
INTRODUCTION
The introduction of personalized therapy transformed the care of selected cancer patients: detection of critical cancer gene somatic mutations in clinical tumor samples better defines patient diagnosis, prognosis and more importantly indicates highly efficient targeted therapies with both health and economic benefits [1].
According to the approved antineoplastic targeted drugs, the mutational status of EGFR, KRAS, NRAS, BRAF, PIK3CA genes is routinely requested by the oncologist for the clinical management of patients with non-small cell lung carcinomas (NSCLC) [2–4], colorectal carcinomas (CRC) [5, 6], melanomas [7], breast carcinomas [8, 9]. In particular EGFR inhibitors for EGFR-mutant NSCLC and the mutation-selective RAF and MEK inhibitors for BRAF-mutant melanoma [10] are examples of successfully targeted therapy selection. Further, the presence of mutations in RAS family oncogenes is associated with a lack of response to targeted therapy: lung and colorectal carcinomas characterized by KRAS mutations and KRAS and NRAS mutations, respectively are unresponsive to treatment with anti-EGFR agents [6, 11].
The routine clinical testing of such alterations faces several challenges. First, routine tumor specimens are usually formalin-fixed and paraffin-embedded (FFPE) and formalin fixation effects are major problems in molecular diagnostics. Formalin induces chemical cross-links to proteins, RNA, and DNA molecules, with concomitant fragmentation of DNA [12, 13] and random nucleotide base changes, which can lead to false-positive results [14]. For these reasons, clinical mutation detection PCR-based assays should include two independent amplifications of DNA extracted from FFPE samples in order to ensure accurate results, as suggested by molecular testing guidelines [15, 16].
Second, when offering optimal patient care, applied technologies should allow fast implementation of available assays in order to respond to new targets development, clinical trials testing, and new rules imposed by drug control agencies. As recently shown in metastatic colorectal cancer treatment, 17% of the KRAS exon 2 wild type patients, who do not respond to anti-EGFR treatment, harbor mutations in KRAS exon 3 and 4 and NRAS exon 2, 3 and 4 [6]. This evidence was rapidly followed by a directive of the European Medicines Agency (EMA) that restricted the use of panitumumab (Vectibix®) and cetuximab (Erbitux®) to patients with KRAS and NRAS (exon 2, 3 and 4) wild type metastatic colorectal cancer [17].
Third, an increasing need is determining the status of multiple clinically relevant genes in single sample-derived tumor. In fact, sequential testing using single gene analysis may be time consuming and need further invasive biopsy procedures, which could be avoided by testing for all informative markers in parallel when the first diagnosis is made [16, 18].
Herein we report the development and validation of a mass spectrometry multiplexed genotyping platform named PentaPanel, that comprises all the criteria defining a molecular diagnostic test: high performance in sensitivity, specificity, spectrum of detected mutations, and turnaround time.
Our approach is designed to concurrently detect 221 recurrent somatic point mutations in the 5 most relevant genes to solid tumors targeted therapies, EGFR, KRAS, NRAS, BRAF, and PIK3CA, applicable to 10 patients in the same run with a 2 day turnaround time.
Results of the PentaPanel genotyping application to the routine molecular pathology diagnostics of 1025 cases at our Institution are also reported.
RESULTS
PentaPanel platform design and characteristics: bidirectional analysis for detection of somatic mutations
We successfully designed 80 multiplexed assays in order to analyze single nucleotide polymorphisms (SNPs) in 56 hotspots of KRAS, NRAS, BRAF, EGFR and PIK3CA. In particular for 24 sites of EGFR, KRAS, NRAS, and BRAF, which are mutated at high frequency as reported by the online Catalogue Of Somatic Mutations in Cancer (COSMIC), we included both duplicate amplifications, in separate wells, and bidirectional single base pair extensions (FIG. 1). Overall, the total 80 assays, variously combined in 8 wells depending on their extension product masses, can simultaneously detect the presence/absence of 221 mutations of KRAS, NRAS, BRAF, EGFR and PIK3CA (FIG. 2).
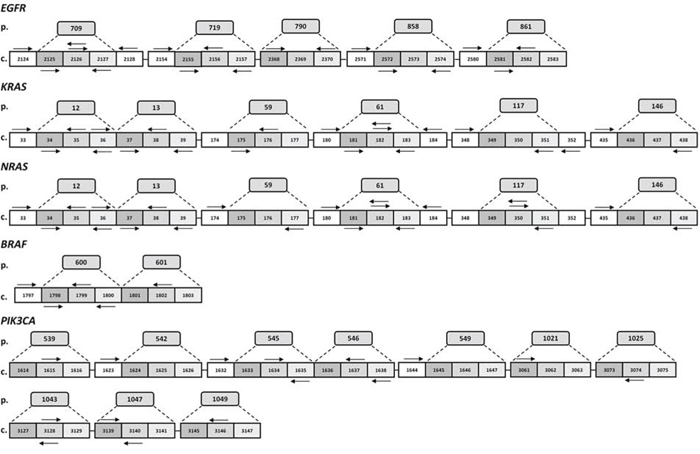
Figure 1: Schematic representation of PentaPanel platform. Hotspot aminoacids (p.) and corresponding coding codons (c.) are schematically represented by boxes for each of the five genes, EGFR, KRAS, NRAS, BRAF, and PIK3CA. Arrows show extension primers adjacent to the targeted mutation sites. Most frequently mutated nucleotides are analyzed by bidirectional approach.

