
Introduction
In recent years there has been a growing appreciation that developing new effective treatments may be facilitated by experimental systems that replicate the multi-component tumour environment [1, 2]. Furthermore, there are serious doubts regarding the relevance of drug testing in established cancer cell lines and transfer of results to the clinic [3]. We have developed a three-dimensional culture system that allows maintenance of complex breast cancer specimens explanted ex vivo for up to four weeks after surgical resection in a supporting matrix of exogenous stromal type I collagen [4, 5]. These ex vivo cultures offer a relatively rapid and quantitative avenue to explore mechanisms of tumour spread and evaluate new treatments.
The metastatic potential of cancer cells is orchestrated by cell-cell adhesion, cell-matrix adhesion, protrusion and contractility, all of which require appropriate regulation and dynamics of the cytoskeleton [6]. The Ras-like Rho family GTPases are key regulators of all of these cellular processes due to their interaction with multiple downstream targets. The ability of Rac1 to induce metastasis has been closely linked to mesenchymal type motility in cancer cell lines [7-9]. This is likely to be due to Rac activity near the cell leading edge and subsequent disruption of cellular organization [10, 11]. Nonetheless, Rac activity must be tightly regulated in order to maintain normal epithelial cell junction formation, polarization and proliferation [12-14].
Rac activation can induce invasion and metastasis of breast cancer cell line models in vitro and in vivo [15]. Over-expression of Rac1 and Rac3 GTPases has been noted in several small cohorts of breast cancers [16-18] and it has been suggested that they may have a role in resistance to endocrine treatment [19, 20]. In breast cancer, this mechanism is best described in the context of HER2 over-expression. As a result of TGFβ stimulation, a complex comprising HER2, Vav2, Rac1, Pak1, actin and actinin is formed at cell protrusions [21]. The induction of Rac1 activity by the guanine nucleotide exchange factor (GEF) Vav2 causes invasion and contributes to cancer cell survival [20] and disruption of correct polarisation of the breast epithelium [22]. Other GEFs may also stimulate Rac1 downstream of HER2, including P-REX1 [22]. Up-regulation of Rac1 activity by P-REX1 and Tiam1 GEFs has been described independently of tumour HER2 status [21], although the latter finding is disputed [23].
Across a panel of 51 established breast cell lines [24], both normal and malignant, similar, detectable levels of Rac1 mRNA were observed. The levels of Rac2 and Rac3 transcripts were more variable, being reliably detected in less than half of the cell lines (our unpublished observations). Studies of Rac function using cell lines already have contributed to understanding of initiation of malignant transformation, but do not inform how intact human tumours may respond to therapeutic intervention [25]. We previously reported that the majority of invasive human breast cancers continue to express E-cadherin and β-catenin correctly at the cell membrane junctions [26]. Therefore, pan-Rac inhibition of the whole, intact breast tumour explant could potentially bring about apparently conflicting effects on epithelial versus mesenchymal cells. It may lead to destruction of epithelial junctions enabling invasiveness, while blocking mesenchymal motility.
In this study we examine the response of authentic primary breast cancers to Rac-based drug treatments. We show that invasive breast cancers are responsive to exposure to EHT1864, a water soluble pan-Rac inhibitor. Consequently, Rac inhibition blocks a key down-stream effector, the Signal Transducer and Activator of Transcription-3 (STAT3) transcription factor. We show that Rac and STAT3 inhibition regimens are effective at blocking tumour invasion ex vivo, independently of known clinical biomarkers such as histological grade and ER status.
Results
Rac1 is over-expressed in invasive human breast cancer
In order to determine whether Rac1 protein might be a viable therapeutic target in breast cancer, we analysed its mRNA expression levels in published gene expression datasets. RAC1 was found to be significantly higher expressed in breast tumours than in normal breast tissue (Figure 1A). We then used a meta dataset of 2999 primary breast tumours (see Methods) to explore whether RAC1 expression associated with particular subtypes of breast tumours. There was no significant variation in RAC1 levels with grade or ER status (Figure 1B-C). RAC1 was significantly higher and associated with poor prognosis in HER2 (ERBB2) over-expressing tumours, although the range of RAC1 expression was similar between HER2+ and HER2- tumours (Figure S1). Importantly, high levels of RAC1 were associated with late recurrence (Figure 1D). Overall, these data suggest that RAC1 may represent a therapeutic target with broad potential in breast cancer, because it does not show a strong association with particular patient groups.
We have recently succeeded in establishing a robust system for long-term culture of human breast cancer explants, successfully growing >90% tumours of all major sub-types [4, 5]. In these cultures, primary breast cancer biopsies from invasive breast cancers are explanted into the centre of a type I collagen matrix. From each tumour cultured ex vivo, 80%-100% of explants are viable and display variable degrees of cellular spread into the surrounding collagen [4]. Growth in mammary epithelial media occurs in all three dimensions, as demonstrated optical projection tomography (Figure 1E; cytokeratin labels epithelial structures (green) invading from the original biopsy material into the surrounding collagen). We found that in many breast cancers grown ex vivo, both Rac1 and its constitutively active isoform Rac1b, are preferentially expressed in invading cells (Figures 1F and S2; n=6) when assessed by immnunohistochemistry. Importantly, both isoforms are expressed in tumour epithelial and mesenchymal cells, indicating wide spread Rac activity.
