
INTRODUCTION
A key point in specific immune responses is the activation and proliferation of antigen-specific T cells. We and others have shown that exogenous cystine (Cys2), the oxidized dimeric form of cysteine (Cys), is dispensable for early T cell activation but indispensable for T cell proliferation [1, 2]. Due to the oxidizing environment, approximately 90% of Cys is found as Cys2 in the extracellular space. Plasma thus contains 50–100 μM Cys2, whereas the concentration of Cys is very low compared to other amino acids [3, 4]. Cys is transported over the cell membrane predominantly by the neutral amino acid transporters ASCT1 (SLC1A4) and ASCT2 (SLC1A5), whereas Cys2 exclusively is transported by the xc− cystine/glutamate antiporter composed of the heavy subunit 4F2 hc (CD98) and the light subunit xCT (SLC7A11) [5, 6]. Inside the cell, Cys2 is rapidly reduced to Cys due to the reducing intracellular milieu [7]. Naïve T cells express no or very low levels of xc− and are deficient in transportation of Cys2 over the plasma membrane [2, 8, 9], but shortly after activation xc− becomes strongly upregulated in both human and mouse T cells and provide the cells with the required amount of Cys2/Cys needed for proliferation [1, 8, 10]. However, the exact mechanisms that are dependent on Cys in order for T cells to proliferate still need to be identified.
Cell proliferation requires doubling of the cell constituents including replication of the DNA. Availability of the deoxyribonucleoside triphosphates (dNTP) DNA building blocks is the limiting factor in DNA synthesis. In resting cells, the dNTP pools are kept low, but during DNA synthesis in the S phase the dNTP pool increases several fold [11–14]. Insufficient levels of dNTP results in an elongation of the S phase [15]. Furthermore, it is essential that the dNTP levels are neither too high nor unbalanced as dysregulated dNTP levels lead to an increased rate of mutagenesis [16]. Thus, DNA synthesis requires a strictly regulated continuous de novo synthesis of dNTPs. Ribonucleotide reductase (RNR) is a key enzyme for dNTP generation. RNR generates deoxyribonucleoside diphosphates (dNDP) through reduction of the corresponding ribonucleoside diphospate (NDP) [11–14]. After conversion from NDP, dNDP is finally phosphorylated to dNTP. RNR is responsible for maintaining the total dNTP pool size and ensuring that the levels of the four dNTPs are balanced. During the catalysis, the 2′-OH group of the NDP ribose ring is reduced to hydrogen. In this process, a disulfide bridge is generated in the active site of RNR [11–14]. In order for RNR to restore its original configuration and be capable of catalyzing a new round of NDP reduction, external thiol-dependent systems are required to reduce the disulfide bridge in the active site. Thioredoxin (Trx) and later glutaredoxin (Grx) were discovered as thiol electron donors for RNR in E. coli [17, 18]. Unlike Trx, Grx was found to be functional as an electron donor only in the presence of glutathione (GSH). In E. coli, the glutaredoxin-glutathione (Grx/GSH) system is the most efficient electron donor for RNR [12, 19], but in yeast the major electron donor is Trx [20]. Which system that is functional in mammalian cells is less clear, although cell-free studies using recombinant mouse RNR have indicated that both Trx and the Grx/GSH system can reduce RNR [21].
GSH is synthesized in the cytosol and consists of the amino acids glutamate, Cys and glycine, where Cys is believed to be the limiting amino acid [22, 23]. Several studies have shown that GSH plays essential roles in T cell function and proliferation [2, 24–29]. These studies indicate that GSH is not required during the early steps of T cell activation, but that it is essential for processes close to or at the level of DNA synthesis. However, the exact role(s) of GSH during T cell proliferation still needs to be determined. Taking into account that Cys is required for GSH synthesis [22, 23], it could be speculated that in order to proliferate, human T cells need exogenous Cys2 to generate sufficient amounts of GSH that subsequently supply the reducing power required for RNR activity and thereby dNTP synthesis and DNA replication. The aim of this study was to identify processes in which exogenous Cys2 is required in order for T cell proliferation to take place.
RESULTS
Exogenous cystine is required for GSH and DNA synthesis
We have recently shown that activation of human T cells as assessed by the upregulation of the activation markers CD25 and CD69 as well as IL-2 production is independent of exogenous Cys2. In contrast, DNA synthesis as measured by 3H-thymidine incorporation is strictly dependent on the presence of Cys2 in the tissue culture medium [1]. As Cys is supposed to be the limiting amino acid in the synthesis of GSH [22, 23] and it has been suggested that GSH can provide the reducing power necessary for RNR to produce dNDP required for dNTP and DNA synthesis [12], we first wanted to study whether a correlation exists between the requirement for Cys and GSH and DNA synthesis in T cells. To study a homogenous T cell population, we purified naïve CD4+ T cells from buffy coats obtained from healthy blood donors. The resulting cell population consisted of approximately 98% CD4+CD45RA+CD25−CD69− T cells [1]. We stimulated the purified T cells with anti-CD3 and anti-CD28-coated Dynabeads for 3 days in Cys2-free DMEM tissue culture medium supplemented with increasing concentrations of Cys2. We found that both GSH levels and DNA synthesis were strictly dependent on Cys2 (Figure 1A). The levels of GSH and DNA synthesis increased in parallel with increasing concentrations of Cys2 in the medium. To determine whether the dependency of Cys2 for DNA synthesis was due to the requirement of Cys2 for GSH synthesis or for other GSH-independent mechanisms, we next stimulated the cells for 3 days in X-VIVO 15 tissue culture medium that contains 290 μM Cys2 with various concentrations of BSO that specifically inhibits synthesis of GSH. BSO concentrations of 100 μM and above completely depleted GSH in the T cells. Likewise, DNA synthesis was impaired by BSO, although we noticed a small displacement between the two curves (Figure 1B). These results suggested that DNA synthesis is dependent on Cys2 due to the requirement of Cys2 for GSH synthesis and not for other mechanisms. To further investigate the requirement of exogenous Cys2 for GSH and DNA synthesis, we inhibited the uptake of Cys2 by incubation of the cells with increasing concentrations of glutamic acid. Glutamic acid is the main exchange substrate in the xc− cystine/glutamate antiporter-mediated uptake of Cys2 and is a highly specific inhibitor of Cys2 uptake [6]. We found that glutamic acid inhibited GSH and DNA synthesis in parallel (Figure 1C). As for BSO-treated cells, we noticed a small displacement between the curves for GSH and DNA synthesis suggesting that some residual DNA synthesis can take place in the absence of GSH. From these experiments we could conclude that T cells require exogenous Cys2 to produce GSH, and that they need GSH for normal DNA synthesis to take place.