Figure 2: KRAS, NRAS, BRAF, EGFR and PIK3CA detectable mutations by PentaPanel platform. Comprehensive list of the 221 mutations covered by PentaPanel in KRAS A. NRAS B. EGFR C. BRAF D. and PIK3CA E. genes; for each hotspot codon both amino acid and nucleotide changes are shown.
Notably, the PentaPanel allowed targeted therapy selection genotyping of 10 patients in the same run with a practical turnaround time of 2 working days, including data analysis, evaluation and reporting (7 hours first day-1 hour hands-on; 2 hours second day -15 min hands-on, 30–60 min for analysis and report generation).
Our PentaPanel analysis was performed using 40ng (5ng/well) of genomic DNA template, although we showed that it is possible to get successful analysis using a minimum amount of 8 ng DNA (1ng/well). The minimum amount was based on validation studies performed on dilution series of FFPE-derived DNA with known mutations: mutation frequency of a sample with high (100 ng) DNA was equivalent to that with low (1ng) DNA (FIG. 3A).
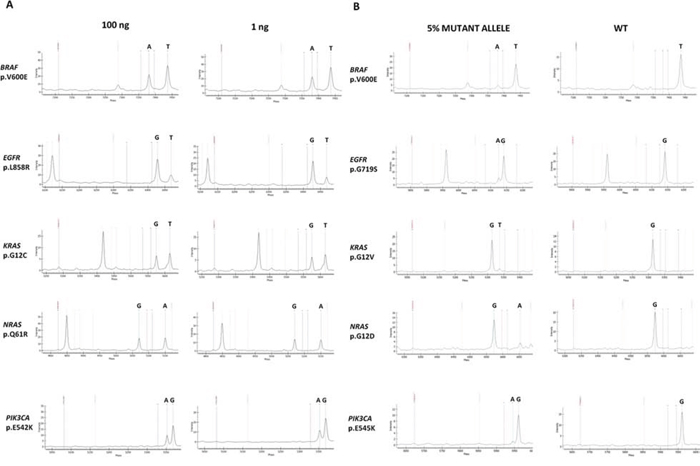
Figure 3: PentaPanel performance. A. Representative spectra of samples with high (100 ng) or low (1ng) DNA template input showing similar frequency of the detected mutations; B. Representative spectra of the sensitivity study showing positive calls for mutations at 5% dilutions.
To determine the analytical sensitivity of the platform we tested serial dilutions of genomic DNA extracted from FFPE samples with known mutant allele frequencies (20%, 10%, 5%, and 2.5%); we analyzed at least one representative mutation in each of the 8 wells: BRAF p.V600E, KRAS p.G12D, KRAS p.G12V, NRAS p.G12D, NRAS p.Q61K, EGFR p.G719S, EGFR p.T790M, EGFR p.L858R, PIK3CA p.E542K, PIK3CA p.E545K mutations. For BRAF p.V600E, KRAS p.G12D, KRAS p.G12V, NRAS p.Q61K, and PIK3CA p.E542K mutations commercially pre-designed reference standards were also utilized.
All the tested mutations were reproducibly (3 replicates) detected at a dilution of 5% (FIG. 3B). For KRAS p.G12V and EGFR p.G719S the detection limit was even lower, 2.5%. Same results were obtained using both FFPE-derived DNA and reference standards.
The reproducibility of the PentaPanel was also evaluated by comparing the results at the two independent laboratory sites and different users: the agreement was 100% for both mutation-positive and wild type specimens.
Importantly, for the first time our assays design allowed the bidirectional analysis of 24 highly frequently mutated sites, meaning that it comprised two separate assays assessing the presence/absence of the mutation in both the forward and the reverse strands. This strategy was critical for the detection of complex mutations such as dinucleotide substitutions occurring in both BRAF and KRAS genes: these mutations when tested by a unidirectional assay were missed as shown in FIG. 4. Moreover, by using the bidirectional approach, each result at a specific site was confirmed by two independent assays (in separated wells of the same run) as recommended by international guidelines [15, 16].