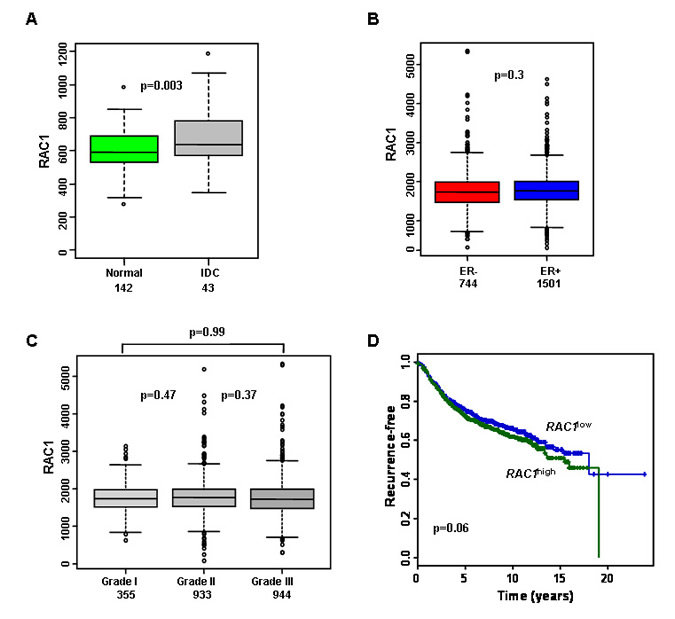
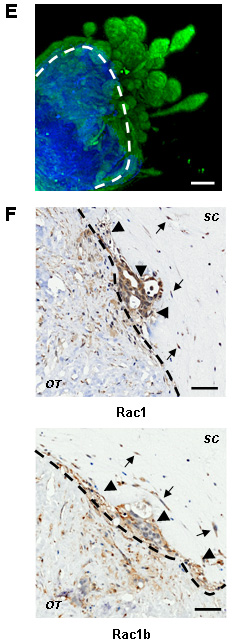
Figure 1: RAC1 is more highly expressed in breast cancer and is associated with poor prognosis. (A) Box plots demonstrating that invasive ductal carcinomas (IDC) have higher RAC1 expression than normal breast ducts, but range is similar. (B-C) RAC1 is not associated with estrogen receptor alpha (ER) status or histological grade of primary breast tumours. (D) Kaplan-Meier survival curves for all breast cancer patients with available data in the meta-dataset show that high levels of RAC1 expression are associated with poorer prognosis (n=1654). RAC1 expression levels were divided into low and high groups at the median. (E) An example of invasion ex vivo: 3D reconstruction of optical projection tomography of a representative ER+ HER2- tumour. The original tumour material is in blue (autofluorescence) and invading epithelial tumour cells are in green (cytokeratin labelled). (F) Immunohistochemistry showing that cells invading ex vivo express Rac1 protein (upper panel) and its short isoform Rac1b (lower panel). Rac1 and Rac1b are expressed in both tumour epithelial (arrowheads) and mesenchymal (arrows) invading cells. Images are from a representative ER- HER2- tumours. Lower magnification images are in Figure S2. Bars, 50 μm. The dotted lines show border between original tumour explant (OT) and surrounding collagen (SC). Brown staining indicates cells positive for Rac1 and Rac1b staining.
Rac inhibition blocks the spread of human breast cancer
We used EHT 1864, a water soluble pan-Rac inhibitor [27], to determine the value of Rac inhibition in the explant model. EHT1864 exhibits high affinity binding to Rac1, as well as the related Rac1b, Rac2, and Rac3 isoforms, inhibiting their activity via a mechanism that involves guanine nucleotide displacement [27]. EHT 1864 has been used previously in cell line models in 2D, especially in the context of Rac1 activity [19, 28]. Continuous treatment of breast cancer explant cultures with EHT 1864 for 14 days resulted in complete block of invasion into the surrounding collagen, and wide spread cell death (Figure 2A). We characterised the effects of Rac inhibition in two regimens: in the first regimen, explants from the same cancer were divided randomly to equally sized treatments groups and treated continuously for 14 days (Figure 2B). In the second regimen, explant cultures were left to grow out for 10 days, and then divided randomly to treatment groups for a further 14 days (Figure 2C). Both regimens resulted in almost complete elimination of tumour outgrowth into the surrounding collagen. Using logistic regression, Rac inhibition with EHT 1864 exhibited an odds ratio of 0.01 of outgrowth compared with vehicle (p < 0.0001, 95% CI 0.003 to 0.04).
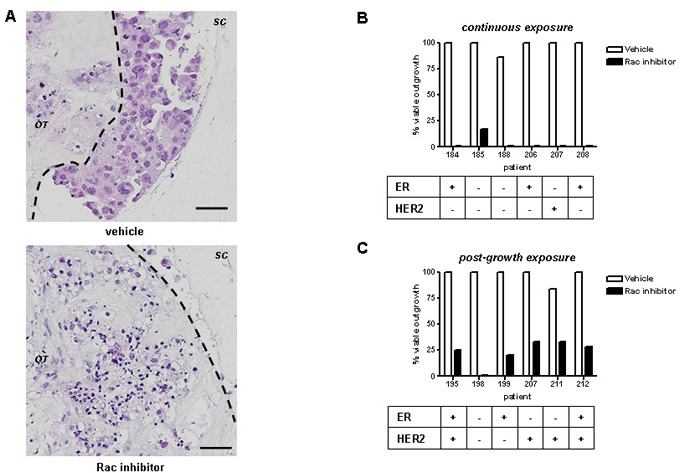
Figure 2: Rac inhibition ex vivo blocks tumour invasion. (A) Tumour invasion into surrounding collagen is readily detectable using H&E staining (upper panel). Continuous Rac inhibition with EHT1864 for 14d (bottom panel) results in blockage of invasion beyond the original tumour material (black outline) and extensive cell death: Bars, 50 µm. Outgrowth of tumour cultures were categorised following treatment with EHT 1864 or vehicle control, as follows: (B) continuous exposure from start of culture, or (C) exposure of existing outgrowths, 10d after start of culture. All inhibitor treatments lasted 14d. Results were obtained from light microscopy examination and subsequently confirmed by H&E staining. Tumour biomarkers shown were determined by a pathologist as part of clinical practice.
