4 receptors triggers the downstream activation of ERK, JNK and p38 MAPK pathways in responding cells [24], we then analyzed the effects of QKI on the three above pathways. Overexpressing QKI impaired the phosphorylation of p38 and Jnk1/Sapk MAPKs, possibly also slightly the phosphorylation of Erk1, 2, during the two first hours of LPS challenge (Figure 3C). In contrast, siQKI respectively increased p38 phosphorylation by 47% at 0.5-hour (P = 0.0011) and by 177% at 1-hour (P = 0.0789) (Figure 3D), as determined by scanning the bands on blots representing three different biological replicates. Noteworthy, QKI overexpression sharply increased the level of total p38, however only residual amount of this kinase was phosphorylated (Figure 3C). As the p38 MAPK pathway is central to the stabilization of many mRNAs encoding factors implicated in the innate and adaptive immune responses, including cytokines [24-26], these results further confirm our hypothesis that the early down-regulation of Qki by LPS signaling pathway is needed for mounting an effective immune response.
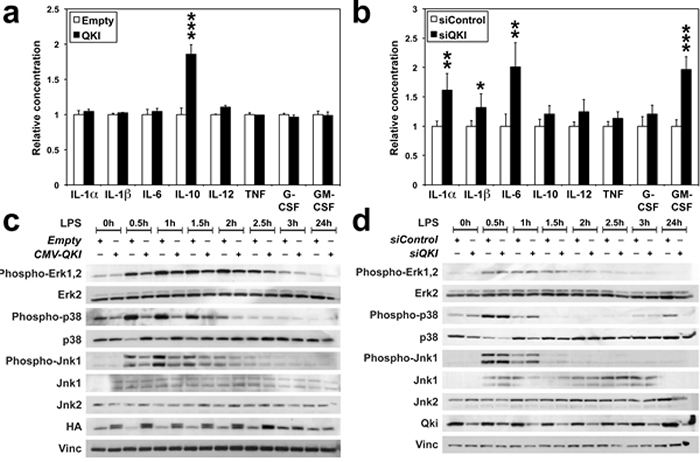
Figure 3: QKI impairs Mapk activation and interleukin production (A-D) RAW-264.7 cells were challenged with LPS 24 hours following transfection with either an empty CMV vector (Empty) or a construct expressing human QKI-5 (QKI) (A, C), or with either a control-siRNA (siControl) or siQKI (B, D). A., B. Supernatants harvested 24 hours after LPS treatment were analyzed by ELISA assay for the indicated cytokines. Values were normalized to Control. *P = 0.069; **P < 0.019; ***P < 0.001. C.-D. Phosphorylation of Erk1/Erk2, Jnk1/Sapk and p38 Mapk following LPS stimulation was followed on Western blots. Stripes in C. and D. come from three different gels prepared from the same extracts. HA: HA-tagged QKI. Vinculin (Vinc) was used as a loading control.
miR-155 also targets QKI in human leukemic B lymphocytes
The expression of miR-155 is high in B-CLL-derived cell lines as well as in CLL patients [27-30], but low in B-cell Burkitt’s lymphomas [31]. We found that QKI and miR-155 expression well discriminate Burkitt’s cell lines, all with lower miR-155 and higher QKI levels, from CLL cell lines, all with higher miR-155 but lower QKI levels (Figure 4A). Accordingly, QKI protein levels in MEC2, Ado-2199 and WAC CLL cells were lower than those in PH3R1, EW36 and BJAB cells (Figure 4B). However, QKI levels in MEC1, Daudi and Raji cells suggest that QKI expression at the protein level is also regulated by other, non-miR-155-dependent mechanisms. Nevertheless, miR-155 mimic reduced QKI expression in BJAB and PH3R1 Burkitt’s cells, but not in MEC2 CLL cells (Figure 4C). In contrast, transfecting MEC2 cells with 155-I slightly but significantly increased QKI levels (Figure 4C). Altogether, these results indicate that QKI is also a target of miR-155 in B cells.