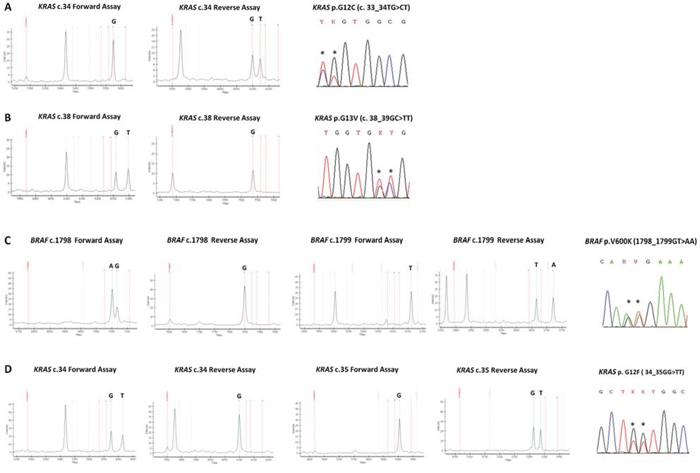
Figure 4: Spectra of bidirectional assays in representative complex mutations. A. KRAS nucleotide c.34, forward assay is WT and reverse assay shows a mutation call (G > T); KRAS p.G12C (c.33_34TG > CT) mutation, as shown by Sanger sequencing. Mutation not yet listed in the COSMIC database B. KRAS nucleotide c.38, forward assay shows a mutation call (G > T) and reverse assay is WT; KRAS p.G13V (c.38_39GC > TT) mutation, as shown by Sanger sequencing C. BRAF nucleotide c.1798, forward assay shows a mutation call (G > A) and reverse assay is WT; BRAF nucleotide c.1799, forward assay is WT and reverse assay shows a mutation call (T > A); BRAF p.V600K (c.1798_1799GT > AA) mutation, as shown by Sanger sequencing. D. KRAS nucleotide c.34, forward assay shows a mutation call (G > T) and reverse assay is WT; KRAS nucleotide c.35, forward assay is WT and reverse assay shows a mutation call (G > T); KRAS p.G12F (c.34_35GG > TT) mutation, as shown by Sanger sequencing. By using a single assay approach all these mutations could be missed, with false negative results.
PentaPanel platform performance and validation
For this study a total of 1025 clinical tumor specimens derived from 6 different cancer types, for which mutation analysis of selected genes had been requested as a part of clinical care, were analyzed using the PentaPanel platform. The samples included adenocarcinomas (n = 783) of the lung, colon, pancreas, and biliary tract, melanomas (n = 62), and breast carcinomas (n = 180). Of the total 1025 specimens, 910 derived from FFPE tissues, 73 were obtained from fresh frozen tissue and 42 were cytological stained smears. Tumor content exceeded 50% in all samples, as estimated by pathological review.
For PentaPanel platform validation, Sanger sequencing was performed for all the study specimens, as the common reference method for detecting somatic mutations in tumors [15]. In particular by Sanger sequencing we analyzed: EGFR (exons 18, 20, and 21) in lung adenocarcinomas and breast carcinomas; KRAS (exons 2, 3, and 4) in colon, lung, pancreas, biliary tract adenocarcinomas; BRAF (exon 15) in colon, lung, pancreas, biliary tract adenocarcinomas, and melanomas; NRAS (exons 2, 3, and 4) in colon adenocarcinomas, and melanomas; PIK3CA (exons 9 and 20) in colon adenocarcinomas and breast carcinomas. EGFR exon 19 deletions were analyzed by sizing assay in lung adenocarcinomas and breast carcinomas.
In five cases (2 lung adenocarcinomas, 1 breast carcinoma, and 2 pancreatic adenocarcinomas, all FFPE tissues), DNA quality was very poor and neither the PentaPanel platform nor the Sanger sequencing protocols were successful in obtaining PCR amplificates; 1020 specimens were then suitable for mutation analysis with an overall success rate of 99.5% (1020/1025) of the samples being profiled. This remarkably high genotype success rate is essentially attributable to the PentaPanel platform: in fact, the number of non-analyzable (NA) samples for Sanger sequencing was 28 (all FFPE tissues), with a success rate of 97.3% (997/1025). Overall, we observed a reduction in the number of NA samples, with 2% of samples rescued by PentaPanel approach versus conventional sequencing. Moreover, by PentaPanel we were able to successfully analyze all types of specimens, including cytological stained smears, as shown in Table 1, for which the low DNA yield is always a limiting factor.
