
INTRODUCTION
Metastatic breast cancer (MBC) is an incurable disease with a 2- to 3-year median overall survival (OS) time [1, 2]. Although marked advances have been made in HER2-targeted therapies [3–6] and strategies to overcome endocrine resistance [7, 8], some patients exhibit refractory MBC, which do not respond to conventional treatments, and are therefore in need of new therapeutic strategies. When developing such strategies, it is important to realize that patients with MBC are a heterogeneous group, and thus the best strategies for treatment may differ between patients.
The results of next-generation sequencing (NGS) approaches in The Cancer Genome Atlas (TCGA) suggest that primary BCs are mutationally heterogeneous [9, 10]. This heterogeneity of MBCs and refractoriness to conventional treatments represent a significant treatment challenge, and there is a pressing need to understand better the biology of these aggressive cancers and to develop more effective therapies. Progress in cancer genomics has raised hopes of more precise identification of patients suitable for targeted therapies tailored to their genotype. However, there remains a feasibility issue before NGS can be used in making decisions about clinical treatment in daily clinical practice, especially for patients with refractory disease who have only limited options for cancer treatments.
The HER pathway plays a critical role in the pathogenesis of breast cancer, especially in patients with HER2 overexpression [11]. HER-2, together with HER-1, HER-3, and HER-4, is member of the human epidermal growth factor receptor family. HER-1, HER-3, and HER-4 can be activated by various ligands, and their activation triggers conformational rearrangement of the receptor molecules to allow homo- or heterodimerization [12]. Although wild-type HER2 overexpression occurs in 20–30% of breast cancers, Bose et al. estimated that about 1.6% of breast cancer patients possess a HER2 mutation [13]. They found that seven HER2 mutations activated the protein, as reflected in its enzyme activity, and downstream HER2 signaling in mouse xenografts. This observation led to a trial of neratinib, a panHER TKI for patients with refractory HER2 positive BC [13].
Considering this role of HER2 mutations as a driver oncogene, we developed a targeted NGS platform, which we call CancerSCAN™, using the 83 most representative genes (Supplemental Table 1). We used the HiSeq 2500 sequencing platform (Illumina, USA) to investigate the mutational profile of tissues from patients with refractory MBC whose disease progressed after conventional treatments. We focused specifically on HER2 mutations. We also performed whole-transcriptome sequencing (RNA-Seq) to evaluate the HER pathway in patients with refractory MBC.
RESULTS
Patients' clinicopathological features (Table 1)
From March 2013 to Nov 2014, 36 patients with MBC that was refractory to conventional treatments were enrolled, and fresh frozen tumour tissues were collected from metastatic sites for targeted sequencing, CancerSCAN™ analysis, and RNA sequencing. Thirty-six tumour samples were analyzed; two patients were excluded because of failure of quality control (QC). The median age at diagnosis of the 36 patients was 45 years (range 26–64). Fourteen patients were hormone receptor (HR)-positive (defined as ER and/or PgR positive). Thirteen BC patients were HER2 positive and 11 patients were triple negative. The median number of chemotherapy regimens before biopsy was 5 (range 3–8) (Table 1). The median follow-up duration from the date of biopsy was 15 months (range 5–20). The median OS time from distant metastasis to death (mOS) was 42.7 months (range 10–81). Among 14 HER2-positive patients, median OS of the patients with HER2 mutation was 20.2 months, which was much shorter than those without HER2 mutation (48.6 months).
Table 1: Clinicopathologic features of 36 refractory MBC patients
NGS using cancerSCAN™
The mean coverage of 36 patients was 941.3× with 99.0% over 100× (Supplemental Table 2). Figure 1 shows the mutational profiles of the 36 patients with refractory MBC. Thirty-five of the 36 patients (97.2%) harboured at least one genetic alteration including copy number variations. A heat-map of mutations of the 83 genes in samples from the 36 patients is shown in Figure 2. A total of 140 genetic alterations were detected in the samples from the 36 patients. Genes in which somatic alterations were detected frequently included TP53 (27 cases, 75%), ERBB2 (17 cases, 47%), PIK3CA (12 cases, 33%), NF1 (five cases, 14%), FGFR (three cases, 8%), PTEN (three cases, 8%), ARID1 (three cases, 8%), RB1 (two cases, 6%), PIK3R1 (one case, 3%), CDH1 (one case, 3%), and AKT1 (one case, 3%), as shown in Figure 1.