Rac inhibition blocks proliferation and induces apoptosis in human breast cancers
Tumour outgrowth resumed if Rac inhibition was removed after 3 days of treatment (Figure 3A; 4 independent experiments), indicating that EHT 1864 treatment is not solely as a result of cytotoxicity. We therefore examined what down-stream effects on apoptosis (assessed by demonstration of cleaved caspase-3) and proliferation (as assessed by Ki67). The number of cells undergoing cell death at the experimental endpoint rose from a basal level of <20% in vehicle-treated tumours [5] to 30%-50% of cells treated with EHT 1864. These cells were found predominantly around the periphery of the original tumour explant (Figure 3B). Vehicle-treated tumours have a wide range of proliferation from 10% to 60% of cells [5], but exposure to EHT 1864 reduced proliferation to less than 5% (Figure 3C). Interestingly, levels of E-cadherin expression were very similar with or without EHT 1864 treatment (Figure 3D).
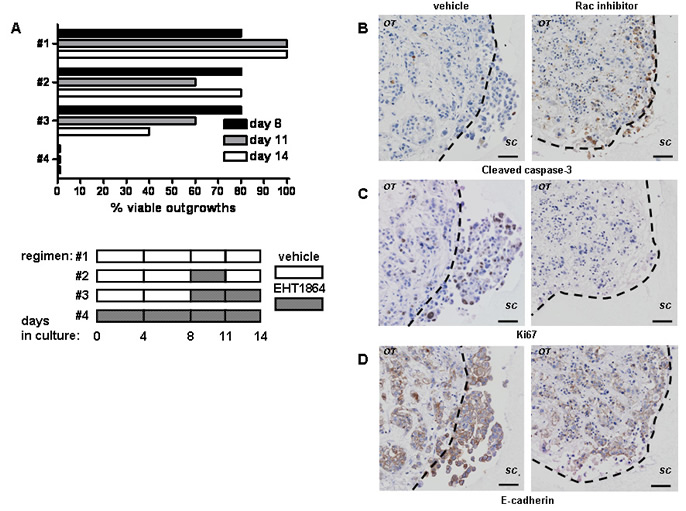
Figure 3: Rac inhibition with of tumour outgrowth ex vivo results in block of proliferation and cell death. (A) The effect of treatment with EHT 1864 or vehicle control on outgrowth of tumour cultures was obtained from light microscopy examination and subsequently confirmed by H&E staining. Shown are reperentative results from ER- HER2- tumour explant culture lasting 14d (n=4). Note that continuous EHT1854 treatment reduces viable outgrowth (regimen #4) in comparison to short-treatment (#2) or vehicle control (#1). Rac inhibition of tumour invasion is accompanied with induction of apoptosis as detected by immunohistochemistry for cleaved caspase-3 (B) and block of proliferation, detected by Ki67 staining (C). (D) E-cadherin levels are unchanged ex vivo. Shown here representative images from a single ER- HER2- tumour, (left panel) without or (right panel) with EHT 1864. The dotted lines show border between original tumour explant (OT) and surrounding collagen (SC). Bars, 50 μm.
STAT3 is a down-stream mediator of Rac activity in breast cancer spread
A cell line 3D model was established to assess the potential contribution of Rac1 down-stream targets in epithelial cell invasion. We modified our collagen-based invasion culture [29] using the HCC1954 cell line, which invades in organised fashion in vivo [30]. In 3D culture, HCC1954 cells express proteins such as E-cadherin in a similar manner to this observed in tumour cells cultured ex vivo (Figure S3). In the absence of serum, HCC1954 cells invade collectively (Figure S4A). Treatment of the cell line culture with EHT 1864 resulted in rapid (<24 h) inhibition of invasion and appearance of apoptotic cells (Figures S5A). Surviving HCC1954 cells were still positive for Ki67, a marker of proliferation (Figure S5B). The effects of pan-Rac inhibition in the cell line model could not be replicated by inhibitors against many known Rac downstream mediators: JNK (SP600125), PAK (IPA3) or p38α (SB203580) (data not shown).
Another well established Rac1 target is phosphorylation of STAT3 at Ser727 is [31]. We examined Rac1 and phospho-Ser727 STAT3 levels by antibody staining in human breast tumours (n=6). Patterns Rac1 and phospho-Ser727 STAT3 staining are similar, especially in epithelial cells (Figure 4A). Phospho-Ser727 STAT3 staining is abundant in the invading cells ex vivo (Figure 4B and S6A). In the HCC1954 model, Rac inhibition resulted in reduction in phospho-Ser727 STAT3 (Figure S6A). Survivin, a well established target of STAT3 transcriptional activity [32], is also down-regulated by EHT 1864 treatment of cell line cultures (Figure S7B). Some HCC1954 cells survived Rac inhibition for 24 h, retaining both phospho-Ser727 STAT3 and Survivin (Figure S5, arrows).