Table 1: FFPE, fresh frozen, and cytological specimens successfully analyzed by PentaPanel
Cancer Type |
Tested samples |
FFPE samples |
Fresh/Frozen samples |
Cytologic samples |
Mutated samples |
Colorectal |
386 |
346 |
40 |
− |
245 (63%) |
Lung |
316 |
261 |
13 |
42 |
158 (50%) |
Melanoma |
62 |
55 |
7 |
− |
44 (71%) |
Breast |
180 |
177 |
3 |
− |
48 (26%) |
Pancreatic |
65 |
55 |
10 |
− |
53 (81%) |
Biliary Tract |
11 |
11 |
− |
− |
4 (36%) |
Total |
1020 |
905 |
73 |
42 |
552 |
Concordance between PentaPanel platform and Sanger sequencing was determined using data from samples that were analyzed by both methods (i.e. we excluded the 23 samples that failed by Sanger sequencing). The overall percentage agreement between the two methods was 97.8% (975/997). In particular 22 samples, which were wild type for all the studied genes by Sanger sequencing, resulted mutated by PentaPanel: twenty were mutated in KRAS codon 12 and two in BRAF codon 600. The 22 negative cases for KRAS and BRAF mutations by standard sequencing were re-evaluated by the more sensitive LNA-PCR/Sequencing method: all the mutations detected by the PentaPanel were confirmed (concordance rate 100%) and no false positives were observed. Thus, we conclude that all the mutations could be detected by PentaPanel with a specificity of 100%.
Frequency and distribution of somatic mutations
In our series of 1020 patients, a total of 605 somatic non-synonymous mutations in the 5 genes were found: 569 single nucleotide variations and 36 complex mutations were observed, as summarized in Table 2. Remarkably, among the total 30 BRAF mutations found in melanomas (30/62, 48%), we detected 6 complex mutations (1 p.V600E2, c.1799_1800TG > AA and 5 pV600K, c.1798_1799TG > AA), 20% (6/30) of the BRAF mutations, showing that the frequency of these events is not low.
Table 2: Single nucleotide and complex mutations identified in the current study
Amino acid |
DNA |
Cancer type |
|||
Lung |
Colorectal |
Melanoma |
Others |
||
KRAS |
108 |
197 |
0 |
57 |
|
p.G12A |
c.35G > C |
10 |
9 |
− |
1 |
p.G12C |
c.34G > T |
35 |
12 |
− |
1 |
p.G12C |
c.33_34TG > CT |
− |
2 |
− |
− |
p.G12D |
c.35G > A |
24 |
65 |
− |
21 |
p.G12F |
c.34_35GG > TT |
3 |
1 |
− |
− |
p.G12H |
c.34_35GG > CA |
− |
− |
− |
1 |
p.G12R |
c.34G > C |
1 |
1 |
− |
5 |
p.G12S |
c.34G > A |
3 |
9 |
− |
− |
p.G12V |
c.35G > T |
18 |
42 |
0 |
23 |
p.G13C |
c.37G > T |
2 |
2 |
− |
1 |
p.G13D |
c.38G > A |
1 |
24 |
− |
− |
p.G13R |
c.37G > C |
1 |
− |
− |
− |
p.G13V |
c.38G > T |
1 |
1 |
− |
− |
p.A59T |
c.175G > A |
− |
1 |
− |
1 |
p.Q61H |
c.183A > C |
5 |
6 |
− |
3 |
p.Q61L |
c.182A > T |
1 |
1 |
− |
− |
p.Q61P |
c.182A > C |
− |
1 |
− |
− |
p.Q61K |
c.180_181TC > AA |
1 |
2 |
− |
− |
p.Q61R |
c.182A > G |
2 |
− |
− |
− |
p.K117R |
c.350 > G |
− |
1 |
− |
− |
p.K117N |
c.351G > C |
− |
3 |
− |
− |
p.A146P |
c.436G > C |
− |
3 |
− |
− |
p.A146T |
c.436G > A |
− |
10 |
− |
− |
p.A146V |
c.437C > T |
− |
1 |
− |
− |
NRAS |
0 |
14 |
14 |
0 |
|
p.G12A |
c.35G > C |
− |
− |
1 |
− |
p.G12C |
c.34G > T |
− |
1 |
− |
− |
p.G12D |
c.35G > A |
− |
6 |
− |
− |
p.G13R |
c.37G > C |
− |
− |
1 |
− |
p.G13V |
c.38G > T |
− |
1 |
− |
− |
p.Q61H |
c.183A > C |
− |
1 |
− |
− |
p.Q61K |
c.181C > A |
− |
3 |
4 |
− |
p.Q61L |
c.182A > T |
− |
1 |
4 |
− |
p.Q61R |
c.182A > G |
− |
1 |
4 |
− |
BRAF |
5 |
22 |
30 |
1 |
|
p.V600E |
c.1799T > A |
5 |
22 |
23 |
1 |
p.V600E2 |
c.1799_1800TG > AA |
− |
− |
1 |
− |
p.V600K |
c.1798_1799GT > AA |
− |
− |
5 |
− |
p.K601E |
c.1801A > G |
− |
− |
1 |
− |
EGFR |
41 |
0 |
0 |
0 |
|
p. E709-T710del insA |
c.2126_2128delAAA |
1 |
− |
− |
− |
p. E746-A750del |
c.2232_2249 > AAA |
14 |
− |
− |
− |
p. E746-T751del insA |
c.2237_2251del15 |
1 |
− |
− |
− |
p. E746-T751del insVA |
c.2237_2251 > TGG |
1 |
− |
− |
− |
p.delL747-A750insP |
c.2238_2248 > GC |
1 |
− |
− |