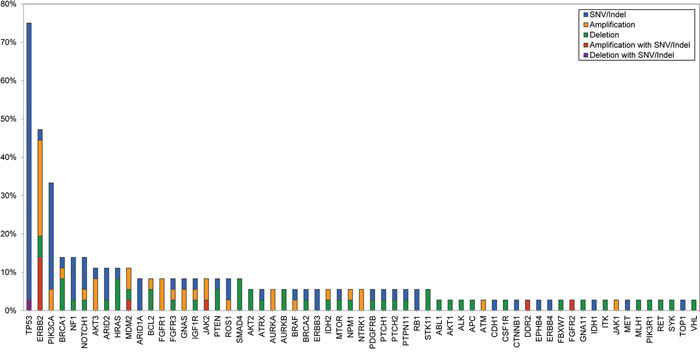
Figure 1: Mutational profile of 36 refractory MBC patients.
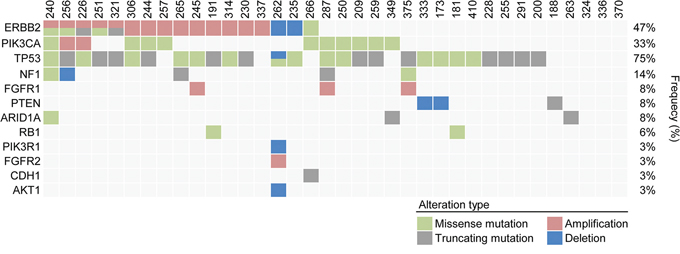
Figure 2: Heat map of patients with genetic alterations of 83 genes among 36 refractory MBC patients.
HER2 mutational status of the 36 refractory MBC patients (Table 2, Figure 2)
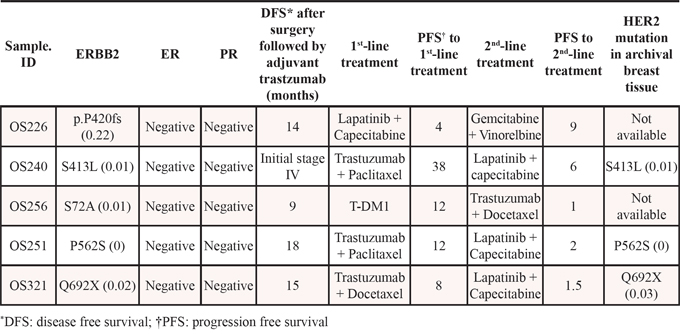
HER2 mutations were found in six of the 36 patients (16.7%, Figure 2). One patient was ER-positive, and the other five patients were HER2-positive. Figure 3 shows the gene maps of the HER2 mutational status of the six patients who harboured HER2 mutations along with the TCGA results. HER2 mutations in three patients were in the receptor ligand domain (RLD). In one patient, the mutation was in the protein tyrosine kinase domain (PTKD), and in the other HER2-negative patient, the mutation was in the growth factor receptor domain (GFRD) (Figure 3). To confirm the mutations in primary tissue, we also genotyped the available archival primary tissue for two patients. We identified one mutation (S413L) with a similar allele frequency to that of metastatic tissue, but none was detected for the other patient.