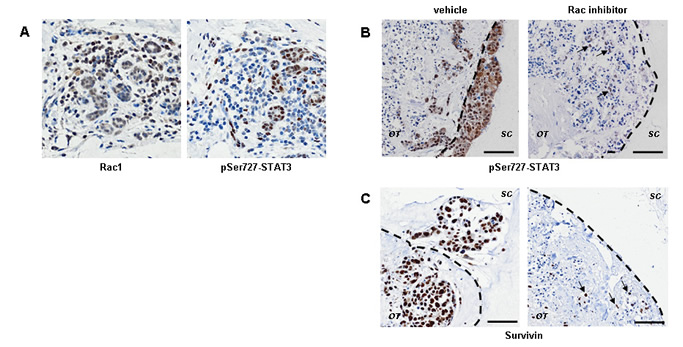
Figure 4: STAT3 signaling is downstream of Rac activity during invasion of human breast cancer. (A) Immunohistochemistry shows that Rac1 protein expression (left panel) and STAT3 phosphorylation on Ser727 (right panel) frequently coincide in human breast cancer. Epithelial cells which are Rac1+ pSTAT3- are also present. (B) STAT3 phosphorylation is associated with invasion ex vivo and is down regulated by Rac inhibition (right panels; inhibition as in Figure 2). Identical patterns are seen with the STAT3 transcriptional target Survivin, ex vivo (C). Even after exposure to EHT 1864, a few cells are still positive for phospho-STAT3 and Survivin (arrows in B and C). Images are shown from a representative ER- HER2- tumour. Lower magnification images are in Figure S6. The dotted lines show border between original tumour explant (OT) and surrounding collagen (SC). Bars, 50 µm.
Block of proliferation by Rac inhibition ex vivo precedes down-regulation of STAT3 activity
We tested whether long-term Rac inhibition also blocks STAT3 activity in the explant tumour cultures. The intensity of phospho-Ser727 STAT3 staining ex vivo was greatly reduced by EHT 1864 (Figure 4B). Consistent with STAT3 activity down-stream of Rac, EHT 1864 treatment resulted in down-regulation of Survivin protein expression (Figures 4C). Interestingly, cells initially surviving Rac inhibition retained both nuclear localization of phospho-Ser727 STAT3 and Survivin ex vivo (arrows).
STAT3 may be indirectly controlled by Rac via an autocrine or paracrine signal [33, 34]. We examined if there are short-term effects of Rac inhibition preceding changes in STAT3 activity ex vivo. The role of STAT3 downstream of Rac activation was confirmed using Stattic, an inhibitor of dimerization and nuclear translocation of STAT3 preventing its constitutive activation [35]. After one round of treatment with EHT 1864, only a partial reduction in phospho-Ser727 STAT3 and Survivin staining can be observed by immunohistochemistry (Figure 5A and not shown). The rapid induction of apoptosis by STAT3 inhibition in comparison to Rac inhibition, supports a more direct effect of STAT3 inhibition (Figure 5B). Importantly, proliferation within the invading cells is greatly reduced by both inhbitors (Figure 5C), suggesting most likely through down-regulation of cyclin D1 (Figure 5D), a known Rac1 and STAT3 target [36-39]. The dramatic induction of apoptosis by Stattic is even more pronounced than this caused by EHT 1864, in line with previous reports in breast cancer cell lines [35, 40-42].
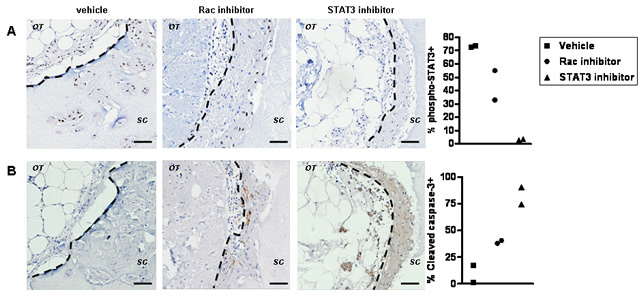
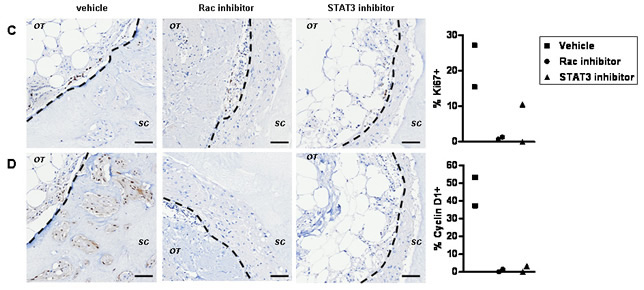
Figure 5: The early cellular response to Rac inhibition ex vivo. (A) Tumour explants were grown ex vivo for 14 days prior to inhibitor treatment in order to define events induced by a single post-growth treatment. Immunohistochemistry suggest that STAT3 phosphorylation (vehicle, left panels) is down regulated by EHT 1864 (middle panels) or Stattic (right panels) within 72 h of treatment. (B) Wide spread apoptosis is detected in Stattic treated cultures (right panel) and to lesser extent in EHT 1864 treated cultures (middle panel). Effects on proliferation are shown by staining with the marker Ki67 and (C) the cell cycle protein cyclin D1 (D): reduced in EHT 1864 (middle panel) or Stattic (right panel) exposed cultures). The dotted lines show border between original tumour explant (OT) and surrounding collagen (SC). Bars, 50 μm. Quantification is shown for percent of positive invading cells from two preparations per treatment, all originating from the same tumour.