− |
p. E747-A751del |
c.2240_2254del15 |
1 |
− |
− |
− |
p. L747-S752del insS |
c.2240_2257del18 |
1 |
− |
− |
− |
p.G719C |
c.2155G > T |
1 |
− |
− |
− |
p.T790M |
c.2369C > T |
2 |
− |
− |
− |
p.L858R |
c.2573T > G |
14 |
− |
− |
− |
p.L861Q |
c.2582T > A |
3 |
− |
− |
− |
p.L861R |
c.2582T > G |
1 |
− |
− |
− |
PIK3CA |
7 |
58 |
1 |
50 |
|
p.E542K |
c.1624G > A |
2 |
10 |
1 |
8 |
p.E545A |
c.1634A > C |
− |
1 |
− |
− |
p.E545G |
c.1634A > G |
− |
1 |
− |
1 |
p.E545D |
c.1635G > T |
− |
1 |
− |
− |
p.E545K |
c.1633G > A |
3 |
21 |
− |
10 |
p.E545Q |
c.1633G > C |
− |
2 |
− |
− |
p.Q546K |
c.1636C > A |
− |
1 |
− |
2 |
p.Q546E |
c.1636C > G |
− |
− |
− |
1 |
p.Q546P |
c.1637A > C |
− |
1 |
− |
− |
p.Q546R |
c.1637A > G |
− |
4 |
− |
− |
p.M1043I |
c.3129G > A |
− |
1 |
− |
− |
p.H1047L |
c.3140A > T |
− |
3 |
− |
6 |
p.H1047R |
c.3140A > G |
2 |
11 |
− |
22 |
p.G1049R |
c.3145G > C |
− |
1 |
− |
− |
In total, we detected 24 KRAS, 9 NRAS, 4 BRAF, 12 EGFR, and 14 PIK3CA different mutations. Notably, among the 24 KRAS different mutations, we reported the occurrence of novel complex mutation in two distinct samples of colon adenocarcinoma: it is a dinucleotide mutation c.33_34TG > CT p.G12C (FIG. 4A), not yet listed in the COSMIC database (COSMIC Release v73).
In our heterogeneous series, mutations most commonly occurred in the KRAS oncogene (362/605, 60%), followed by PIK3CA (116/605, 19%), BRAF (58/605, 10%), EGFR (41/604, 7%), and NRAS (28/605, 5%). Mutation occurrences in most genes had strong tendency toward mutual exclusivity, except for PIK3CA in colon, lung and pancreatic cancers that, on the opposite, tended toward co-occurrence: out of 67 PIK3CA mutations, 47 were co-occurring mutations (70%). In particular, 45 adenocarcinomas (41 colon, 2 lung, 2 pancreas) exhibited co-occurring KRAS and PIK3CA mutations; two colon adenocarcinomas had concomitant BRAF and PIK3CA mutations. A melanoma case had NRAS mutation co-occurring with PIK3CA mutation. The PIK3CA tendency toward co-occurrence was not observed in breast carcinomas: out of 47 PIK3CA mutations, only one case showed the presence of concomitant KRAS and PIK3CA mutations.
In colorectal carcinomas, 1 case presented concomitant KRAS and NRAS mutations and 2 cases had concomitant G12D and G13D KRAS mutations.
Overall, KRAS mutations were found in 195/386 colorectal cancers (50%), 108/316 lung cancers (34%), 52/65 pancreatic cancers (80%), 3/11 biliary tract cancers (27%), and 2/180 (1%) breast cancers; BRAF mutations were found in 30/62 melanomas (48%), 22/386 colorectal cancers (6%), 5/316 lung cancers (2%), and 1/65 pancreatic cancer (1%); NRAS mutations were found in 14/62 melanomas (23%), and 14/386 colorectal cancers (4%); PIK3CA mutations were found in 61/406 colorectal cancers (15%), 47/180 breast cancers (26%), 7/316 lung cancers (2%), 2/65 pancreatic cancer (3%), 1/62 melanomas (2%), and 1/11 biliary tract cancers (9%); EGFR mutations were found in 41/316 lung cancers (13%).
DISCUSSION
In this study we report the rapid development of a high throughput, cost-effective and simple strategy for the detection of clinically relevant mutations in a variety of solid tumors such as carcinomas of the lung, colon, breast, pancreas, biliary tract, and melanomas.
Given the increasingly critical role of molecular investigations in the management of cancer patients, there is an immediate need for robust, high-quality diagnostic tests. The complexities of NGS technologies and data analysis are slowing their wide spread availability in the molecular pathology laboratories, which must readily comply with regulatory, quality and professional standards [19–21]. While working to optimize and validate this revolutionary approach, diagnostics laboratories need a robust genotyping platform which can carry-on their daily routine testing.