Table 2: Fourteen HER2-positive and one HER2-negative with HER2 mutation patients
Sample.ID |
ERBB2 |
ERBB2 |
Mutation |
Amplification value |
GRB2 mRNA |
HER2–3 pathway |
OS191 |
Amp. (14.53) |
No |
14.53 |
0.674 |
0.447 |
|
OS226 |
p.P420fs (0.22) |
Amp. (13.99) |
Yes |
13.99 |
0.144 |
−0.591 |
OS230 |
Amp. (22.38) |
No |
22.38 |
0.897 |
0.178 |
|
OS240 |
S413L (0.01) |
Amp. (16.39) |
Yes |
16.39 |
0.431 |
−0.136 |
OS244 |
Amp. (12.36) |
No |
12.36 |
0.286 |
-0.037 |
|
OS245 |
Amp. (16.04) |
No |
16.04 |
N/A |
N/A |
|
OS256 |
S72A (0.01) |
Amp. (25.93) |
Yes |
25.93 |
N/A |
N/A |
OS257 |
Amp. (30.13) |
No |
30.13 |
1.716 |
−0.470 |
|
OS265 |
Amp. (14.09) |
No |
14.09 |
1.189 |
−0.156 |
|
OS306 |
Amp. (13.8) |
No |
13.8 |
N/A |
N/A |
|
OS251 |
P562S (0) |
Amp. (20) |
yes |
20 |
−0.024 |
−0.593 |
OS314 |
Amp. (9.52) |
No |
9.52 |
1.388 |
−0.485 |
|
OS321 |
Q692X (0.02) |
Amp. (5.39) |
Yes |
5.39 |
0.541 |
−0.206 |
OS337 |
Amp. (3.99) |
No |
3.99 |
0.301 |
0.040 |
|
OS266 |
L755S (0.19) |
None |
Yes |
2 |
0.144 |
−0.591 |
*NA: not available
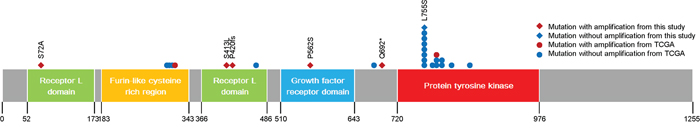
Figure 3: Gene map of ERBB2 mutations.
Comparison of HER2 mutations with TCGA data (Supplemental Table 3)
Supplemental Table 3 shows the six HER2 mutations found in our series compared with TCGA data. The VAF percentages were much lower in our series than in the TCGA dataset.
ERBB2 mutation validation by digital PCR (Table 3)
To confirm the low-frequency HER2 mutation, we performed digital PCR assays with negative controls. Among the five samples with HER2 mutations, all mutant alleles in the NGS were detected by digital PCR with similar allele frequency (Figure 4). In each negative control, the signals were much lower in the mutant alleles than in the positive samples (Table 3).
Table 3: Digital PCR results of HER2 mutations in five patients with HER2 mutation using NGS
Sample ID |
NGS |
dPCR* % (FAM/VIC) |
||
Amino acid change |
VAF% (Alt/Ref) |
Negative control |
Sample |
|
226 |
P420fs |
0.2172 (16/7365) |
0.0103 (3.752/36312) |
0.4523 (11.05/2442.6) |
240 |
S413L |
1.1954 (96/8668) |
0.0670 (35.173/52471) |
1.2859 (558.09/45731) |
256 |
S72A |
0.7457 (42/5590) |
0.0766 (17.506/22848) |
0.8343 (190.36/22186) |
251 |
P562S |
0.4345 (38/8475) |
0.0197 (7.409/37529) |
0.2603 (143.46/55095) |
321 |
Q692X |
1.6919 (38/2208) |
0.0023 (0.601/26319) |
1.2574 (433.51/34475) |
*dPCR: digital PCR
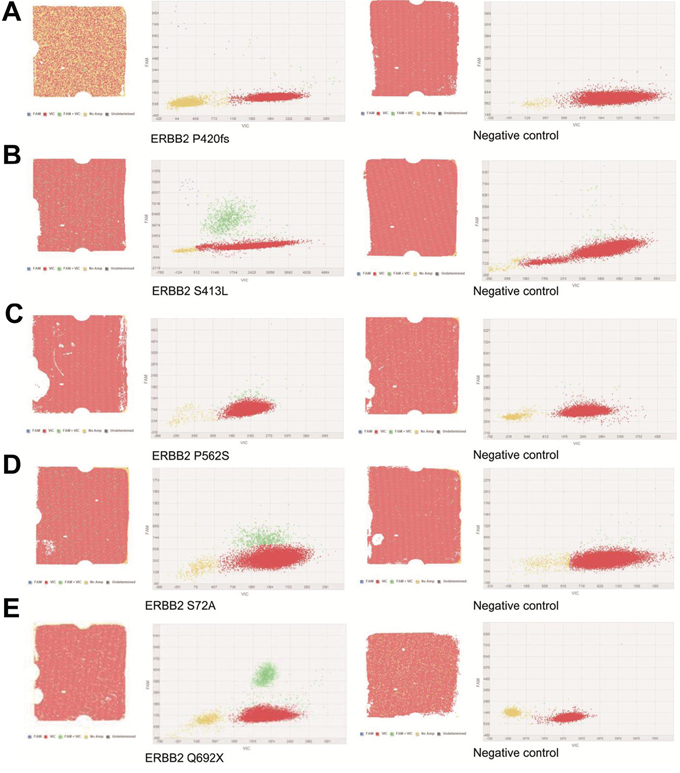
Figure 4: Digital PCR results of the patients who were HER2-positive and HER2 mutation. A. Patient 226. B. Patient 240. C. Patient 256. D. Patient 251. E. Patient 321.