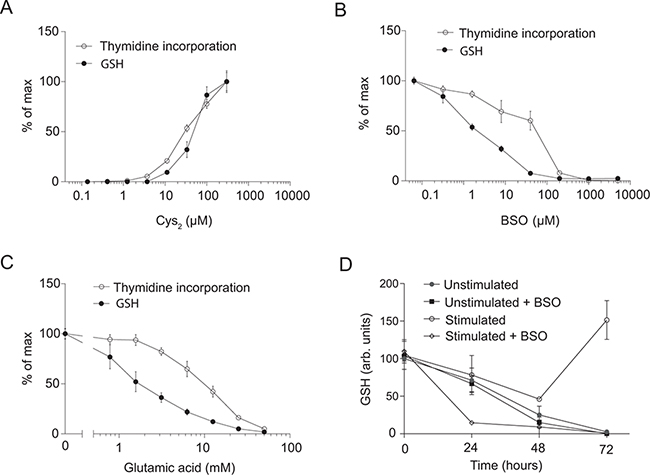
Figure 1: Exogenous cystine is required for GSH and DNA synthesis. A. Thymidine incorporation and GSH levels of CD4+ T cells activated for 3 days in Cys2-free DMEM medium supplemented with increasing amounts of Cys2. B. Thymidine incorporation and GSH levels of CD4+ T cells activated for 3 days in X-VIVO 15 medium supplemented with increasing levels of BSO. C. Thymidine incorporation and GSH levels of CD4+ T cells activated for 3 days in DMEM medium supplemented with the indicated concentrations of glutamic acid. (A–C) Data are shown as percentage of maximum 3H-thymidine incorporation and GSH levels, respectively. D. GSH levels of CD4+ T cells either left unstimulated or activated for 0–72 hours in the presence or absence of 100 μM BSO. (A–D) Data are given as mean ± SEM from at least two experiments, each carried out in triplicates.
To further study GSH occurrence and production, we isolated naïve CD4+ T cells and left them either unstimulated or stimulated them with anti-CD3/CD28 beads in X-VIVO 15 tissue culture medium with or without 100 μM BSO for 0 to 72 hours. We found that freshly purified naïve CD4+ T cells contain GSH, and that the GSH level slowly declines in unstimulated T cells independently of the presence of BSO (Figure 1D). This indicated that naïve unstimulated T cells consume or lose GSH to the tissue culture medium and that they do not actively produce GSH in vitro. Stimulation of the T cells in the presence of BSO lead to a rapid decline in GSH indicating that T cell stimulation accelerates the consumption or loss of GSH. T cell stimulation in the absence of BSO initially led to a decline in GSH concentration, but after 72 hours the GSH concentration exceeded the initial GSH concentration in naïve T cells (Figure 1D), indicating that de novo synthesis of GSH takes place in activated CD4+ T cells.
Thioredoxin and GSH can partly substitute for each other in DNA synthesis
From the experiments shown in Figure 1A–1C we could conclude that exogenous Cys2 is required for GSH production and that GSH is required for optimal DNA synthesis in activated CD4+ T cells. However, we also noted that some residual DNA synthesis took place even in cells completely depleted of GSH (Figure 1B and 1C). This indicated that GSH can be replaced by other reducing agents during DNA synthesis. It has been suggested that Trx and the Grx/GSH system can substitute for each other in providing the reducing power required for DNA synthesis [12, 21], and we wanted to see whether this could also be the case in human T cells. We therefore determined the expression of Trx in human CD4+ T cells stimulated for 0 to 72 h and compared it with Trx expression in the human leukemic T cell line Jurkat. We found that naïve CD4+ T cells express very low levels of Trx and that T cell stimulation induces significant Trx upregulation. Following 72 h of stimulation, primary T cells expressed Trx levels similar to those of Jurkat cells (Figure 2A).
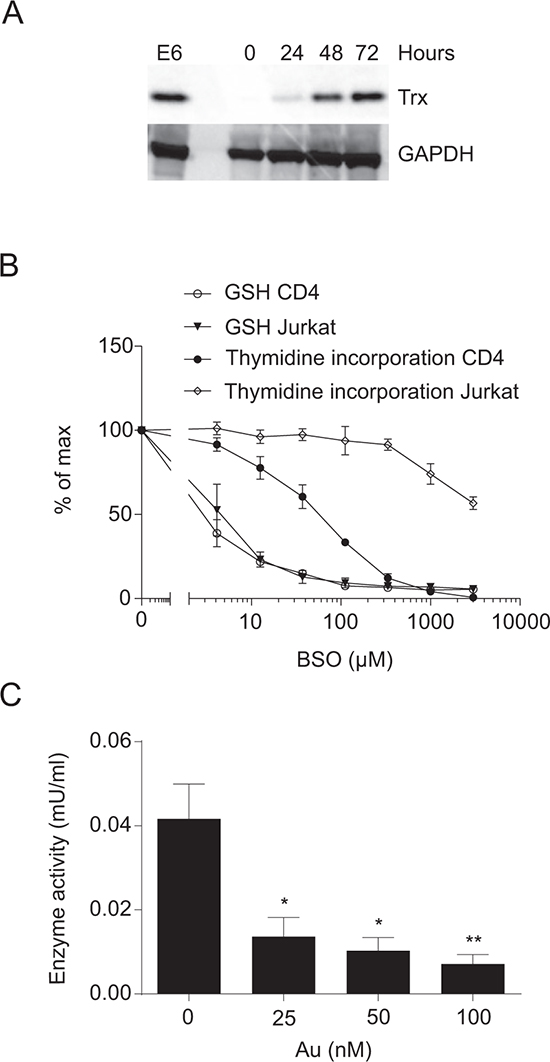
Figure 2: Thioredoxin in primary CD4+ T cells and Jurkat cells. A. Representative Western blot of Trx and GAPDH (loading control) in Jurkat cells (E6) and CD4+ T cells activated for 0–72 hours in X-VIVO 15 medium. B. Thymidine incorporation and GSH levels of CD4+ T cells activated in X-VIVO 15 medium and Jurkat cells cultured in RPMI-1640 medium for 48 hours in the presence of the indicated concentrations of BSO. Data show mean ± SEM of two experiments carried out in duplicates. C. Thioredoxin reductase activity of CD4+ T cells activated for 3 days in X-VIVO 15 medium with the indicated concentrations of Au. Data show mean ± SEM of four experiments.