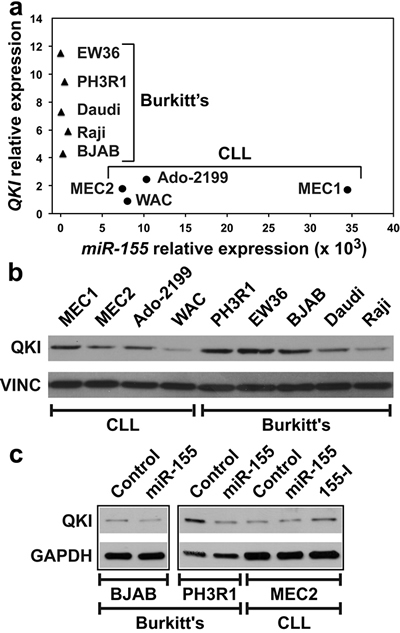
Figure 4: Burkitt’s and CLL cell lines display different levels of QKI and miR-155 expression. A. QKI and miR-155 levels were measured by qRT-PCR in five Burkitt’s lymphoma cell lines (filled triangles) and in four CLL-derived cell lines (filled circles). B. QKI protein levels in the same cells as in A.. C. Indicated cells were transfected with either a Control RNA or miR-155. MEC2 cells were also transfected with 155-I. Western blot was performed 48 hours following transfection. Panels are from the same blot.
QKI regulates FAS expression and Caspases 3/7 activity in B cells
A bioinformatics analysis showed that the 3’-UTR of FAS transcripts contains the consensus sequence for QKI binding [32]. siQKI, functional in both MEC2 and BJAB cell lines (Figure 5A), significantly reduced FAS levels and the percentage of Fas-expressing cells in both cell lines (Figure 5B, 5C). Furthermore, siQKI decreased FAS transcript levels in MEC2 but not BJAB cells (Figure 5D), suggesting that different molecular mechanisms control FAS expression in these two types of hematological malignancies. Finally, siQKI decreased the activity of Caspases 3/7 in both cell lines (Figure 5E), suggesting that QKI might act as a TSG also in B cells.
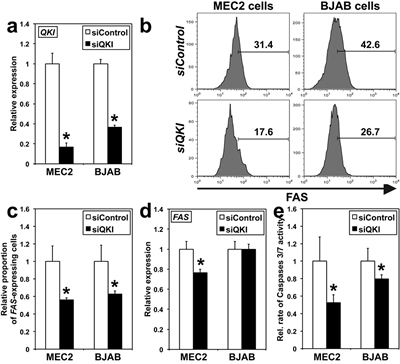
Figure 5: QKI effects on FAS expression and Caspases 3/7 activity in BJAB and MEC2 cells. Analyses were conducted 48 hours following transfection. A. QKI transcripts levels were determined using qRT-PCR. *siQKI different from siControl, P < 0.0003 (n = 3). B. Representative histograms of FAS staining on MEC2 and BJAB cells as determined by flow cytometry (n = 3). C. Graphical presentation of triplicate results of flow cytometry run in B.. *P < 0.026 (n = 3). D. FAS expression was analyzed using qRT-PCR. *P < 0.01. E. The relative rate of Caspases 3/7 activity was measured in cells transfected either with siQKI or siControl. *P < 0.031. In A. and C.-E., values for siControl were arbitrarily set to 1.
QKI is implicated in lymphocyte function and homeostasis
We then analyzed transcriptome modifications induced by siQKI in MEC2 and BJAB cells using Affymetrix arrays (MIAME: E-MTAB-2375) and Ingenuity software. Expectedly, pathways implicated in the regulation of lymphocyte differentiation, proliferation, function and/or signaling were affected in both cell lines (not shown). However, siQKI had also cell specific effects (Tables 1 and 2). Of interest, the p38 pathway, and particularly IRAK3 expression were affected by siQKI in MEC2 cells (Table 1). This result and those of Figures 3C, 3D, suggest that p38 pathway is regulated by QKI in different types of hematopoietic cells. Of note, 30% of the 100 transcripts, most significantly affected by siQKI in MEC2 cells were predicted targets of miR-155 (Supplementary Table 2), versus 23% in BJAB cells (Supplementary Table 3). Thus, variations in QKI levels in B cells (Figure 4) are likely to be instrumental in both Burkitt’s and CLL cell transformation.
Table 1: The twenty most significant pathways affected by siQI transfection in MEC2 CLL cells.