Stattic treatment has similar long-term effects to those seen with EHT 1864 on tumour invasion ex vivo (Figure 6). Using logistic regression, STAT3 inhibition with Stattic exhibited an odds ratio of 0.003 (p < 0.0001, 95% CI 0.001 to 0.018) compared with vehicle.
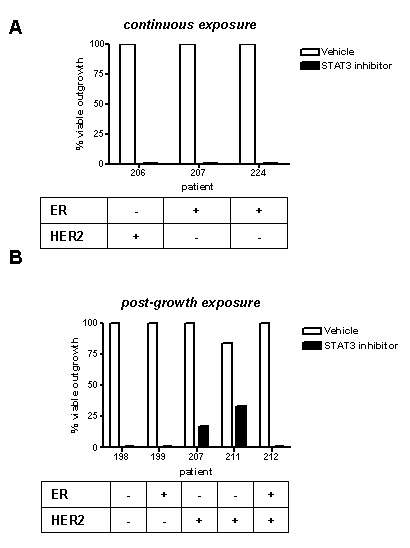
Figure 6: STAT3 inhibition blocks tumour outgrowth ex vivo. Outgrowth of tumour cultures were categorised following treatment with Stattic or vehicle control, as follows: (A) continuous exposure from start of ex vivo culture, or (B) exposure of existing outgrowths, 10d after start of culture. All inhibitor treatments lasted 14d. Results were obtained from light microscopy examination and subsequently confirmed by H&E staining. Tumour biomarkers shown were determined by a pathologist as part of clinical practice.
Discussion
Although endocrine (for ER+ tumours), cytotoxic chemotherapy (for ER- tumours) or Herceptin (for HER2+ tumours) regimens are successful treatments for some breast cancer patients, mortality due to recurrence of cancers which develop drug resistance and metastases is still common [43]. Therefore, identification of novel treatment for metastatic breast cancer is important to enhance long term patient survival [44]. As direct measurements of enzymatic activity in human tumours are difficult and possibly inaccurate due to clinical constrains in sample collection, other methods are need to evaluate such novel treatments.
The over-expression of Rho GTPases has previously been implicated in the promotion of various cancer types [16, 45]. It is also well established that Rho, Cdc42 and particularly Rac1 have critical roles in experimental tumour metastasis [33, 38]. It is usually thought that Rac activity in cancer is associated with the loss of epithelial cell-cell junctions and gain of mesenchymal phenotype [46]. Many signaling pathways lie downstream of Rac proteins, although their relationship to malignancy is not always clearly understood. These include the activation of JNK and p38 proteins [47], as well as non-canonical (β-catenin-independent) WNT signaling pathways [48]. It has been suggested that the constitutively active isoforms Rac1b and Rac3 show pathway preference, which is distinct from that of the GEF-dependent isoforms Rac1 and Rac2 [8, 16, 49].
Here, we have shown that the spread of breast tumours in an ex vivo model is sensitive to Rac inhibition. This 3D culture model recapitulates epithelial invasion of human breast cancer tumours in vivo, in particular maintaining E-cadherin membrane expression, unlike invasive breast cancer cell lines [26, 29]. Recent evidence links proliferation and invasion to the spread of cancer, both of which are inhibited in our model system upon EHT 1864 exposure [33, 50]. Moreover, a study in mouse melanoblasts suggested a role for Rac1 in proliferation of motile cells, although the exact mechanism was not identified [51].
Our experiments demonstrate that an approach involving pan-Rac inhibition has the potential to be applied to the clinic. In the current study, inhibition of all Rac isoforms by EHT1864 in explants of intact primary cancers prevented spread into collagen, reduced proliferation and subsequently caused apoptosis. The reduction in cyclin D1 levels by Rac inhibition did not result in greater invasiveness, as reported in MDA-MB-231 cells [52]. EHT 1864 blocks the constitutively active Rac1b and Rac3 isoforms [27], unlike the more widely used Rac GEF inhibitor NSC23766. EHT 1864 is likely to be superior to using a Rac GEF inhibitor, which will not have any effect on the Rac1b or Rac3, long considered to be associated with breast cancer aggressiveness [8, 16]. To date, a range of in vitro and in vivo models has not reported any significant cytotoxicity associated with EHT 1864. This represents a therapeutic opportunity as breast cancers should have higher over-all Rac activity than this of the surrounding normal breast.
We have demonstrated, at the breast cancer tissue level, a link between STAT3 and Rac proteins. STAT3 is a critical transcription factor regulating cell death during breast involution [53], but it can also function as an oncogene [39]. STAT3 activity (in absence of STAT5 activity) has been shown previously to be present in 40% of human breast cancers and has been linked to higher grade and lymph node metastasis [54]. STAT3 inhibition has a clear anti-tumour effect as demonstrated by a variety of reagents [42, 55]. It is conceivable that disruption of the feedback loop between Rac1 and E-cadherin adherens junctions could lead to aberrant STAT3 activation in breast cancer [56].
The current study suggests that invasion of breast cancer is associated with up-regulation of Rac and STAT3 activity. The challenge now is to understand which breast cancer patients may be most suited for Rac-based treatment, once a drug is available for human use. Our finding that patients with HER2high RAC1high tumours have the worst prognosis, suggests that Rac targeting in the clinic may particularly benefit patients with HER2+ tumours. This is supported by recent experiments in mice suggesting that gp130/STAT3 signalling is critical in HER2-induced tumourigenesis [57]. Our findings that even after exhaustive Rac inhibition in intact breast tumour, STAT3 activity is detected in rare live cells suggest that optimal treatment may require a combination of drugs inhibiting Rac and STAT3 activities.