If Trx can substitute for GSH, it would be expected that DNA synthesis in Jurkat cells with a constitutive high level of Trx would be more resistant to GSH depletion than primary T cells. We consequently treated primary T cells and Jurkat cells in parallel with increasing concentrations of BSO for 48 h and subsequently measured GSH levels and DNA synthesis. We found that even though GSH levels decreased at equal rates in the two cell types in response BSO treatment, DNA synthesis in Jurkat cells was much more resistant to BSO treatment than DNA synthesis in primary T cells (Figure 2B). This supported that Trx can substitute for GSH in DNA synthesis in human T cells.
Trx-mediated reduction of other proteins results in oxidation of Trx [30, 31]. In order for Trx to be capable of catalyzing a new round of reduction, Trx must it-self be reduced. The Trx reductases (TrxR) are the only enzymes known to reduce oxidized Trx, and inhibition of TrxR impairs the redox function of Trx [30–32]. Auranofin (Au) is an irreversible inhibitor of TxrR [33]. To determine the concentration of Au required to inhibit TrxR in primary T cells, we stimulated naïve CD4+ T cells with CD3/CD28 beads for 3 days in the presence of increasing concentrations of Au and subsequently measured the TxrR activity. We found that Au already at 25 nM significantly inhibited TrxR activity (Figure 2C).
To further investigate whether GSH and Trx can substitute for each other during DNA synthesis in primary T cells, we next stimulated CD4+ T cells in a checkerboard titration of BSO and Au for 3 days. We subsequently measured GSH levels, DNA synthesis as 3H-thymidine and BrdU incorporation, and T cell division as dilution of CFSE. We found that the low dose of Au (25 nM) did not significantly affect DNA synthesis or cell division in the absence of BSO (Figure 3A–3D). Treatment with the low dose of BSO (100 μM) without Au completely abrogated GSH expression and reduced 3H-thymidine and BrdU incorporation and cell division, although it did not affect the percentage but only the mean fluorescence intensity (MFI) of cells that incorporated BrdU (Figure 3A–3D). Interestingly, the combination of the low dose of Au (25 nM) and BSO (100 μM) completely abolished 3H-thymidine and BrdU incorporation and cell division (Figure 3B–3D). These experiments demonstrated that inhibition of either GSH or Trx still allows some DNA synthesis, whereas simultaneous inhibition of GSH and Trx completely abolishes DNA synthesis. This further supported that GSH and Trx can partly substitute for each other during DNA synthesis.
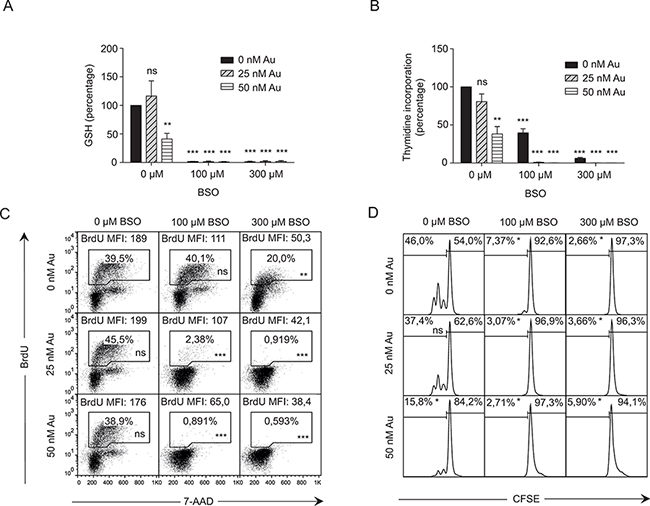
Figure 3: Thioredoxin and GSH can partly substitute for each other in DNA synthesis. A. GSH levels and B. thymidine incorporation of CD4+ T cells activated for 3 days in X-VIVO 15 medium in the presence of the indicated concentrations of BSO and Au. Data show mean ± SEM of three experiments carried out in duplicates. C. DNA synthesis as measured by BrdU incorporation and 7-AAD staining and D. cell division as measured by CFSE dilution of CD4+ T cells activated for 3 days in X-VIVO 15 medium in the presence of the indicated concentrations of BSO and Au. Data show representative results from two experiments carried out in duplicates.
GSH and/or Trx is required for dNTP production
A key molecule in DNA synthesis is RNR that converts NDP into dNDP. First, we determined the expression of the regulatory subunit of RNR (R1) in human primary T cells compared to Jurkat T cells. We stimulated naïve CD4+ T cells for 72 hours with anti-CD3/CD28 beads and determined R1 expression by Western blot analysis. R1 is only expressed in very low amounts in naïve CD4+ T cells. However, upon stimulation R1 is upregulated and within 48 hours the expression level in primary T cells is comparable to that seen in Jurkat T cells (Figure 4A). In the conversion of NDP to dNDP, RNR is oxidized and its further function is dependent on reduction by external electron donors such as Grx/GSH or Trx [21]. If RNR is not reduced, dNDP and thereby dNTP production halts. Therefore dNTP levels can be used as an output for RNR activity [34]. To obtain a sufficient number of cells, we used Jurkat T cells for these analyses. Since dNTP levels fluctuate during the different stages of the cell cycle, we initially synchronized the cells by serum starvation. Upon release from serum starvation, we either left the synchronized cells untreated or treated them with BSO or Au alone or with a combination of both that resulted in a 50% reduction of cells in the S phase as detemined by BrdU staining (Figure 4B). After 16 hours of incubation when the cells had entered S phase, we extracted dNTP and measured the dCTP concentrations as a representative for dNTP. Treatment with either BSO or Au alone did not significantly affect the amount of dCTP synthesized. However, the combination of BSO and Au significantly reduced the amount of dCTP produced (Figure 4C). This indicated that the RNR activity is significantly impaired in T cells when both GSH and Trx are inhibited. Alternatively, inhibition of GSH and Trx might affect the RNR levels in the cells. To rule out this possibility, we determined R1 expression in T cells activated in the presence of different combinations of BSO and Au concentrations. The combination of low concentrations of BSO and Au, which completely abolished DNA synthesis (Figure 3B and 3D), did not affect the expression of R1 (Figure 4D, top row). Taken together, these experiments indicated that the inability of T cells to synthesize DNA when GSH and Trx are inhibited is not caused by a reduced level of RNR but rather by impaired activity of RNR.