Rank |
Ingenuity Canonical Pathways |
P-value |
Transcripts |
1 |
Role of Oct4 in Mammalian Embryonic Stem Cell Pluripotency |
1.05E-03 |
FAM208A, RARA, MEF2A, IGF2BP1 |
2 |
T Helper Cell Differentiation |
5.13E-03 |
TGFBR1, CD80, HLA-DOB, CXCR5 |
3 |
Notch Signaling |
6.46E-03 |
ADAM17, DTX3, JAG1 |
4 |
Cardiac Hypertrophy Signaling |
7.41E-03 |
ROCK2, GNAS, TGFBR1, IGF1, IRS1, SOS1, MEF2A |
5 |
Role of NFAT in Regulation of the Immune Response |
7.59E-03 |
GNAS, CD80, SOS1, MEF2A, HLA-DOB, ITPR1 |
6 |
PPARα/RXRα Activation |
8.71E-03 |
GNAS, TGFBR1, GPD2, IRS1, SOS1, ACVR2B |
7 |
TGF-β Signaling |
9.77E-03 |
TGFBR1, SOS1, ACVR2B, SMAD5 |
8 |
Role of NFAT in Cardiac Hypertrophy |
1.02E-02 |
GNAS, TGFBR1, IGF1, SOS1, MEF2A, ITPR1 |
9 |
Chronic Myeloid Leukemia Signaling |
1.48E-02 |
TGFBR1, CTBP2, SOS1, E2F5 |
10 |
IGF-1 Signaling |
1.62E-02 |
NEDD4, IGF1, IRS1, SOS1 |
11 |
Cholecystokinin/Gastrin-mediated Signaling |
1.86E-02 |
ROCK2, SOS1, MEF2A, ITPR1 |
12 |
β-alanine Degradation I |
2.00E-02 |
ABAT |
13 |
Glycine Degradation (Creatine Biosynthesis) |
2.00E-02 |
GATM |
14 |
Antiproliferative Role of TOB in T Cell Signaling |
2.75E-02 |
TGFBR1, TWSG1 |
15 |
Glycerol-3-phosphate Shuttle |
2.95E-02 |
GPD2 |
16 |
4-aminobutyrate Degradation I |
2.95E-02 |
ABAT |
17 |
GDNF Family Ligand-Receptor Interactions |
3.09E-02 |
IRS1, SOS1, ITPR1 |
18 |
p38 MAPK Signaling |
3.09E-02 |
TGFBR1, MAPT, MEF2A, IRAK3 |
19 |
IL-4 Signaling |
3.80E-02 |
IRS1, SOS1, HLA-DOB |
20 |
B Cell Development |
4.07E-02 |
CD80, HLA-DOB |
Transcripts whose levels also changed in BJAB Burkitt’s cells are given in bold letters.
Table 2: The twenty most significant pathways affected by siQI transfection in BJAB Burkitt’s cells.
Rank |
Ingenuity Canonical Pathways |
P-value |
Transcripts |
1 |
Dendritic Cell Maturation |
2.69E-05 |
COL1A1, CD80, PIK3CG, IL32, FSCN1, AKT3, LTB, IL1B, TLR3, CCR7 |
2 |
Lymphotoxin β Receptor Signaling |
3.02E-04 |
VCAM1, PIK3CG, AKT3, LTB, TRAF1 |
3 |
Crosstalk between Dendritic Cells and Natural Killer Cells |
4.57E-04 |
CD80, CD69, FSCN1, LTB, TLR3, CCR7 |
4 |
NF-κB Signaling |
4.90E-04 |
TGFBR1, HDAC2, PIK3CG, IGF1R, AKT3, IL1B, TLR3, TNFRSF17 |
5 |
Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis |
5.50E-04 |
VCAM1, C5AR1, PIK3CG, IL32, RAC1, AKT3, LTB, IL1B, TLR3, WNT5A, TRAF1 |
6 |
Docosahexaenoic Acid (DHA) Signaling |
8.51E-04 |
PIK3CG, AKT3, IL1B, APP |
7 |
Hepatic Fibrosis / Hepatic Stellate Cell Activation |
8.51E-04 |
COL1A1, VCAM1, TGFBR1, TIMP1, IGF1R, IL1B, CCR7 |
8 |
Role of Tissue Factor in Cancer |
1.38E-03 |
PIK3CG, EGR1, RAC1, ITGA6, AKT3, IL1B |
9 |
Altered T Cell and B Cell Signaling in Rheumatoid Arthritis |
2.51E-03 |
CD80, LTB, IL1B, TLR3, TNFRSF17 |
10 |
RAR Activation |
3.09E-03 |
GNAS, DHRS9, PIK3CG, RAC1, AKT3, SMAD5, RXRA |
11 |
Communication between Innate and Adaptive Immune Cells |
3.24E-03 |
CD80, IL1B, TLR3, CCR7, TNFRSF17 |
12 |
Human Embryonic Stem Cell Pluripotency |
3.55E-03 |
GNAS, TGFBR1, PIK3CG, AKT3, SMAD5, WNT5A |
13 |
Regulation of the Epithelial-Mesenchymal Transition Pathway |
3.80E-03 |
TGFBR1, PIK3CG, EGR1, ZEB2, PARD6B, AKT3, WNT5A |
14 |
Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses |
4.07E-03 |
CLEC7A, C5AR1, PIK3CG, IL1B, TLR3 |
15 |
CXCR4 Signaling |
6.76E-03 |
MYL12A, GNAS, PIK3CG, EGR1, RAC1, AKT3 |
16 |
Sphingosine-1-phosphate Signaling |
7.24E-03 |