HER2 pathway score analysis using RNA-Seq (Figure 5)
Standardized mRNA expression values of HER2 show a linear relationship with HER2 copy numbers. The mutation status of the HER2 gene did not alter the mRNA level expression of the HER2 gene in the patients (Figure 5A). Among the patients with an increased HER2 copy number, the average mRNA expression level of the HER2-HER3 pathway differed between the patients with a HER2 mutation and those with the wild-type HER2 (Figure 5B). This was related to down-regulation of several components in the pathway, such as GRB2, PIK3CB, and RAF1 (Supplemental Table 4). Growth Receptor Biding protein 2 (GRB2) was the most significantly down-regulated gene in the HER2-mutated samples. The treatment history of the patients and their GRB2 expression levels are summarized in Tables 2 and 4.
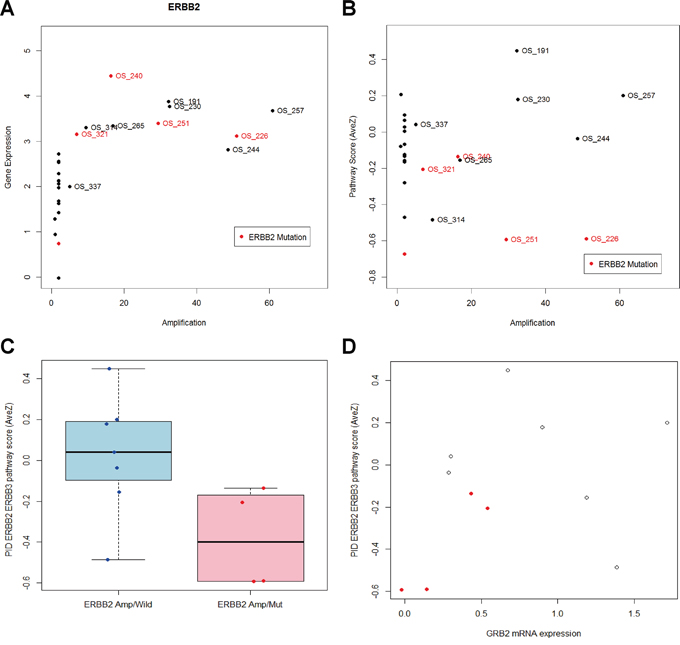
Figure 5: HER2 pathway analysis. A. ERBB2 copy number and gene expression of twenty seven metastatic breast cancer patients. ERBB2 gene expression is significantly higher in patients with increased copy number. Patients with ERBB2 mutation is red colored. B. Averaged mRNA expression of genes in the PID ERBB2 ERBB3 pathway is not associated with ERBB2 amplification. C. Among eleven patients with ERBB2 amplification, pathway score is significantly different between patients with ERBB2 mutation and patients without ERBB2 mutation (P-value = 0.0413). D. ERBB2 amplified and mutated samples yield less expression of GRB2 and pathway level gene expression than ERBB2 amplified and wild.