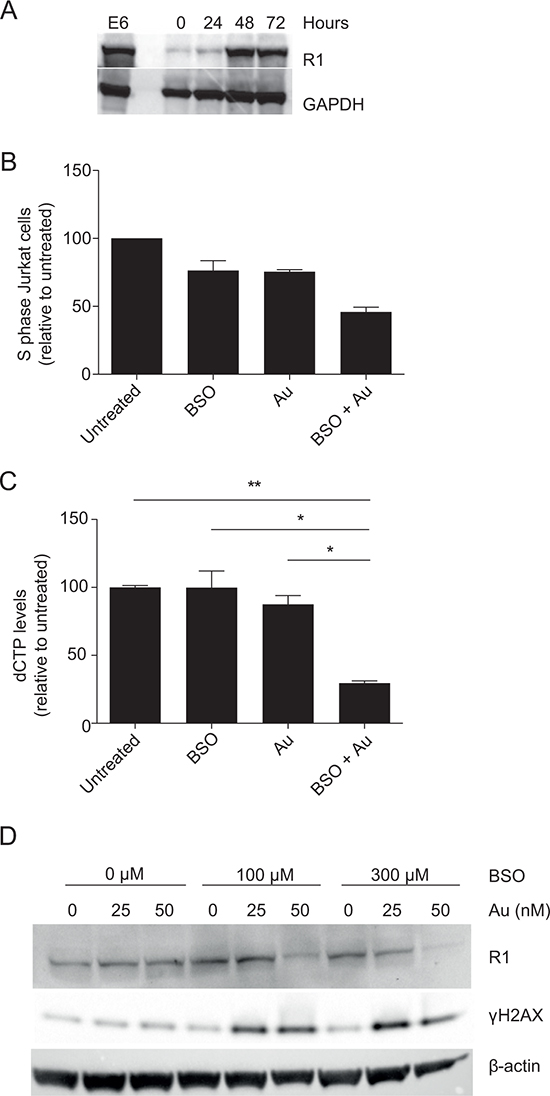
Figure 4: Inhibition of GSH and Trx impairs dNTP production and induces γH2AX. A. Representative Western blot of R1 and GAPDH (loading control) in Jurkat cells and CD4+ T cells activated for 0–72 hours. The same gel as shown in Figure 2A was stripped and reblotted with antibodies against R1. B. The fraction of Jurkat cells in S phase relative to untreated cells 16 hours after release from serum starvation. The cells were either left untreated, treated with 300 μM BSO, 200 nM Au or a combination of both. C. dCTP levels in cell cycle-synchronized Jurkat cells treated as in (B) dCTP measurements are shown as mean ± SEM of two experiments carried out in triplicates. D. Representative Western blot of R1, γH2AX and β-actin (loading control) in CD4+ T cells activated for 3 days in the presence of the indicated concentrations of BSO and Au.
If RNR activity and thereby dNTP production are impaired in cells with reduced GSH and Trx, DNA double-stranded breaks (DSB) due to DNA replication stress would be expected in these cells [35, 36]. H2AX, a member of the histone family, is rapidly phosphorylated at serine 139 in response to DSB, and this serine 139-phosphorylated form of H2AX is denoted γH2AX [37, 38]. In order to study whether the cells showed signs of DSB in response to inhibition of GSH and Trx, we stimulated purified CD4+ T cells in a checkerboard titration of BSO and Au for 48 h. The cells were subsequently lyzed and their levels of γH2AX determined. Whereas γH2AX was not increased in cells treated with either BSO or Au alone compared to untreated cells, the γH2AX levels were clearly increased in cells treated with the combination of BSO and Au (Figure 4D, middle row). These observations indicated that DNA replication is impaired in cells with reduced GSH and Trx activity and supported that GSH and/or Trx is required for RNR activity and thereby dNTP production.
DISCUSSION
In this study, we show that exogenous Cys2 is indispensable for proliferation of activated human T cells because it is required for production of GSH that subsequently is required for optimal RNR activity and thereby dNDP and dNTP production. For several years, the general paradigm was that T cells cannot import Cys2. This paradigm built on early measurements of Cys and Cys2 uptake, which indicated that lymphocytes express the ASCT1 and/or ASCT2 transporters for Cys but not the xc− transporter for Cys2 [7, 9, 39]. Based on these observations it was suggested that a sufficient high concentration of exogenous Cys in the microenvironment is provided to T cells by activated antigen presenting cells [40, 41]. Later it was found that although naïve T cells do not express xc−, T cell activation strongly induces the expression of xc−, and it was demonstrated that xc− can provide activated T cells with the required amount of Cys2 needed for T cell proliferation [1, 8, 10]. One remaining question was why T cell proliferation is dependent on exogenous Cys2. Cys together with glutamate and glycine constitute the building blocks of GSH, and it has therefore been suggested that T cells require exogenous Cys2/Cys to generate GSH [42, 43]. As GSH is required for optimal T cell proliferation [2, 26–29], a dependency of exogenous Cys2 for GSH synthesis would explain the central role of exogenous Cys2 in T cell proliferation. However, it has been proposed that T cells can provide Cys for GSH synthesis by an alternative route via the transsulfuration pathway [8] in which methionine is converted to Cys inside the cell completely independently of exogenous Cys2. Whether a correlation between the levels of exogenous Cys2 and intracellular GSH exists in activated T cells has to our knowledge not been published previously. Here we show that a direct correlation exists between the levels of exogenous Cys2 and intracellular GSH in activated human T cells, and that this correlation also encompasses DNA synthesis. Furthermore, T cells activated in tissue culture medium containing Cys2 concentrations below 3 μM were completely depleted of GSH. Thus, although the transsulfuration pathway has been described in T cells, this pathway clearly cannot supply T cells with sufficient amounts of Cys for GSH production as recently described for hepatocytes [44]. That transsulfuration cannot supply human T cells for the amount of Cys required for GSH and DNA synthesis was further supported by the observation that blocking Cys2 uptake completely inhibited GSH production and DNA synthesis although the cells were cultured in medium containing methionine. In accordance with previous studies [26–28], we found that BSO inhibited GSH production and DNA synthesis in parallel. Thus, when GSH synthesis was blocked, DNA synthesis was halted although Cys2/Cys was freely available for the T cells. From these results it could be concluded that in order to proliferate, activated T cells need exogenous Cys2 to produce GSH that subsequently is required for optimal DNA synthesis. This is in good agreement with previous studies which found that proliferation and survival of human natural killer cells and primary and malignant B cells are dependent on exogenous Cys2/Cys [45–48].