GNAS, PIK3CG, RAC1, AKT3, ASAH1 |
17 |
Role of NANOG in Mammalian Embryonic Stem Cell Pluripotency |
7.41E-03 |
PIK3CG, AKT3, SMAD5, WNT5A, TCL1A |
18 |
Small Cell Lung Cancer Signaling |
7.59E-03 |
PIK3CG, AKT3, RXRA, TRAF1 |
19 |
NF-κB Activation by Viruses |
8.71E-03 |
PIK3CG, ITGA6, AKT3, CXCR5 |
20 |
PTEN Signaling |
9.33E-03 |
TGFBR1, PIK3CG, IGF1R, RAC1, AKT3 |
Transcripts whose levels also changed in MEC2 CLL cells are given in bold letters.
QKI is downregulated in B cells of CLL patients
QKI being a target of miR-155 at least in certain conditions, we would expect its expression to be reduced in leukemias presented with high miR-155 levels, such as CLL or AML [27-30, 5]. Indeed, the levels of QKI transcripts were significantly lower in CLL patients (Figure 6A). In agreement with this result, publicly available array data show that in both, CLL and AML patients, the expression of QKI is significantly reduced as compared with healthy donors (data not shown). Finally, Qki levels were also lower in B cells of Eµ-miR-155 transgenic mice at the most advanced stage of leukemia (Figure 6B). These results suggest that the disruption of cross-regulations between QKI, miR-155 and factors implicated in the immune response, may generally be associated with B cell leukemic transformation.
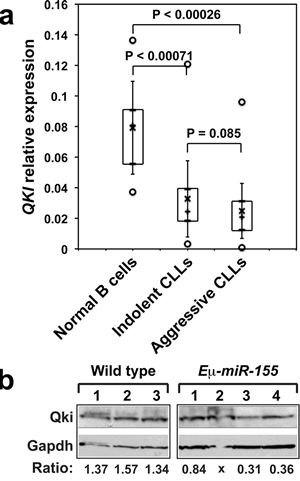
Figure 6: A. RNAs extracted from B cells purified from 10 healthy donors (HD), 38 patients with indolent CLL (IND) and 72 patients with aggressive CLL (AGG) were analyzed by qRT-PCR. Boxes include values from the first to the third quartiles; o, extreme data points; +, median; x, mean ± SD. B. Qki expression in B cells purified from the spleens of wild type or Eµ-miR-155 transgenic mice was analyzed by Western blotting. Panels are from the same blot. Qki/Gapdh ratios are given under each lane. Spleens from Eµ-miR-155 mice were: pre-leukemic (1 and 2); and leukemic (3 and 4).
DISCUSSION
We found that Qki levels are reduced at the onset of immune response to LPS, when miR-155 levels are on the rise, positioning Qki as an immune factor and a target of miR-155. In addition, we found that QKI overexpression increases IL-10 production, as well as the total amount of p38, while markedly impairing p38 phosphorylation. It is highly probable that following LPS stimulation, cells sensing the lack of p38 phosphorylation in the presence of excess QKI increase their levels of total p38 to compensate for this deficiency. Of note, K-Ras similarly was previously shown to induce p38 expression but not its phosphorylation, and higher levels of unphosphorylated p38 promote Ras transformation in rat intestinal epithelium through an increased complex formation with Erk-1, -2 kinases [33]. As the reduction of Qki levels only took place at the first hours of the immune response, we hypothesize that miR-155 targeting of Qki is restricted only at the beginning of the response, to allow p38 MAPK required activation following LPS challenge. It is thus very likely that during the first hours of LPS signaling, dose-dependent effects take place between Qki, miR-155 and MAPKs activity, paralleling the oscillatory activity of “early” genes, such as NF-κB and AP-1 activity [34]. Similarly, low miR-155 expression at the early phase of dendritic cells maturation enables the activation of the p38 pathway, thus favoring IL-1 expression and signaling cascade [35].