Materials and Methods
Gene Expression Analysis
Raw .cel files from seventeen Affymetrix U133A/plus 2 gene expression datasets were downloaded from NCBI GEO (GSE12276, GSE21653, GSE3744, GSE5460, GSE2109, GSE1561, GSE17907, GSE2990, GSE7390, GSE11121, GSE16716, GSE2034, GSE1456, GSE6532, GSE3494 and GSE19615) or the caBIG (geral-00143) repositories, summarised with Ensembl alternative CDF [58] and normalised with RMA [59], before integration using ComBat [60] to remove dataset-specific bias as previously described [61]. The most conservative measure available of recurrence-free, disease-free and distant-metastasis-free survival was used. Cox proportional hazards regression was performed using SPSS version 14.
Ethical approval
The use of tissue from invasive breast cancer treated at the Edinburgh Breast Unit at the Western General Hospital was approved by the Lothian Research Ethics Committee (06/S1103/65). The clinical parameters of all tumours used in this study are listed in Table S1.
Collagen-based 3D cultures
The set-up of collagen type I-based cultures, ex vivo and cell line, was described in detail previously [4, 29].
For cell line cultures, 75 μL or 150 μL cell-Matrigel plugs were made using 2 x 106 cells/ml and 5mg/ml Matrigel (BD bioscience), in a U-shaped 96-well plate. After overnight incubation, cell plugs were carefully removed from their 96-well plate and embedded in 1 ml of rat collagen I (1 mg/ml) in a 24-well plate or 2 ml of collagen in a 12-well plate. These cultures were incubated for a further 1 h and then carefully freed from the edges of the well (to allow contraction of the collagen) and supplemented with 1 ml or 2 ml RPMI media (without serum). The cells were then left to invade. Media was changed every 3-5 days. Rac inhibitor (EHT 1864, Tocris #3872, 20 μM) or STAT3 inhibitor (Stattic, Tocris #2798, 10 μM) were added at the last 24 h or 96 h.
For ex vivo cultures, breast tumour biopsy materials (cut to 1mm pieces) were explated instead of Matrigel plugs in a 24-well plate format. The media used ex vivo is complete MEGM (Lonza). Continuous inhibitor treatment lasted between days 1-14. For late addition assays, assays with viable growth at day 6 or 8 (as observed using a light microscope) were divided to equal size groups (> 4 assays per treatment). Treatment with inhibitor or appropriate vehicle control lasted for subsequent 12 days. Inhibitors were added at concentrations as above every 3 days. In experiments represented in Figure 4, tumours (n=4) were allowed to grown for 7-14 d prior to inhibitor treatment lasting 3 additional days. Quantification of positive nuclei in invading cells was performed on entire sections obtained from the same tumour (>90 cells/preparation).
To terminate all experiments, cultures were fixed in 10% phosphate buffered formalin and wax embedded.
Staining protocol of 3D ex vivo cultures for optical projection tomography
Large-scale specimen staining for optical projection tomography has followed a previously published protocol [4]. Briefly, fixed and permebialised preparations were incubated with primary antibody to identify the epithelial content using rabbit pan-cytokeratin (Cell Signalling), and subsequently with a fluorescein-conjugated secondary antibody (Invitrogen). The preparation was mounted in 1% low melting agarose and dehydrated in 100% Methanol for 24 h. The specimen was cleared in BABB solution (1:2 Benzyl alcohol, Benzyl benzoate) for 24 h and scanned using the Optical Projection Tomograph (Bioptonics 3001M). Both target and auto-fluorescence were captured using a GFP+ filter and GFP1 filter respectively.
Immunohistochemistry
Antigen retrieval for all staining was performed using sodium citrate buffer (18 μM Citric Acid, 82 μM sodium citrate, pH 6.0). Standard immunohistochemistry protocol was performed using the REAL EnVision mouse or rabbit kit (Dako), according to manufacturer’s instructions. Primary antibodies used were: β-catenin, BD #610153, Mouse, 1:500; CD44, AbD serotec #MCA2504, Mouse, 1:10,000; Cleaved caspase-3, Cell Signaling #9661, 1:400; Cyclin D1, Dako #M3635, Rabbit 1:100 E-cadherin (intracellular), BD #610181, Mouse, 1:1500; G3BP2, Sigma #HPA018304, Rabbit, 1:5000; Ki67, Dako #M7240, Mouse, 1:400; phosphorylated STAT3 (Ser727), Eurogentec, #65367, Rabbit, 1:200-1:600; Rac1, GeneTex, #GTX100761, Rabbit, 1:100-1:200.
Categorisation of effects on explant outgrowths ex vivo
After 14 d of treatment with either Rac inhibitior (EHT 1864) or STAT3 inhibitor (Stattic), all preparations were inspected using live microscopy. Cell invasion from the original tumour explant into the surrounding collagen is typically observed after 6-8 d from start of culture [5]. At each experimental time point, media was removed and cellular outgrowths ware determined by light microscopy (x10 magnification). The viability of outgrowths was confirmed subsequently using H&E sections from the fixed preparations.
Acknowledgements
We thank Elaine McLay (Breakthrough Research Unit, University of Edinburgh) for technical support, Lorna Renshaw (WGH Breast Unit) for consenting patients and collecting tumour materials, Matthew Pearson (MRC-HGU), Joanne Farrell and Lisa Mitchell (MRC Technology) for help with imaging and Linda Williams (Centre for Population Health Sciences, University of Edinburgh) for statistical support.