DNA synthesis is, among other factors, dependent on a balanced presence of dNTP generated from RNR-produced dNDP [49, 50]. RNR activity requires an electron donor, which in bacteria, yeast and plants is usually either Trx or Grx [12]. From test tubes experiments using recombinant mouse RNR, it has been shown that both Trx and Grx can reduce mammalian RNR, and that the Grx function is dependent on GSH [21]. We found that GSH was required for optimal DNA synthesis and T cell proliferation; however, we noted some residual DNA synthesis and proliferation in T cells completely depleted of GSH, indicating that other electron donors might be able to substitute for the Grx/GSH pathway. We found a high constitutive expression of Trx in the human leukemic T cell line Jurkat. Combined with the observation that DNA synthesis in Jurkat cells is much more resistant to BSO treatment than DNA synthesis in primary T cells, although the GSH production is equally sensitive to BSO, this supported that Trx can substitute for GSH as electron donor for RNR in human T cells. The synergistic effect of BSO and Au on dNTP production and phosphorylation of H2AX further supported that both GSH and Trx play a role in DNA synthesis. Thus, our study extended previous experiments using recombinant mouse RNR and indicated that Grx/GSH is the primary electron donor for RNR in intact T cells, but that Trx also plays a role and can substitute for Grx/GSH in this process. That either Grx/GSH or Trx can act as electron donor for RNR in intact mammalian cells is supported by the observation that hepatocyte proliferation in vivo requires either GSH or at least one functional allele of the TrxR 1 gene [51].
The significance of plasma Cys2 and intracellular GSH levels for CD4+ T cells in connection with diseases has primarily been studied in relation to HIV. Several studies have shown that HIV infected persons tend to have lower plasma Cys2 and intracellular GSH levels compared to healthy controls [52]. In light of this, supplementation with the drug N-acetyl-cysteine (NAC), which increases plasma Cys levels, has been studied in HIV infected persons. Such trials showed that oral supplementation with NAC replenished GSH in lymphocytes and improved the overall T cell function [53]. The importance of Cys2 and GSH in relation to HIV infection is underscored by the fact that the decrease in CD4+ T cell numbers in the late asymptomatic stage of HIV seemed to coincide with a decrease in plasma Cys2 [4], and likewise the concentrations of glutathione reductase was found to be lower in patients with symptomatic HIV than in asymptomatic HIV patients [54]. Whether HIV causes lowered glutathione reductase and GSH levels in CD4+ T cells or the lowered levels allow for HIV replication is not known. An increased knowledge of the roles of antioxidants in CD4+ T cell biology could help to understand the connection between decreased antioxidant levels and exacerbation of HIV. Interestingly, studies have shown that both BSO and Au could potentially be used in treatment of HIV. BSO in combination with an HDAC inhibitor was shown to be useful in killing latently infected CD4+ T cells, as BSO made the infected cells more susceptible to treatment with the HDAC inhibitor. This allowed the use of both drugs at concentrations that were non-toxic for uninfected cells [55]. Au was shown to be more effective in inducing apoptosis in memory CD4+ T cells than in naïve CD4+ T cells [56]. As memory CD4+ T cells are a major reservoir for HIV, Au, like BSO, might be effective in killing latently infected cells when used in combination with other drugs.
In conclusion, in this study we show that exogenous Cys2 is required for proliferation of activated human T cells, because Cys2 is required for GSH production and GSH is essential for optimal RNR activity and thereby for dNTP synthesis and DNA replication.
MATERIALS AND METHODS
Chemicals
L-cystine dihydrochloride (Cys2) (C6727), L-buthionine-sulfoximine (BSO) (B2515), 2-mercaptoethanol (2-ME) (M3148) and L-methionine (M5308) were from Sigma-Aldrich Denmark ApS, Brondby, Denmark. Auranofin (EI-206) was from Enzo Life Sciences, Aarhus, Denmark.
Primary cells
Buffy coats were obtained from anonymous healthy blood donors. Written informed consent was obtained from blood donors at the Department of Clinical Immunology, University Hospital Rigshospitalet, Copenhagen and used without the possibility to identify case specific information. Use of these buffy coats for research was approved by the ethical committee, Region H, The Capital Region of Denmark. Mononuclear cells were isolated from the buffy coats by Lymphoprep™ (Axis-Shield, Oslo, Norway) density gradient centrifugation. Naïve CD4+ T cells were subsequently isolated from the mononuclear cell preparation by negative selection using the naïve CD4+ T cell isolation kit II (130-094-131, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and MACS® Separation Columns (130-042-401, Miltenyi Biotec GmbH) according to the manufacturer's instructions. The purified T cells were cultured in either Dulbecco's Modified Eagle Medium (DMEM, 21013, Invitrogen, Paisly, UK) or X-VIVO 15 medium (1041, Lonza, Verviers, Belgium). DMEM 21013 contains no Cys2, methionine, glutamine, glutamic acid or GSH. It was supplemented to a final concentration of 10% fetal bovine serum (FBS), 2 mM glutamine, 0.5 IU/l penicillin, 500 mg/l streptomycin, 50 μM methionine and various concentrations of Cys2. X-VIVO 15 medium contains 290 μM Cys2, 200 μM methionine, 4 mM glutamine and no GSH and with L-glutamic acid concentrations only known by the company. The cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (111.31D, Invitrogen) at 37°C in 5% CO2 at a cell concentration of 0.5 × 106 cells/ml at a bead to cell ratio of two to five.