Later on, the miR-155 targeting of Qki transcript was impaired. This might be due to the fact that proliferating cells, including human monocytes stimulated with LPS and IFNγ, tend to express mRNAs with shortened 3’-UTR and consequently fewer miRNA target sites [36]. Alternatively, Qki 3’-UTR might act as a sponge for miR-155. Finally, miR-155 overexpression in MCF7 breast cancer cells increases the levels of some of its target transcripts through the progressive shortening of their 3’-UTR, including p38 MAPK [37].
Our results show that QKI overexpression increased IL-10 expression and affected p38 MAPK, while siQKI modulated the expression of IRAK3, a negative regulator of TLR signaling, including p38 MAPK [38]. Altogether, these results suggest that QKI, miR-155, p38 and IL-10 [39] are involved in a common regulatory circuitry in B cells and macrophages, and that QKI may represent a new factor of hematopoietic cell transformation. This hypothesis is also supported by previous findings showing that the expression of QKI isoforms, changes significantly during hematopoietic differentiation, suggesting for a critical role of QKI in hematopoiesis and function [23].
Our results further show an inverse correlation between QKI and miR-155 expression in CLL and Burkitt’s cell lines, and establish that QKI might render B cells prone to cell death by increasing Caspase3/7 signaling and FAS expression, suggesting that QKI may behave as a TSG in B cells. Importantly, QKI expression was low in CLL patients, known to have high levels of miR-155 [27-30], as well as in leukemic B cells isolated from the spleens of Eµ-miR-155 mice, results further supported by data available in public databases.
Finally, QKI unusually long 3’-UTR contains target sites for most miRNAs (targetscan.org). Thus, QKI may represents a regulatory hub directing many cellular processes. As human QKI 3’-UTR contains three miR-155 target site, the third one being present only in apes, where QKI came to represent the first most probable target of miR-155, one can expect miR-155 - QKI cross-regulatory interactions to have gained a critical importance in this group.
Mice with spontaneous deletion of Qki have marked rapid tremor and seizures, and their entire central nervous system is severely depleted in myelin [40]. As miR-155 expression is elevated in neuro-inflammatory pathologies such as MS or ALS [41, 42], our data further suggest that a lack of QKI anti-inflammatory input may result in the deleterious dominance of miR-155 activity, therefore, miR-155-QKI interactions could prove to be significantly important for future therapies aimed at neuro-degenerative pathologies presented with high levels of miR-155.
In conclusion, while miR-155 is critical for mounting an effective immune response, its prolonged expression under chronic inflammatory conditions drives immune pathologies and leukemias. Therefore, a better understanding of miR-155/QKI functions and their control of expression in immune cells should allow to design new miR-155-based cancer immunotherapies. Our findings suggest that when QKI-miR-155 reciprocal regulation becomes dysfunctional, enhanced miR-155 activity drives tumor development and evasion of the immune response.
Supplementary information is available at Oncotarget website.
Materials and METHODS
Affymetrix microarray (Santa Clara, CA, USA) analyses were submitted to the MIAME database (accession number E-MTAB-2375). Purified CD19+ B cells were purchased from Sanguine BioSciences (Sherman Oaks, CA, USA). Eμ-miR-155 transgenic mice were previously described [6]. B cells from mice were isolated using B-cell purification kit from Miltenyi Biotech (San Diego, CA, USA).
Cells were grown following standard procedures. Information for miRNAs and siRNAs is found in Supplementary Table 1. The fragments of QKI 3’UTR and the Luciferase reporter construct containing the promoter of QKI gene were purchased from SwitchGear. Each miR-155 site was subsequently mutated using the Quick-Exchange Mutagenesis kit (Agilent; Santa Clara, CA, USA). Human QKI-5 cDNA was purchased from Genecopeia (Rockville, MD, USA) and recloned into the pCMV-HA expression vector.