This study was funded by Scottish Funding Council and Breakthrough Breast Cancer (DJH and RRM). Clinical specimens were obtained through the Cancer Research UK supported Edinburgh Experimental Cancer Medicine Centre.
All authors declare no conflicting interests.
Reference
1. Wirtz D, Konstantopoulos K and Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011; 11(7):512-522.
2. Sonnenschein C and Soto AM. The death of the cancer cell. Cancer Res. 2011; 71(13):4334-4337.
3. Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V, Padmanabhan R, Davidson B, Ganapathi R, Sood AK, Rueda BR, Ambudkar SV, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(46):18708-18713.
4. Leeper AD, Farrell J, Dixon JM, Wedden SE, Harrison DJ and Katz E. (2011). Long-term culture of human breast cancer specimens and their analysis using optical projection tomography. J Vis Exp, pp. e3085.
5. Leeper AD, Farrell J, Williams LJ, S. TJ, Dixon JM, Wedden SE, Harrison DJ and Katz E. Determining tamoxifen sensitivity using primary breast cancer tissue in collagen-based three-dimensional culture. Biomaterials. 2012; 33(3):907-915.
6. Montell DJ. Morphogenetic cell movements: diversity from modular mechanical properties. Science. 2008; 322(5907):1502-1505.
7. Zuo Y, Shields SK and Chakraborty C. Enhanced intrinsic migration of aggressive breast cancer cells by inhibition of Rac1 GTPase. Biochem Biophys Res Commun. 2006; 351(2):361-367.
8. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z and Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005; 436(7047):123-127.
9. Koh MS and Moon A. Activation of H-Ras and Rac1 correlates with epidermal growth factor-induced invasion in Hs578T and MDA-MB-231 breast carcinoma cells. Biochem Biophys Res Commun. 2011; 406(1):25-29.
10. Weber GF, Bjerke MA and DeSimone DW. Integrins and cadherins join forces to form adhesive networks. J Cell Sci. 2011; 124(Pt 8):1183-1193.
11. Yagi S, Matsuda M and Kiyokawa E. Suppression of Rac1 activity at the apical membrane of MDCK cells is essential for cyst structure maintenance. EMBO reports. 2012.
12. Arulanandam R, Vultur A, Cao J, Carefoot E, Elliott BE, Truesdell PF, Larue L, Feracci H and Raptis L. Cadherin-cadherin engagement promotes cell survival via Rac1/Cdc42 and signal transducer and activator of transcription-3. Mol Cancer Res. 2009; 7(8):1310-1327.
13. Akhtar N and Streuli CH. Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia. J Cell Biol. 2006; 173(5):781-793.
14. Jeanes AI, Wang P, Moreno-Layseca P, Paul N, Cheung J, Tsang R, Akhtar N, Foster FF, Brennan K and Streuli CH. Specific β-containing integrins exert differential control on proliferation and 2D collective cell migration in mammary epithelial cells. The Journal of Biological Chemistry. 2012 [Epub ahead of print].
15. Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS and Kazanietz MG. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal. 2011; 24(2):353-362.
16. Mira JP, Benard V, Groffen J, Sanders LC and Knaus UG. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc Natl Acad Sci U S A. 2000; 97(1):185-189.
17. Fritz G, Brachetti C, Bahlmann F, Schmidt M and Kaina B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer. 2002; 87(6):635-644.
18. Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M and Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000; 19(26):3013-3020.
19. Rosenblatt AE, Garcia MI, Lyons L, Xie Y, Maiorino C, Desire L, Slingerland J and Burnstein KL. Inhibition of the Rho GTPase, Rac1, decreases estrogen receptor levels and is a novel therapeutic strategy in breast cancer. Endocr Relat Cancer. 2011; 18(2):207-219.
20. Walker MP, Zhang M, Le TP, Wu P, Laine M and Greene GL. RAC3 is a pro-migratory co-activator of ERalpha. Oncogene. 2011; 30(17):1984-1994.
21. Wang SE, Shin I, Wu FY, Friedman DB and Arteaga CL. HER2/Neu (ErbB2) signaling to Rac1-Pak1 is temporally and spatially modulated by transforming growth factor β. Cancer Res. 2006; 66(19):9591-9600.
22. Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, Gutkind JS, Parsons RE and Kazanietz MG. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010; 40(6):877-892.
23. Stebel A, Brachetti C, Kunkel M, Schmidt M and Fritz G. Progression of breast tumors is accompanied by a decrease in expression of the Rho guanine exchange factor Tiam1. Oncol Rep. 2009; 21(1):217-222.
24. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10(6):515-527.
25. Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E and Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008; 135(3):510-523.
26. Dubois-Marshall S, Thomas JS, Faratian D, Harrison DJ and Katz E. Two possible mechanisms of epithelial to mesenchymal transition in invasive ductal breast cancer. Clin Exp Metastasis. 2011; 28(8):811-818.
27. Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F and Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007; 282(49):35666-35678.
28. Nethe M, Anthony EC, Fernandez-Borja M, Dee R, Geerts D, Hensbergen PJ, Deelder AM, Schmidt G and Hordijk PL. Focal-adhesion targeting links caveolin-1 to a Rac1-degradation pathway. J Cell Sci. 2010; 123(Pt 11):1948-1958.