Jurkat cells
Jurkat cells (E6) were cultured in RPMI 1640 medium (R5886, Sigma-Aldrich). RPMI 1640 contains 208 μM Cys2, 100 μM methionine, 118 μM glutamic acid, 3.25 μM GSH and was supplemented to a final concentration of 10% FBS, 2 mM glutamine, 0.5 IU/l penicillin and 500 mg/l streptomycin. Cells were incubated at 37°C in 5% CO2. For cell cycle synchronization, cells were cultured in serum-free RPMI 1640 medium for 48 hours.
GSH and thioredoxin reductase assays
Measurements of intracellular GSH levels were performed using the Promega GSH-Glo™ Glutathione Assay (V6911, Promega Biotech AB, Nacka, Sweden). This is a luminescence-based assay in which a luciferin derivative is converted into luciferin in the presence of GSH in a reaction that is catalyzed by glutathione S-transferase. The amount of light generated in the firefly luciferase-coupled reaction is proportional to the amount of GSH present in the sample. The assay was performed according to the manufacturer's instructions. We measured only GSH (also called reduced glutathione) and did not include measurements of the oxidized form of glutathione (GSSG). In brief, at the indicated times, the cells were resuspended in 50 μl PBS and transferred to Nunc-Immuno™ MicroWell™ 96 well polystyrene plates. 50 μl GSH-Glo™ Reagent 2X was added to each well, and the plate was incubated for 30 min at room temperature under gentle shaking. 100 μl Luciferin Detection Reagent was added and the luminescence was subsequently measured on a Wallac 1420 Victor2TM Workstation from PerkinElmer.
Measurements of thioredoxin reductase (TrxR) activity were performed using the Abcam® Thioredoxin Reductase (TrxR) Assay Kit (ab83463, Abcam, Cambridge, United Kingdom). This is a colorimetric assay in which reduction of 5,5′-dithiobis (2-nitrobenzoic) acid (DTNB) to 5-thio-2-nitrobenzoic acid (TNB) by TrxR generates a yellow color (λmax 412 nm). The assay was performed according to the manufacturer's instructions. In brief, cells were lyzed in 100 ul assay buffer + Protease/Phosphatase Inhibitor Cocktail (5872S, Cell Signaling) and 12.5 μl lysate was used for each reaction. As other enzymes, such as glutathione reductase and glutathione peroxidase, can also reduce DTNB, two measurements were performed for each sample; the first measures total DTNB reduction in the sample, and the second measures DTNB reduction in the presence of a TrxR specific inhibitor. The absorbance was measured at 405 nm several times for up to 45 minutes following setup of the assay. A TNB standard curve and a TrxR positive control were included for each assay. Enzyme activity in each sample was calculated from the standard curve.
Western blot analysis
Western blot analysis was carried out as previously described [57, 58]. In short, cells were stimulated with Dynabeads Human T-Activator CD3/CD28 for the time indicated at 37°C, lysed in 1% Triton X-100 lysis buffer + Protease/Phosphatase Inhibitor Cocktail (5872S, Cell Signaling) and run on 10% polyacrylamide gels. The proteins were transferred to Amersham Hybond ECL nitrocellulose sheets (RPN2020D, GE Healthcare, Brondby, Denmark) and visualized using ECL technology (RPN2232, GE Healthcare). As primary antibodies we used rabbit polyclonal anti-H2AX phospho S139 (1:10000) (ab11174, Abcam, Cambirdge, UK), rabbit polyclonal anti-GAPDH (1:5000) (ab9485, Abcam), rabbit monoclonal anti-Trx (1:1000) (2429, Cell Signaling, (via BioNordika Denmark A/S, Herlev, Denmark)), rabbit polyclonal anti-R1 (1:1000) (3388, Cell Signaling), and mouse monoclonal anti-β-actin (1:1000) (3700S, Cell Signaling). Secondary antibodies included HRP-conjugated polyclonal swine anti-rabbit Ig (1:1000) (P0399, DAKO, Glostrup, Denmark) and HRP-conjugated polyclonal rabbit anti-mouse Ig (1:2000) (P0260, DAKO).
DNA synthesis
DNA synthesis was measured by 3H-thymidine and 5-bromo-2′-deoxyuridine (BrdU) incorporation. The 3H-thymidine incorporation assay was carried out as previously described [59]. Briefly, cells were incubated for 48 or 72 hours at 37°C in 5% CO2, and 20 μl medium (X-VIVO, D-MEM or RPMI-1640) containing 50 μCi/ml [Methyl-3H]-thymidine (NET027005MC, Perkin Elmer) was subsequently added to each well resulting in a specific activity of 1 μCi/well. The incubation was continued for another 6 hours before the cells were harvested and 3H-thymidine incorporation was quantified as counts per minute (cpm) using a scintillation counter. BrdU incorporation assays were performed using the FITC BrdU Flow Kit (BD Biosciences, San Diego, CA, USA) according to supplier's protocol and as previously described [60]. In short, cultured cells were incubated with 17.7 μM BrdU for 1 hour and subsequently fixed, permeabilized and treated with DNase. Afterwards the cells were stained with FITC anti-BrdU and 7-AAD and analyzed by flow cytometry on a FACS Calibur.