Luciferase assays were run 48 hours after transfection as previously described [43]. Caspase-Glo 3/7 kit (Promega; Madison, WI, USA) was used to measure cell death. The assays were performed three times in quadruplicate, and the mean ± S.D. of caspase 3/7 activation (expressed as arbitrary units) is shown.
RNAs were extracted either with TRIzol (Life Technologies) or the RNA purification kit from Norgen (Thorold, ON, Canada). They were subsequently subjected to DNase digestion (Turbo-DNase- Life Technologies). MiRNA and gene qRT-PCRs were respectively performed using the corresponding Assays from Life Technologies (Supplementary Table 1). RT-PCRs were run in triplicates. Values were normalized using one of the following normalizers: RNU-44, RNU-48, RNU-6B or U6 for MiRNA assays and OAZ1, β-Actin or GAPDH for Gene Expression assays.
Antibodies and other related information are listed in Supplementary Table 1. Flow cytometry staining was done following standard procedures and run on a Calibur (BD-Biosciences) machine. The data were analyzed using FloJow (Ashland, OR, USA) software. Cytokine production was measured using the ELISA kit (Qiagen) following manufacturer’s instruction. Statistical analysis were done using the Student t test, and P values are provided in the Figure legends.
ACKNOLEDGMENTS
This work was supported by R01CA151319 grant to C.M.C. Authors want to thank Dr. Stefan Costinean (OSU) and Dr. Nicola Zanesi (OSU) for their help with experiments in mice.
AUTHORSHIP
Contribution: E.T. and C.M.C. designed the strategy of the study and experimental flow; E.T. M.C. and D.P. performed experiments and analyzed data; M.B., H.-L.S., T.K., T.S. and T.R. performed qRT-PCR, Luciferase and Western blot experiments; R.C. performed qRT-PCR analyses for QKI on CLL-patients and cell lines; S.V. and C.F. performed Affymetrix analyses, while D.V and A.L. performed Ingenuity pathway analyses; L.Z.R. and T.J.K. performed patient diagnostic analyses; J.-J.M. performed mutagenesis experiments; M.C. and M.B. performed luciferase assays; J.-J.M. H.A. E.T. and C.M.C. conceived the idea and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Tili E, Michaille JJ, Croce CM. MicroRNAs play a central role in molecular dysfunctions linking inflammation with cancer. Immunol Rev. 2013; 253:167-184.
2. Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009; 28:264-284.
3. Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunol Rev. 2013; 253:146-157.
4. Greither T, Grochola L, Udelnow A, Lautenschlager C, Wurl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumours associates with poorer survival. Int J Cancer. 2010; 126:73–80.
5. Marcucci G1, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrózek K, Nicolet D, Kohlschmidt J, Whitman SP, Mendler JH, Schwind S, Becker H, Eisfeld AK, et al. Bloomfield CD. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J Clin Oncol. 2013; 31:2086-2093.
6. Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci. USA 2006; 103:7024-7029.
7. Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, Slack FJ. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci USA. 2012; 109:1695-1704.
8. Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007; 179:5082-5089.
9. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007; 104:1604-1609.
10. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, Ciarlariello D, Neviani P, Harb J, Kauffman LR, Shidham A, Croce CM. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009; 114:1374-1382.
11. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009; 106:7113-7118.
12. Pedersen IM, Otero D, Kao E, Miletic AV, Hother C, Ralfkiaer E, Rickert RC, Gronbaek K, David M. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med. 2009; 1:288-295.
13. Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009; 3:220-231.
14. Larocque D, Richard S. QUAKING KH domain proteins as regulators of glial cell fate and myelination. RNA Biol. 2005; 2:37-40.
15. Pilotte J, Larocque D, Richard S. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev. 2001; 15:845-858.
16. Feng Y, Bankston A. The star family member QKI and cell signaling. Adv Exp Med Biol. 2010; 693:25-36.
17. Chen AJ, Paik JH, Zhang H, Shukla SA, Mortensen R, Hu J, Ying H, Hu B, Hurt J, Farny N, Dong C, Xiao Y, Wang YA, et al. STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev. 2012; 26:1459-1472.
18. Bian Y, Wang L, Lu H, Yang G, Zhang Z, Fu H, Lu X, Wei M, Sun J, Zhao Q, Dong G, Lu Z. Downregulation of tumor suppressor QKI in gastric cancer and its implication in cancer prognosis. Biochem Biophys Res Commun. 2012; 422:187-193.