The presented approach uses the MALDI-TOF mass spectrometry technology, associated with iPLEX assays, that allows analysis of multiple mutations in a single investigation [22]. This technology utilizes small (80 base pairs) PCR products which are optimal for amplification of FFPE DNA templates.
The PentaPanel platform assesses single nucleotide polymorphisms in 56 hotspots of the 5 most clinically relevant cancer genes, KRAS, NRAS, BRAF, EGFR and PIK3CA for a total of 221 detectable mutations. Indeed, the PentaPanel genotyping approach covers point mutations of the mandatory biomarkers for non-small cell lung carcinomas, colorectal carcinomas, melanomas, and satisfactorily meets the needs of a high quality throughput pathology laboratory, such as accuracy, sensitivity, hands-on time, and costs. This is a cost effective method, especially when complex testing of numerous mutations is requested: for example the RAS-testing (KRAS and NRAS mutations in exon 2, 3 and 4) in colorectal cancer performed by PentaPanel produced a significant reagent cost saving of 30% over real-time PCR or pyrosequencing analysis. Moreover, the high levels of multiplexing and automation of PentaPanel genotyping reduce personnel costs below those of other standard methods.
EGFR exon 19 deletions were not included in this platform and they were detected by sizing electrophoresis. Efficient approaches for point mutations analysis are generally not optimal for insertions/deletions [23]. In fact, investigating the entire mutation spectrum of deletions within the EGFR gene is feasible by this method but both a high number of assays and an increased amount of DNA sample would be required for the test.
The presented results highlight the PentaPanel advantages: it was successfully used to interrogate different DNAs isolated from routinely processed specimens (FFPE, frozen, and cytological samples); it required small amount (as low as 8ng) of DNA template for the assessment of multiple genes status simultaneously; it could genotype 10 patients in the same run with a practical turnaround time of 2 days, including data analysis and evaluation; and its hand-on timing easily allowed more than a run a day.
Importantly, the PentaPanel platform analyzed 24 highly frequently mutated sites bidirectionally, meaning that it comprised two separate assays assessing the presence of the mutation in both forward and reverse strands in separate wells. This is crucial for the detection of complex mutations such as dinucleotide substitutions occurring in both BRAF and KRAS genes.
Particularly, in melanomas the BRAF complex mutations were 20% of the total BRAF detected mutations (6/30: 1 p.V600E2 and 5 pV600K), revealing that such events are not rare and they must be taken into account when performing BRAF testing. The correct BRAF status identification is crucial since evidence of clinical benefit to vemurafenib treatment is accumulating for patients with mutations other than V600E [24]. In fact, the spectrum of tested mutations is another critical issue successfully addressed by the PentaPanel compared to the other targeted methods (real-time PCR-based methods), currently used for routine diagnostics. The real-time PCR assays are rapid and highly sensitive [15] but they often require a rather significant amount of DNA, not always available in limited biopsy/cytologic samples, and more importantly, underestimate not pre-designed mutations. Therefore, these tests might not cover all clinically relevant mutations [25–27], consequently excluding patients from the potential benefits of targeted therapy.
The PentaPanel bidirectionally strategy also allows to confirm each result at a specific site by two assays in different wells in the same run and no additional validation steps are needed. Notably, this approach markedly reduces the possibility of both false-negative and false-positive results: the presented data showed a concordance of 100% between PentaPanel and the Sanger sequencing integrated with the more sensitive LNA-PCR/Sequencing results. Indeed, we found that the PentaPanel achieves a specificity of 100% and no false positive mutation calls were observed in our series; the analytical sensitivity of detecting mutant alleles was as low as 5%.
The application of the PentaPanel to routine molecular analysis of 1025 samples showed a genotyping high success rate (99,5%), with a 2% rescue of successfully analyzed samples by this approach compared to Sanger sequencing.
In conclusion, this well established and robust high throughput technology was overall easy to be set up and rapidly introduced in the routine molecular diagnostics of our Institution; importantly the presented data support its valid clinical application, and compliance to molecular testing guidelines.
Although the mass spectrometry based genotyping will not be a definitive cancer diagnostics platform as being able to detect only targeted mutations, the PentaPanel can provide the immediate and accurate multiplex approach for clinically relevant gene mutation analysis in many solid tumors; moreover, its extreme flexibility and limited costs can allow both rapid implementation and application to newly identified biomarkers for target therapies selection, and its effectiveness across many diseases can be particularly relevant in multiple clinical trials, including the new basket trial approach, aiming to identify appropriate targeted drug combination strategies.
MATERIALS AND METHODS
Tumor samples
We retrospectively collected anonymized solid tumor samples from 1025 consecutive patients that had undergone routine diagnostic somatic mutation analysis at the San Raffaele Hospital Pathology Unit (Milan, Italy) between September 2013 and December 2014.