Cell division assay
CFSE (C34554, Life Technologies, Naerum, Denmark) was used to stain cells and measure cell division as previously described [61, 62]. In short, purified naïve CD4+ T cells were resuspended in 4 ml 37°C PBS. CFSE was added to the cells at a final concentration of 2.5 μM CFSE. The cells were then incubated for 9 minutes at 37°C. FBS was added to terminate the incorporation of CFSE. Following CFSE loading, the cells were incubated in X-VIVO medium at 37°C in 5% CO2 with anti-CD3/CD28 beads and the indicated inhibitors. After 72 hours, the cells were analyzed by flow cytometry on a FACS Calibur.
dNTP extraction and quantification
dNTP extraction and quantification was performed as previously described [63]. For dNTP extraction, cells were washed in Hanks Balanced Salt solution and resuspended in 60% ice-cold methanol before being lysed using ultrasound. The cell debris was pelleted by centrifugation at 16.000 g for 15 min, 4°C. Supernatants were transferred to Amicon Ultra centrifugal filters (UFC500324, Merck Milipore, Hellerup, Denmark) and centrifuged at 14.000 g for 30 min, 4°C to remove macromolecules larger than 3 kDa. Liquid was removed from the sample by freeze drying, and the resultant pellet was resuspended in 12 μl nuclease-free H2O. The samples were either used directly for the assay or stored at −80°C. For dNTP quantification, 5 μl of each sample was mixed with 20 μl mastermix (1 μl nucleotide detection primer (10 μM), 1 μl dCTP detection template (10 μM), 1 μl of FAM-dCTP probe (10 μM), 2 μl of MgCl2 (25 mM), 1 μl of dNTPs (2.5 mM), 2.5 μl of 10X PCR Buffer II, 0.175 μl of AmpliTaq Gold Polymerase (5U/μl) (4311816, Applied Biosystems, Life Technologies), and 11.325 μl of nuclease-free H2O). The qPCR program included a 10 minute hot start at 95°C followed by 75 minutes at 60°C, while measuring the fluorescence intensity every 1 minute. The raw fluorescence spectra for each well were exported to excel and analyzed. A dCTP standard curve was included for each assay using dCTP from Life Technologies (10297–018). The following primers were used: Nucleotide detection primer 5′-CCG CCT CCA CCG CC-3′, dCTP detection template 5′-CCA CTC ACT CTT ACC TCA ATC CTT TGT TTG GCG GTG GAG GCG G-3′, and FAM-dCTP probe 5′-/6FAM/AGG ATT GAG/ZEN/GTA AGA GTG AGT GG/IABkFQ/-3'. 6FAM (6-carboxyfluorescein), ZEN (non-abbreviation), IABkFQ (Iowa black fluorescein quencher).
Statistical analysis
Data is shown as mean ± SEM, significance is calculated by using unpaired Student's t-test. * indicates p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
The Danish Medical Research Council, The Lundbeck Foundation, The Novo Nordisk Foundation and The A.P. Møller Foundation for the Advancement of Medical Sciences.
REFERENCES
1. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, von Essen MR, Woetmann A, Odum N, Bonefeld CM, Geisler C. Activated human CD4 T cells express transporters for both cysteine and cystine. Sci Rep. 2012; 2:266.
2. Messina JP, Lawrence DA. Effects of 2-mercaptoethanol and buthionine sulfoximine on cystine metabolism by and proliferation of mitogen-stimulated human and mouse lymphocytes. Int J Immunopharmacol. 1992; 14:1221–1234.
3. Saetre R, Rabenstein DL. Determination of cysteine in plasma and urine and homocysteine in plasma by high-pressure liquid chromatography. Anal Biochem. 1978; 90:684–692.
4. Hack V, Schmid D, Breitkreutz R, Stahl-Henning C, Drings P, Kinscherf R, Taut F, Holm E, Droge W. Cystine levels, cystine flux, and protein catabolism in cancer cachexia, HIV/SIV infection, and senescence. FASEB J. 1997; 11:84–92.
5. Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 2004; 447:469–479.
6. Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008; 215:593–602.
7. Bannai S. Transport of cystine and cysteine in mammalian cells. Biochim Biophys Acta. 1984; 779:289–306.
8. Garg SK, Yan Z, Vitvitsky V, Banerjee R. Differential dependence on cysteine from transsulfuration versus transport during T cell activation. Antioxid Redox Signal. 2011; 15:39–47.
9. Ishii T, Sugita Y, Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987; 133:330–336.
10. Castellani P, Angelini G, Delfino L, Matucci A, Rubartelli A. The thiol redox state of lymphoid organs is modified by immunization: role of different immune cell populations. Eur J Immunol. 2008; 38:2419–2425.
11. Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006; 75:681–706.
12. Holmgren A, Sengupta R. The use of thiols by ribonucleotide reductase. Free Radic Biol Med. 2010; 49:1617–1628.
13. Hofer A, Crona M, Logan DT, Sjoberg BM. DNA building blocks: keeping control of manufacture. Crit Rev Biochem Mol Biol. 2012; 47:50–63.
14. Guarino E, Salguero I, Kearsey SE. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin Cell Dev Biol. 2014; 30:97–103.
15. Koc A, Mathews CK, Wheeler LJ, Gross MK, Merrill GF. Thioredoxin is required for deoxyribonucleotide pool maintenance during S phase. J Biol Chem. 2006; 281: 15058–15063.
16. Niida H, Shimada M, Murakami H, Nakanishi M. Mechanisms of dNTP supply that play an essential role in maintaining genome integrity in eukaryotic cells. Cancer Sci. 2010; 101:2505–2509.
17. Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV. Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli. J Biol Chem. 1964; 239:3436–3444.
18. Holmgren A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc Natl Acad Sci U S A. 1976; 73:2275–2279.
19. Potamitou A, Holmgren A, Vlamis-Gardikas A. Protein levels of Escherichia coli thioredoxins and glutaredoxins and their relation to null mutants, growth phase, and function. J Biol Chem. 2002; 277:18561–18567.
20. Camier S, Ma E, Leroy C, Pruvost A, Toledano M, Marsolier-Kergoat MC. Visualization of ribonucleotide reductase catalytic oxidation establishes thioredoxins as its major reductants in yeast. Free Radic Biol Med. 2007; 42:1008–1016.
21. Zahedi Avval F, Holmgren A. Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for Mammalian s phase ribonucleotide reductase. J Biol Chem. 2009; 284:8233–8240.
22. Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983; 52:711–760.
23. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009; 30:42–59.
24. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013; 13:349–361.
25. Yan Z, Banerjee R. Redox remodeling as an immunoregulatory strategy. Biochemistry. 2010; 49:1059–1066.
26. Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989; 143:1974–1981.
27. Hamilos DL, Zelarney P, Mascali JJ. Lymphocyte proliferation in glutathione-depleted lymphocytes: direct relationship between glutathione availability and the proliferative response. Immunopharmacology. 1989; 18:223–235.
28. Suthanthiran M, Anderson ME, Sharma VK, Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc Natl Acad Sci U S A. 1990; 87:3343–3347.