19. Ji S, Ye G, Zhang J, Wang L, Wang T, Wang Z, Zhang T, Wang G, Guo Z, Luo Y, Cai J, Yang JY. miR-574-5p negatively regulates Qki6/7 to impact β-catenin/Wnt signalling and the development of colorectal cancer. Gut. 2013; 62:716-726.
20. D’Urso PI, D’Urso OF, Storelli C, Mallardo M, Gianfreda CD, Montinaro A, Cimmino A, Pietro C, Marsigliante S. miR-155 is up-regulated in primary and secondary glioblastoma and promotes tumour growth by inhibiting GABA receptors. Int J Oncol. 2012; 41:228-234.
21. Poltronieri P, D’Urso PI, Mezzolla V, D’Urso OF. Potential of anti-cancer therapy based on anti-miR-155 oligonucleotides in glioma and brain tumours. Chem Biol Drug Des. 2013; 81:79-84.
22. Yang G, Fu H, Zhang J, Lu X, Yu F, Jin L, Bai L, Huang B, Shen L, Feng Y, Yao L, Lu Z. RNA-binding protein quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology. 2010; 138:231-240.
23. Fu H, Yang G, Wei M, Liu L, Jin L, Lu X, Wang L, Shen L, Zhang J, Lu H, Yao L, Lu Z. The RNA-binding protein QKI5 is a direct target of C/EBPα and delays macrophage differentiation. Mol Biol Cell. 2012; 23:1628-1635.
24. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013; 13:679-692.
25. Bode JG, Ehlting C, Häussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012; 24:1185-1194.
26. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012; 33:449-458.
27. Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005; 353:1793-1801.
28. Ferrajoli A, Shanafelt TD, Ivan C, Shimizu M, Rabe KG, Nouraee N, Ikuo M, Ghosh AK, Lerner S, Rassenti LZ, Xiao L, Hu J, Reuben JM, et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood. 2013; 122:1891-1899.
29. Tili E, Michaille JJ, Luo Z, Volinia S, Rassenti LZ, Kipps TJ, Croce CM. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood. 2012; 120:2631-2638.
30. Li P, Grgurevic S, Liu Z, Harris D, Rozovski U, Calin GA, Keating MJ, Estrov Z. Signal transducer and activator of transcription-3 induces MicroRNA-155 expression in chronic lymphocytic leukemia. PLoS One. 2013; 8:e64678.
31. Kluiver J, Haralambieva E, de Jong D, Blokzijl T, Jacobs S, Kroesen BJ et al. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer. 2006; 45:147-153.
32. Galarneau A, Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol. 2005; 12:691-698.
33. Tang J, Qi X, Mercola D, Han J, Chen G. Essential role of p38gamma in K-Ras transformation independent of phosphorylation. J Biol Chem. 2005; 280:23910-23917.
34. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science. 2002; 298:1241-1245.
35. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci USA. 2009; 106:2735-2740.
36. Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008; 320:1643-1647.
37. Martin EC, Krebs AE, Burks HE, Elliott S, Baddoo M, Collins-Burow BM, Flemington EK, Burow ME. miR-155 induced transcriptome changes in the MCF-7 breast cancer cell line leads to enhanced mitogen activated protein kinase signaling. Genes Cancer. 2014; 5:353-364.
38. Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002; 110:191-202.
39. McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, O’Neill LA. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010; 285:20492-20498.
40. Mitrovic N, Caboche J; Carre JB; Besson MJ; Maurin Y. The quaking mouse: an epileptic mutant with alterations affecting the modulatory mechanisms of the NMDA receptor complex. Brain Res. 1991; 566:248-254.
41. Moore CS, Rao VT, Durafourt BA, Bedell BJ, Ludwin SK, Bar-Or A, Antel JP. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann Neurol. 2013; 74:709-720.
42. Koval ED, Shaner C, Zhang P, du Maine X, Fischer K, Tay J, Chau BN, Wu GF, Miller TM. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013; 22:4127-4135.
43. Tili E, Michaille JJ, Liu CG, Alder H, Taccioli C, Volinia S, Calin GA, Croce CM. GAM/ZFp/ZNF512B is central to a gene sensor circuitry involving cell-cycle regulators, TGF{beta} effectors, Drosha and microRNAs with opposite oncogenic potentials. Nucleic Acids Res. 2010; 38:7673-7688.