29. Katz E, Dubois-Marshall S, Sims AH, Gautier P, Caldwell H, Meehan RR and Harrison DJ. An in vitro model that recapitulates the epithelial to mesenchymal transition (EMT) in human breast cancer. PLoS ONE. 2011; 6(2):e17083.
30. Clinchy B, Gazdar A, Rabinovsky R, Yefenof E, Gordon B and Vitetta ES. The growth and metastasis of human, HER-2/neu-overexpressing tumor cell lines in male SCID mice. Breast Cancer Res Treat. 2000; 61(3):217-228.
31. Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH and Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000; 290(5489):144-147.
32. Yoshida T, Zhang Y, Rivera Rosado LA, Chen J, Khan T, Moon SY and Zhang B. Blockade of Rac1 activity induces G(1) cell cycle arrest or apoptosis in breast cancer cells through downregulation of cyclin D1, survivin, and X-linked inhibitor of apoptosis protein. Mol Cancer Ther. 2010; 9(6):1657-1668.
33. Arulanandam R, Geletu M, Feracci H and Raptis L. Activated Rac1 requires gp130 for Stat3 activation, cell proliferation and migration. Exp Cell Res. 2010; 316(5):875-886.
34. Faruqi TR, Gomez D, Bustelo XR, Bar-Sagi D and Reich NC. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci U S A. 2001; 98(16):9014-9019.
35. Schust J, Sperl B, Hollis A, Mayer TU and Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chemistry & biology. 2006; 13(11):1235-1242.
36. Yang C, Liu Y, Leskow FC, Weaver VM and Kazanietz MG. Rac-GAP-dependent inhibition of breast cancer cell proliferation by {β}2-chimerin. J Biol Chem. 2005; 280(26):24363-24370.
37. Yang C, Klein EA, Assoian RK and Kazanietz MG. Heregulin β1 promotes breast cancer cell proliferation through Rac/ERK-dependent induction of cyclin D1 and p21Cip1. Biochem J. 2008; 410(1):167-175.
38. Klein EA, Campbell LE, Kothapalli D, Fournier AK and Assoian RK. Joint requirement for Rac and ERK activities underlies the mid-G1 phase induction of cyclin D1 and S phase entry in both epithelial and mesenchymal cells. J Biol Chem. 2008; 283(45):30911-30918.
39. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C and Darnell JE, Jr. Stat3 as an oncogene. Cell. 1999; 98(3):295-303.
40. Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D, Yoder S, Enkemann S, Eschrich S, Lee JH, Beam CA, Cheng J, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006; 12(1):11-19.
41. Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, Cen L, Li C and Lin J. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia (New York, NY. 2010; 12(1):39-50.
42. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM and Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007; 104(18):7391-7396.
43. Lu J, Steeg PS, Price JE, Krishnamurthy S, Mani SA, Reuben J, Cristofanilli M, Dontu G, Bidaut L, Valero V, Hortobagyi GN and Yu D. Breast Cancer Metastasis: Challenges and Opportunities. Cancer Res. 2009; 69(12):4951-4953.
44. Ito Y, Iwase T and Hatake K. Eradication of breast cancer cells in patients with distant metastasis: the finishing touches? Breast Cancer. 2011.
45. Benitah SA, Espina C, Valerón PF and Lacal JC. Rho GTPases in human carcinogenesis: a tale of excess. Rev Oncol. 2003; 5(2):70-78.
46. Lozano E, Frasa MA, Smolarczyk K, Knaus UG and Braga VM. PAK is required for the disruption of E-cadherin adhesion by the small GTPase Rac. J Cell Sci. 2008; 121(Pt 7):933-938.
47. Turkson J, Bowman T, Adnane J, Zhang Y, Djeu JY, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S and Jove R. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol Cell Biol. 1999; 19(11):7519-7528.
48. McDonald SL and Silver A. The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009; 101(2):209-214.
49. Matos P and Jordan P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res. 2008; 6(7):1178-1184.
50. Lee HO, Silva AS, Concilio S, Li YS, Slifker M, Gatenby RA and Cheng JD. Evolution of tumor invasiveness: the adaptive tumor microenvironment landscape model. Cancer Res. 2011; 71(20):6327-6337.
51. Li A, Ma Y, Yu X, Mort RL, Lindsay CR, Stevenson D, Strathdee D, Insall RH, Chernoff J, Snapper SB, Jackson IJ, Larue L, Sansom OJ and Machesky LM. Rac1 Drives Melanoblast Organization during Mouse Development by Orchestrating Pseudopod- Driven Motility and Cell-Cycle Progression. Dev Cell. 2011; 21(4):722-734.
52. Tobin NP, Sims AH, Lundgren KL, Lehn S and Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer. 2011; 11:417.
53. Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW and Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011; 13(3):303-309.
54. Walker SR, Nelson EA, Zou L, Chaudhury M, Signoretti S, Richardson A and Frank DA. Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol Cancer Res. 2009; 7(6):966-976.
55. Yue P and Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009; 18(1):45-56.
56. Raptis L, Arulanandam R, Geletu M and Turkson J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp Cell Res. 2011; 317(13):1787-1795.
57. Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, Morse MA, Clay TM and Lyerly HK. HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011; 71(13):4380-4391.
58. Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ and Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005; 33(20):e175.
59. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B and Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003; 31(4):e15.
60. Johnson WE, Li C and Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8(1):118-127.
61. Sims AH, Smethurst GJ, Hey Y, Okoniewski MJ, Pepper SD, Howell A, Miller CJ and Clarke RB. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets - improving meta-analysis and prediction of prognosis. BMC Med Genomics. 2008; 1:42.