Table 4: HER2 mutation status of five HER2-positive MBC patients
Clinical and mutational characteristics of the six patients with HER2 mutations
To evaluate the role of HER2 mutations in HER2-positive refractory MBC, clinical outcomes and the disease course of the five HER2-positive patients with HER2 mutations were investigated. As shown in Figures 1 and 2 and Table 2, these five patients had HER2-positive BC with HER2 amplification as well as HER2 mutations. However, these mutations did not involve the same site: three were missense and two were truncation mutations, and there was no mutation in the PTKD (Figure 3). Interestingly, no patient was both ER-positive and HER2-positive. All five patients with HER2 mutations in the HER2-amplified BC were HR-negative. Most importantly, four of the five patients did not show any durable clinical response to trastuzumab- and/or lapatinib-containing chemotherapies for more than 12 months (Table 4). They were HER2-positive patients with MBC whose disease was refractory to targeted therapies including trastuzumab-DM1.
DISCUSSION
Our genomic study results show a higher incidence (16.7%) of HER2 mutations in patients with refractory MBCs than in other series, which is merely 1.6% [13, 16]. This difference may relate to this population of refractory patients or to the deep sequencing method used. Most importantly, our study suggests that HER2 mutations in patients with HER2-amplified BC may contribute to resistance to anti-HER2-directed therapy with chemotherapies, even though only low frequencies of the mutations were found (Table 2), which imply HER2 mutation could be one of the predictive markers of HER2 directed therapy and new resistance mechanism of HER2 pathway in breast cancer. Furthermore, this finding was supported by GRB2 downregulation, which is down-stream of HER2 pathway even though it did not validated CancerSCAN™ (GRB2 is not included in CancerSCAN™ panel). Lim et al., reported down-regulation of GRB2 in heregulin-stimulated-HER2-overexpressing breast cancer cells that lead to reduced proliferation through inactivation of the Akt pathway [14]. It is unclear whether the down-regulation of GRB2 is a consequence of HER2 mutation. It is also possible that the down-regulation of GRB2 might be caused by drug exposure. Decreased expression of GRB2 in lapatinib-treated BC cell lines has been reported [15]. As most (or all) of the HER2-amplified patients in our study had been exposed to HER2-targeting drugs, it is also possible that down-regulation of the GRB2 gene and HER2 pathways might be caused by the treatment, coincidently overlapped with the mutational status of the patient sample. Further functional study of these mutations using an in-vitro model may reveal this relationship. Insufficient amount of GBR2 product makes it hard to deliver an HER2-triggered oncogenic signal, therefore patients may be less dependent on HER2-targeted drugs. In spite of this limitation, we decided to try to improve the accuracy of the method to identify HER2 mutations. To exclude false-positive results and to evaluate whether these HER2 mutations are recurrent, we performed digital PCR with the same tumour tissues and CancerSCAN™ from archival breast tumour tissues. Our results suggest that there may be a patient population that receives little of no benefit from HER2-targeted therapies even though they have HER2-overexpressing BCs. HER2 mutations may be a main reason for this primary resistance to HER2-directed therapies.
Several mechanisms are thought to be responsible for resistance to HER2-targeted therapies. Several mechanisms of resistance to both trastuzumab and lapatinib have been identified in preclinical studies [17–23]. However, few of these have been validated prospectively in the clinic [6, 24, 25]. Unfortunately, the identification of a robust clinical or molecular predictor of trastuzumab benefit, including HER2 itself, has proven challenging [26–29]. There are no reports that HER2 somatic mutations play a role in primary resistance in HER2-amplified BCs, especially for patients with heavily pretreated disease, and there is insufficient scientific evidence to support this rationale.
Our results suggest that HER2 mutations may be useful as a predictive marker to identify which patients will not benefit from HER2-directed therapy. Emerging clinical data suggest that combinations of therapies targeting the HER2-signaling network at multiple points early in the natural history of HER2-positive breast cancer can abrogate drug resistance. For this reason, double-blockades may be regarded as an alternative for overcoming resistance. Obviously, patients with HER2 mutations will not be offered this therapeutic option because of the low activity of HER2 pathway in this population. Paradoxically, HER2 mutations are not considered to be the main driver of genetic alterations to override tumour aggressiveness, unlike in other malignancies, such as NSCLC and colorectal carcinomas [30–34]. HER2 somatic mutations have been shown recently to drive tumorigenesis in HER2-negative breast cancers [13]. We found a HER2 mutation in one HER2-negative patient (1/36, 2.6%) in the same site found in a previous report [13]. However, the other HER2 mutations were found mainly in patients with HER2-amplification (Table 2, Figure 3). Additionally, most of these mutations were recurrent mutations, although they only occurred in low frequencies. This result may derive from the deep targeted sequencing of CancerSCAN™, which may explain why deep targeted sequencing is needed for all exon sites as well as hot spots.