29. Smyth MJ. Glutathione modulates activation-dependent proliferation of human peripheral blood lymphocyte populations without regulating their activated function. J Immunol. 1991; 146:1921–1927.
30. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014; 66:75–87.
31. Gromer S, Urig S, Becker K. The thioredoxin system— from science to clinic. Med Res Rev. 2004; 24:40–89.
32. Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000; 346:1–8.
33. Saccoccia F, Angelucci F, Boumis G, Carotti D, Desiato G, Miele AE, Bellelli A. Thioredoxin reductase and its inhibitors. Curr Protein Pept Sci. 2014; 15:621–646.
34. Zhou BS, Ker R, Ho R, Yu J, Zhao YR, Shih J, Yen Y. Determination of deoxyribonucleoside triphosphate pool sizes in ribonucleotide reductase cDNA transfected human KB cells. Biochem Pharmacol. 1998; 55:1657–1665.
35. Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 35:7545–7556.
36. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. H2AX and cancer. Nat Rev Cancer. 2008; 8:957–967.
37. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273:5858–5868.
38. Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007; 6:1239–1248.
39. Gmunder H, Eck HP, Droge W. Low membrane transport activity for cystine in resting and mitogenically stimulated human lymphocyte preparations and human T cell clones. Eur J Biochem. 1991; 201:113–117.
40. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di TG, Clarke F, Sitia R, Rubartelli A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A. 2002; 99:1491–1496.
41. Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nat Chem Biol. 2009; 5:721–723.
42. Zmuda J, Friedenson B. Changes in intracellular glutathione levels in stimulated and unstimulated lymphocytes in the presence of 2-mercaptoethanol or cysteine. J Immunol. 1983; 130:362–364.
43. Gmunder H, Eck HP, Benninghoff B, Roth S, Droge W. Macrophages regulate intracellular glutathione levels of lymphocytes. Evidence for an immunoregulatory role of cysteine. Cell Immunol. 1990; 129:32–46.
44. Eriksson S, Prigge JR, Talago EA, Arner ES, Schmidt EE. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat Commun. 2015; 6:6479.
45. Yamauchi A, Bloom ET. Requirement of thiol compounds as reducing agents for IL-2-mediated induction of LAK activity and proliferation of human NK cells. J Immunol. 1993; 151:5535–5544.
46. Yamauchi A, Bloom ET. Control of cell cycle progression in human natural killer cells through redox regulation of expression and phosphorylation of retinoblastoma gene product protein. Blood. 1997; 89:4092–4099.
47. Vene R, Delfino L, Castellani P, Balza E, Bertolotti M, Sitia R, Rubartelli A. Redox remodeling allows and controls B-cell activation and differentiation. Antioxid Redox Signal. 2010; 13:1145–1155.
48. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W, Keating MJ, Huang P. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012; 14:276–286.
49. Thelander L, Reichard P. Reduction of ribonucleotides. Annu Rev Biochem. 1979; 48:133–158.
50. Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988; 57:349–374.
51. Prigge JR, Eriksson S, Iverson SV, Meade TA, Capecchi MR, Arner ES, Schmidt EE. Hepatocyte DNA replication in growing liver requires either glutathione or a single allele of txnrd1. Free Radic Biol Med. 2012; 52:803–810.
52. Droge W, Breitkreutz R. Glutathione and immune function. Proc Nutr Soc. 2000; 59:595–600.
53. Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007; 7:355–359.
54. Osuji FN, Onyenekwe CC, Ifeanyichukwu M, Ahaneku JE, Ezeani M, Ezeugwunne IP. Antioxidant activity in HIV and malaria co-infected subjects in Anambra State, southeastern Nigeria. Asian Pac J Trop Med. 2012; 5:841–847.
55. Savarino A, Mai A, Norelli S, El DS, Valente S, Rotili D, Altucci L, Palamara AT, Garaci E. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology. 2009; 6:52.
56. Chirullo B, Sgarbanti R, Limongi D, Shytaj IL, Alvarez D, Das B, Boe A, DaFonseca S, Chomont N, Liotta L, Petricoin EI, Norelli S, Pelosi E, et al. A candidate anti-HIV reservoir compound, auranofin, exerts a selective ‘antimemory’ effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis. 2013; 4:e944.
57. Nielsen M, Nissen MH, Gerwien J, Zocca MB, Rasmussen HM, Nakajima K, Ropke C, Geisler C, Kaltoft K, Odum N. Spontaneous interleukin-5 production in cutaneous T-cell lymphoma lines is mediated by constitutively activated Stat3. Blood. 2002; 99:973–977.
58. Kongsbak M, von Essen MR, Boding L, Levring TB, Schjerling P, Lauritsen JP, Woetmann A, Odum N, Bonefeld CM, Geisler C. Vitamin D up-regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4+ T cells. PLoS One. 2014; 9:e96695.
59. Nielsen M, Svejgaard A, Skov S, Dobson P, Bendtzen K, Geisler C, Odum N. IL-2 induces beta2-integrin adhesion via a wortmannin/LY294002-sensitive, rapamycin-resistant pathway. Phosphorylation of a 125-kilodalton protein correlates with induction of adhesion, but not mitogenesis. J Immunol. 1996; 157:5350–5358.
60. Larsen JM, Geisler C, Nielsen MW, Boding L, Von EM, Hansen AK, Skov L, Bonefeld CM. Cellular dynamics in the draining lymph nodes during sensitization and elicitation phases of contact hypersensitivity. Contact Dermatitis. 2007; 57:300–308.
61. Bonefeld CM, Haks M, Nielsen B, von Essen MR, Boding L, Hansen AK, Larsen JM, Odum N, Krimpenfort P, Kruisbeek A, Christensen JP, Thomsen AR, Geisler C. TCR down-regulation controls virus-specific CD8+ T cell responses. J Immunol. 2008; 181:7786–7799.
62. Boding L, Bonefeld CM, Nielsen BL, Lauritsen JP, von Essen MR, Hansen AK, Larsen JM, Nielsen MM, Odum N, Geisler C. TCR down-regulation controls T cell homeostasis. J Immunol. 2009; 183:4994–5005.
63. Wilson PM, Labonte MJ, Russell J, Louie S, Ghobrial AA, Ladner RD. A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 2011; 39:e112.