A comprehensive written informed consent was signed for the procedures (fine needle aspiration, biopsies and surgical resections) that produced the tissue samples and the diagnostic workup. All information regarding the human material was managed using anonymous numerical codes. Clinical data and follow up information were not used for this study. According to our country's legislation, since it was a retrospective study, with no direct patient involvement, the ethical approval and patients consent for the study were not required (Official Gazette No. 301 of December 30, 2014).
All samples were handled in compliance with the Helsinki declaration.
The study included 386 colon adenocarcinomas, 316 lung adenocarcinomas, 62 melanomas and 256 other malignancies (180 breast carcinomas, 65 pancreatic and 11 biliary tract adenocarcinomas). 910 samples derived from formalin-fixed, paraffin-embedded (FFPE) specimens, 73 were obtained from fresh frozen tissues (both biopsies or surgical specimens) and 42 were cytological stained smears.
All the diagnosis were confirmed by two pathologists (GA and DC) and tumor-rich areas (>50%) were selected in order to perform manual macrodissection prior DNA extraction. Coverslips were removed from stained cytological samples using xylene followed by hydration and air-drying.
For tissue blocks, both FFPE and frozen OCT-embedded, a variable number (5–10) of 5 μm unstained sections were prepared, depending on the tumor size.
For all samples total genomic DNA was isolated by automated extraction using the Magcore Nucleic Acid Extractor (RBC Bioscience, Taiwan) following the manufacturer's protocols. Quality and quantity of isolated DNA was assessed by a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Mutation analysis using sequenom massARRAY genotyping assays
Briefly, the Sequenom MassARRAY approach included designing multiplexed specific assays that use primers flanking the mutation site and extension primers that bind adjacent to the mutation site. After the amplification of the region of interest, a primer extension reaction was carried out. The extension reaction included sequence-specific hybridization and sequence-dependent single base termination (iPLEX) that generated different products for the mutated and wild type alleles, each with its unique mass, then identified using mass spectrometry.
Assay amplification primers and extension oligos were designed using MassARRAY Assay Design software v. 4.0 (Sequenom, USA) with a maximum of 12 multiplexed assays per well. In particular, 8-well multiplexed assays were designed to assess single nucleotide polymorphisms (SNPs) involving 56 hotspots of the KRAS, NRAS, BRAF, EGFR and PIK3CA genes (PentaPanel). For 24 sites of EGFR, KRAS, NRAS, and BRAF, which are mutated at high frequency as reported by the Catalogue Of Somatic Mutations In Cancer (COSMIC), we included both duplicate amplifications, in separate wells, and bidirectional single base pair extension (Figure 1); the PentaPanel comprised a total of 80 assays, 48 of them present in both forward and reverse direction, covering 221 mutations in the 5 genes (Figure 2).
Amplification, nucleotide dephosphorylation and single base primer extension by i-PLEX® Gold chemistry were performed according to the manufacturer's protocol (Sequenom). A MALDI-TOF mass spectrometer (MassARRAY Compact, Sequenom) was used to resolve extension products; data analysis was performed utilizing MassARRAY Typer Analyzer software (Sequenom).
Dilution series of FFPE-derived DNA with known mutations in a background of wild type FFPE-derived DNA (20%, 10%, 5%, 2.5% mutant to wild type DNA ratios) together with commercially available pre-designed reference standards (Horizon Diagnostics, UK) were used to detect the allelic analytical sensitivity of the assays.
The PentaPanel platform was then used to analyze the KRAS, NRAS, BRAF, EGFR and PIK3CA genes status in the previously described series of 1025 cases, utilizing 40ng of genomic DNA template for all the samples. Using a 96-well plate it was possible to investigate 10 cases in the same run, including both positive and negative controls, with a turnaround time of 2 days.
SPSS version 17.0 was used for statistical analysis.
Mutation analysis by standard Sanger sequencing
We analyzed the entire coding region of exon 2, exon 3, and exon 4 of both KRAS and NRAS, exon 15 of BRAF, exon 18, exon 20, exon 21 of EGFR, exon 9 and exon 20 of PIK3CA. PCR primers sequences and thermal conditions are summarized in supplemental data (Supplementary Table 1).
Amplified products were purified using MinElute PCR Purification Kit (Qiagen Gmbh, Germany) and sequenced in both directions using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, USA), according to the manufacturer's protocol, on an ABI Prism 3130 Genetic Analyze running ABI Prism DNA Sequence Analysis Software. To increase the sensitivity of standard Sanger sequencing we modified the standard PCR sequencing assay by the addition of 20 pmol of Locked Nucleid Acid (LNA) probe (Exiqon, Denmark) complementary to the wild type sequence of the KRAS (codons 12–13) and BRAF (codons 598–601), as previously described [28].