What remains a challenge is determining the precise resistance mechanism(s) in this particular type of patient. Answering this question will lead to the development of individualized and effective therapies for refractory MBC. This will require commitment to in-depth functional studies and molecular analysis of the tumors. Alternatively, the increasing use of preoperative therapy should provide a clinical research platform for the prediction of the response to combinations of anti-HER2 agents with cytotoxic chemotherapy to stratify patients for the following treatment(s) to improve therapeutic outcomes for patients with refractory HER2-positive BC. These residual cancers may be interrogated with open-ended molecular approaches to select those patients who might need to avoid conventional HER2-directed therapies.
In conclusion, HER2 mutations in patients with refractory BC may help explain the resistance to conventional HER2-directed therapies in HER2-positive BC.
MATERIALS AND METHODS
Patients
We conducted a prospective study at the Samsung Medical Center (SMC) from March 2013 to November 2014. The patients with breast cancer who progressed after or were refractory to conventional treatments, including endocrine therapies or chemotherapies such as anthracycline- and taxane-containing regimens, were recruited into this trial. Patients with HER2-positive BC had received at least two HER2-targeted therapies for 2 years before they enrolled in this trial.
Genomic DNA extraction and quality measurement
Genomic DNA was extracted from fresh frozen tumour tissue and matched normal blood specimens using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Genomic DNA quality and quantity were analyzed using a NanoDrop 8000 UV-Vis spectrometer (Thermo Scientific Inc., Willington, DE, USA), Qubit 2.0 Fluorometer (Life Technologies Inc., Grand Island, NY, USA), and 2200 TapeStation instrument (Agilent Technologies, Santa Clara, CA, USA).
Sequencing using a customized cancer panel (CancerSCAN™)
Genomic DNA (250 ng) from each tissue was sheared in a Covaris S220 ultrasonicator (Covaris, Woburn MA, USA) and used for the construction of a library using CancerSCAN™ probes and a SureSelect XT reagent kit, HSQ (Agilent Technologies) according to the manufacturer's protocol. This panel is designed to enrich exons of 83 genes (Supplemental Table 1), covering 366.2kb of the human genome. After enriched exome libraries were multiplexed, the libraries were sequenced on a HiSeq 2500 sequencing platform (Illumina). Briefly, a paired-end DNA sequencing library was prepared through gDNA shearing, end-repair, A-tailing, paired-end adaptor ligation, and amplification. After hybridization of the library with bait sequences for 27 hours, the captured library was purified and amplified with an index barcode tag, and the library quality and quantity were assessed. Sequencing of the exome library was performed using the 100 bp paired-end mode of the TruSeq Rapid PE Cluster Kit and TruSeq Rapid SBS Kit (Illumina).
Variants detection using the customized cancer panel (CancerSCAN™)
Sequence reads were mapped to the human genome (hg19) using Burrows-Wheeler Aligner (BWA) [35]. Duplicate read removal was performed using Picard and SAMtools [36]. Local alignment was optimized using the Genome Analysis Toolkit (GATK) [37]. Variant calling was done only in regions targeted in CancerSCAN™. To detect single nucleotide variants, we integrated the results of three kinds of variant caller, which increased the sensitivity [38–40]. We used Pindel to detect indels [41]. Copy number variations were calculated for targeted regions by dividing the read depth per exon by the estimated normal reads per exon using an in-house reference. Among the variants obtained from the tumour tissue, germ-line events were filtered using the results of the matched blood sample.
Gene expression and pathway analysis
FASTQ files from RNA sequencing of the SMC's sample from MBC patients were mapped to the human genome reference (hg19) using the bow-tie method [42]. TopHat was used to generate read counts per gene [43]. We normalized each patient's gene expression data to publically available breast cancer data. Gene expression data from the TCGA BRCA project (tumour = 526, normal = 61) were used for normalization. The statistical algorithm COMBAT was applied to reduce the platform and batch effects on data analysis [44]. After reducing the batch effect, we standardize each patients' gene expression value using the mean and standard deviation for gene expression obtained from normal breast tissue. An individualized pathway alteration score was obtained using the IPAS method [45].
Bioinformatic analysis
All statistical analyses were performed by the Biostatistics and Clinical Epidemiology Center in our institute. We implemented the method found in the R “compound.Cox” package.
REMARK guidelines
In reporting our study, we have adhered to the guidelines of the important 2005 methodological paper entitled “Reporting recommendations for tumor marker prognostic studies (REMARK guidelines)” [46, 47].
Digital PCR (polymerase chain reaction)
Digital PCR was performed using a QuantStudio 3D Digital PCR System platform comprising a Gene Amp 9700 PCR machine including a chip adapter kit, an automatic chip loader, and the QuantStudio 3D Instrument (Life Technologies, Inc.). The assay IDs of the primers and TaqMan probes are listed in Supplemental Table 5. We prepared 18 reaction mixtures containing 9 ul of two-fold QuantStudio 3D Digital PCR Master Mix (Life Technologies, Inc.), 0.9 ul of 20-fold TaqMan Assay by Design primer-probe mix, 2 ul diluted gDNA (100 ng/l), and 6.1 ul nuclease-free water (Biosesang, Korea). We loaded 14.5 ul of the reaction mixture onto a QuantStudio 3D Digital PCR 20K Chip (Life Technologies, Inc.) using an automatic chip loader according to the manufactures' instructions. Loaded chips underwent amplification in the Gene Amp 9700 PCR System under the following conditions: 96°C for 10 min, 42 cycles at 60°C for 2 min and at 98°C for 30 s, followed by a final extension step at 60°C for 2 min. After amplification, the chips were imaged on the QuantStudio 3D Instrument, which assesses raw data and calculates the estimated concentration of the nucleic acid sequence targeted by the FAM and VIC dye-labelled probes according to the Poisson distribution. The resulting data are reported in copies/ul along with the results of the quality assessment. For deeper analysis of the obtained chip data, QuantStudio 3D Analysis Suite Cloud Software (Life Technologies, Inc.) was used for relative and quantitative data analysis.
ACKNOWLEDGMENTS
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : HI13C2096). This work was also supported by the Samsung Biomedical Research Institute (grant number SMO1131841).
CONFLICTS OF INTEREST
The authors have declared that no conflict of interest exists.
Authors' contributions
YHP, YHI, HTS, and WYP conceived the project ideas. HTS and TJA analysed the NGS data. HHJ and JYK analysed clinical data supervision from YHP. KJA and KHP analysed the transcriptome data under the supervision of YLC, KA, and WYP. JSA, YHP, and YHI provided patient-related materials or clinical data used in this study. Statistical analyses were performed by KHP and HTS in consultation with TJA. Histopathological analyses were performed by IGD. YHP provided overall supervision for the project and wrote the manuscript with HTS and KHP and TJA, with input from HHJ and JYK.
REFERENCES
1. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics. CA Cancer J Clin. 2014; 64:252–271.
2. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013; 49:1374–1403.
3. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987; 235:177–182.
4. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007; 357:39–51.
5. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344:783–792.
6. Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012; 366:109–119.
7. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366:520–529.
8. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015; 16:25–35.
9. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512:155–160.
10. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
11. Monteiro Ide P, Madureira P, de Vasconscelos A, Pozza DH, de Mello RA. Targeting HER family in HER2-positive metastatic breast cancer: potential biomarkers and novel targeted therapies. Pharmacogenomics. 2015; 16:257–271.
12. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005; 5:341–354.
13. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3:224–237.
14. Lim SJ, Lopez-Berestein G, Hung MC, Lupu R, Tari AM. Grb2 downregulation leads to Akt inactivation in heregulin-stimulated and ErbB2-overexpressing breast cancer cells. Oncogene. 2000; 19:6271–6276.
15. Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, Gilmer TM. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther. 2007; 6:1629–1640.
16. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
17. Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, Arribas J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. Embo J. 2006; 25:3234–3244.
18. Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, Di Cosimo S, Matias-Guiu X, Ramon y Cajal S, Arribas J, Baselga J. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007; 99:628–638.
19. Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD, Sanchez G, Guzman M, Parra JL, Ellis C, Gagnon R, Koehler M, Gomez H, et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010; 16:2688–2695.
20. Xia W, Liu Z, Zong R, Liu L, Zhao S, Bacus SS, Mao Y, He J, Wulfkuhle JD, Petricoin EF, 3rd, Osada T, Yang XY, Hartman ZC, et al. Truncated ErbB2 expressed in tumor cell nuclei contributes to acquired therapeutic resistance to ErbB2 kinase inhibitors. Mol Cancer Ther. 2011; 10:1367–1374.
21. Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006; 13:221–232.
22. Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009; 8:2152–2162.
23. Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, Arteaga CL. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007; 13:4909–4919.
24. Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, Bianchi G, Cortes J, McNally VA, Ross GA, Fumoleau P, Gianni L. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010; 28:1138–1144.
25. Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, Lluch A, Staroslawska E, de la Haba-Rodriguez J, Im SA, Pedrini JL, Poirier B, Morandi P, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012; 13:25–32.
26. Perez EA, Dueck AC, McCullough AE, Chen B, Geiger XJ, Jenkins RB, Lingle WL, Davidson NE, Martino S, Kaufman PA, Kutteh LA, Sledge GW, Harris LN, et al. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 trial. J Clin Oncol. 2013; 31:2115–2122.
27. Perez EA, Reinholz MM, Hillman DW, Tenner KS, Schroeder MJ, Davidson NE, Martino S, Sledge GW, Harris LN, Gralow JR, Dueck AC, Ketterling RP, Ingle JN, et al. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J Clin Oncol. 2010; 28:4307–4315.
28. Moreno-Aspitia A, Hillman DW, Dyar SH, Tenner KS, Gralow J, Kaufman PA, Davidson NE, Lafky JM, Reinholz MM, Lingle WL, Kutteh LA, Carney WP, Dueck AC, et al. Soluble human epidermal growth factor receptor 2 (HER2) levels in patients with HER2-positive breast cancer receiving chemotherapy with or without trastuzumab: results from North Central Cancer Treatment Group adjuvant trial N9831. Cancer. 2013; 119:2675–2682.
29. Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med. 2008; 358:1409–1411.
30. Lee JW, Soung YH, Seo SH, Kim SY, Park CH, Wang YP, Park K, Nam SW, Park WS, Kim SH, Lee JY, Yoo NJ, Lee SH. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006; 12:57–61.
31. Buttitta F, Barassi F, Fresu G, Felicioni L, Chella A, Paolizzi D, Lattanzio G, Salvatore S, Camplese PP, Rosini S, Iarussi T, Mucilli F, Sacco R, et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer. 2006; 119:2586–2591.
32. Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, Wistuba II, Fong KM, Toyooka S, Shimizu N, Fujisawa T, Minna JD, Gazdar AF. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005; 65:1642–1646.
33. Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, Barthorpe A, Blow M, Brackenbury L, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004; 431:525–526.
34. Willmore-Payne C, Holden JA, Layfield LJ. Detection of epidermal growth factor receptor and human epidermal growth factor receptor 2 activating mutations in lung adenocarcinoma by high-resolution melting amplicon analysis: correlation with gene copy number, protein expression, and hormone receptor expression. Hum Pathol. 2006; 37:755–763.
35. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595.
36. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079.
37. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303.
38. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213–219.
39. Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012; 40:11189–11201.
40. Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011; 39:e132.
41. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009; 25:2865–2871.
42. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25.
43. Pollier J, Rombauts S, Goossens A. Analysis of RNA-Seq data with TopHat and Cufflinks for genome-wide expression analysis of jasmonate-treated plants and plant cultures. Methods Mol Biol. 2013; 1011:305–315.
44. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8:118–127.
45. Ahn T, Lee E, Huh N, Park T. Personalized identification of altered pathways in cancer using accumulated normal tissue data. Bioinformatics. 2014; 30:i422–429.
46. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Reporting recommendations for tumor marker prognostic studies. J Clin Oncol. 2005; 23:9067–9072.
47. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat. 2006; 100:229–235.