Length analysis of fluorescently labelled PCR products for EGFR deletions in exon 19
Deletions in exon 19 of EGFR gene were determined by fragment length analysis after PCR amplification with the use of FAM-labeled primer as previously described [3]. Separation was done with a four-color laser-induced fluorescence capillary electrophoresis system (ABI Prism 3130 Genetic Analyzer, Applied Biosystems). The collected data were evaluated with the Gene Scan Analysis Software. All mutant were confirmed by DNA direct sequencing.
CONFLICTS OF INTEREST
Paola Bartocci and Gianluca Barbieri are employees of Diatech Pharmacogenetics. The remaining authors declare no conflicts of interest.
REFERENCES
1. Gonzalez de Castro D, Clarke PA, Al-Lazikani B, Workman P. Personalized cancer medicine: molecular diagnostics, predictive biomarkers, and drug resistance. Clin Pharmacol Ther. 2013; 93:252–259.
2. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361:947–957.
3. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009; 361:958–967.
4. Dienstmann R, Martinez P, Felip E. Personalizing therapy with targeted agents in non-small cell lung cancer. Oncotarget. 2011; 2:165–177.
5. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008; 359:1757–1765.
6. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013; 369:1023–1034.
7. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–2516.
8. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH, Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30:777–782.
9. Abramson VG, Cooper Lloyd M, Ballinger T, Sanders ME, Du L, Lai D, Su Z, Mayer I, Levy M, LaFrance DR, Vnencak-Jones CL, Shyr Y, Dahlman KB, Pao W, Arteaga CL. Characterization of breast cancers with PI3K mutations in an academic practice setting using SNaPshot profiling. Breast Cancer Res Treat. 2014; 145:389–399.
10. Arnedos M, Vielh P, Soria JC, Andre F. The genetic complexity of common cancers and the promise of personalized medicine: is there any hope? J Pathol. 2014; 232:274–282.
11. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008; 9:962–972.
12. Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002; 161:1961–1971.
13. Wong SQ, Li J, Tan AY, Vedururu R, Pang JM, Do H, Ellul J, Doig K, Bell A, MacArthur GA, Fox SB, Thomas DM, Fellowes A, Parisot JP, Dobrovic A. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 2014; 7:23.
14. Williams C, Ponten F, Moberg C, Soderkvist P, Uhlen M, Ponten J, Sitbon G, Lundeberg J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999; 155:1467–1471.
15. Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M College of American Pathologists International Association for the Study of Lung C and Association for Molecular P. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013; 15:415–453.
16. Cree IA, Deans Z, Ligtenberg MJ, Normanno N, Edsjo A, Rouleau E, Sole F, Thunnissen E, Timens W, Schuuring E, Dequeker E, Murray S, Dietel M, Groenen P, Van Krieken JH European Society of Pathology Task Force on Quality Assurance in Molecular P, et al. Guidance for laboratories performing molecular pathology for cancer patients. J Clin Pathol. 2014; 67:923–931.
17. Boleij A, Tops BB, Rombout PD, Dequeker EM, Ligtenberg MJ, van Krieken JH, Initiative DR. RAS testing in metastatic colorectal cancer: excellent reproducibility amongst 17 Dutch pathology centers. Oncotarget. 2015.
18. Garraway LA. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013; 31:1806–1814.
19. Lohmann K, Klein C. Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics. 2014; 11:699–707.
20. Salto-Tellez M, Gonzalez de Castro D. Next-generation sequencing: a change of paradigm in molecular diagnostic validation. J Pathol. 2014; 234:5–10.
21. Heuckmann JM, Thomas RK. A new generation of cancer genome diagnostics for routine clinical use: overcoming the roadblocks to personalized cancer medicine. Ann Oncol. 2015.
22. Fumagalli D, Gavin PG, Taniyama Y, Kim SI, Choi HJ, Paik S, Pogue-Geile KL. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer. 2010; 10:101.
23. Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, Chung CH, Massion PP, Vnencak-Jones CL, Iafrate AJ, Pao W. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011; 13:74–84.
24. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, Garbe C, Testori A, Maio M, Lorigan P, Lebbe C, Jouary T, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014; 15:323–332.
25. Ihle MA, Fassunke J, Konig K, Grunewald I, Schlaak M, Kreuzberg N, Tietze L, Schildhaus HU, Buttner R, Merkelbach-Bruse S. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014; 14:13.
26. Qu K, Pan Q, Zhang X, Rodriguez L, Zhang K, Li H, Ho A, Sanders H, Sferruzza A, Cheng SM, Nguyen D, Jones D, Waldman F. Detection of BRAF V600 mutations in metastatic melanoma: comparison of the Cobas 4800 and Sanger sequencing assays. J Mol Diagn. 2013; 15:790–795.
27. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, Yao H, Lazar AJ, Aldape KD, Medeiros LJ, Luthra R. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013; 15:220–226.
28. Arcila M, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagn. 2011; 13:64–73.