
INTRODUCTION
An advanced viral vector gene delivery platform based upon a recombinant adenovirus serotype-5 (Ad5), referred to as Ad5 [E1-, E2b-], can be utilized as a vaccine and immunotherapy modality even in the presence of pre-existing immunity against adenovirus [1–10]. The platform consists of a replication defective Ad5 in which portions of the early 1 (E1), early 2 (E2b) and early 3 (E3) Ad5 gene regions have been deleted [11, 12]. The deletions in the E2b region, specifically the DNA polymerase and preterminal protein, have been reported to result in a dramatic decrease of late gene expression, such as Ad5 fiber [12], which results in a marked reduction in host inflammatory responses to the vector and the associated toxicity [13]. Human cells transfected with these Ad5 [E1-, E2b-] constructs were shown to express the encoded transgene(s) for prolonged times in vivo compared to other vector platforms [4, 5], as the lack of Ad5 late gene expression in the proprietary platform renders infected antigen-presenting cells (APCs) less vulnerable to anti-Ad5 immunity, and permits them to produce and express inserted transgenes for extended periods of time [14]. Administration of these vaccines resulted in specific immunization and immunotherapy against infectious diseases and cancers [1–10]. In a Phase I/II clinical trial, cohorts of patients with metastatic colorectal cancer (mCRC) were vaccinated with escalating doses of the Ad5 [E1-, E2b-] platform carrying a gene for carcinoembryonic antigen (CEA) [1, 10]. CEA represents an attractive target for immunotherapy since it is overexpressed in the majority of human carcinomas [15, 16]. Ad5 [E1-, E2b-]-CEA was well tolerated in mCRC patients and CEA-directed T-cell responses were induced in a dose-responsive manner [10]; no significant changes in Treg:Teffector cell ratios were noted in this trial [1]. Patients in this study exhibited evidence of a favorable survival probability, with all 25 patients treated at least two times with Ad5 [E1-, E2b-]-CEA exhibiting a 12-month overall survival probability of 48%, with a mean overall survival of 11 months [1, 10].
The phenotypic heterogeneity in terms of expression of different tumor-associated antigens (TAAs) in a given primary or metastatic tumor mass is a well-established phenomenon [17–21]. One can speculate that the use of an immunotherapeutic vaccine regimen targeting three distinct TAAs, each of which is widely expressed on the majority of human carcinomas, would be potentially therapeutically advantageous over the use of a vaccine targeting only one TAA. With the safety and immunogenicity of Ad5 [E1-, E2b-]-CEA established in patients as a single agent, we now investigate a multi-target approach. We previously reported that a human immunodeficiency virus (HIV) vaccine containing four adenovirus constructs expressing Gag, Pol, Nef or Env could elicit an immune response to all four antigens when given simultaneously, even in the presence of Ad5 immunity [3].
Brachyury is a member of the T-box family of transcription factors that play key roles during early development, mostly in the formation and differentiation of normal mesoderm, which is characterized by a highly conserved DNA binding domain designated as the T-box [22]. Recently, the epithelial-mesenchymal transition (EMT) has been recognized as a key step during the progression of primary tumors into a metastatic state, in which brachyury plays a crucial role [23–25]. Brachyury expression is undetectable or minimally expressed in most normal adult human tissues and is overexpressed in multiple human cancers [24]. In addition, expression of brachyury has been shown to be associated with poor prognosis of colorectal [26], lung [27], prostate [28], hepatocellular [29], and breast [30] carcinomas. Brachyury overexpression in human tumor cells has also been associated with drug resistance [31, 32]. Transcription factors have been considered “difficult to drug” due to their primary location in the nucleus and lack of a hydrophobic groove for drug attachment. Studies have shown, however, that brachyury-specific T cells can be generated both in vitro and in vivo and that these T cells have the ability to lyse human tumors endogenously expressing brachyury [22, 33, 34]. Patients generate brachyury-specific T cells post-vaccination using vaccines expressing CEA and prostate-specific antigen (PSA), indicating the potential immunogenicity of brachyury in humans [35, 36]. A recently completed Phase I study [37] with a recombinant Saccharomyces cerevisiae brachyury vaccine also revealed the generation of brachyury-specific T cells, thus providing further evidence of immunogenicity.
MUC1 (CD227) is a TAA that is overexpressed on a majority of human carcinomas and several hematologic malignancies [38–41]. MUC1 is normally expressed at the surface of glandular epithelial cells [42] and, in carcinomas, it is overexpressed and aberrantly hypoglycosylated [38, 42, 43]. Several clinical trials have been and are being performed to evaluate the use of MUC1 in immunotherapeutic vaccines [44–48]. Some of these trials have indicated that targeting MUC1 is safe and may provide survival benefit [45, 47, 49]. We have previously identified multiple enhancer agonist epitopes, several of which are in the MUC1 C-terminus region [50, 51]. This is potentially important because numerous studies [52–56] have demonstrated that the C-terminus of MUC1 has oncogenic potential, associates with poor prognosis and drug resistance, and induces “stemness” features in a range of human carcinomas. The human T-cell lines generated using these MUC1 agonist epitopes were more efficient than those generated with the corresponding native epitopes in terms of antigen-specific interferon (IFN)–γ production and lysis of tumor cells endogenously expressing native MUC1 [50, 51]. Therefore, we believe that MUC1 containing modified agonist epitopes has a greater potential as an immunogenic agent for vaccine development.
The Ad5 [E1-, E2b-]-CEA vector, which encoded the entire CEA sequence modified to express an enhancer T-cell epitope, has been described previously [1, 6, 10]. Studies were undertaken to determine whether recombinant Ad5 [E1-, E2b-]-MUC1 and Ad5 [E1-, E2b-]-brachyury constructs could be developed that have the ability to generate MUC1- and brachyury-specific T-cell responses. Ad5 [E1-, E2b-]-brachyury was constructed to encode the entire brachyury gene devoid of 25 amino acids involved in DNA binding and modified to express an enhancer T-cell epitope; Ad5 [E1-, E2b-]-MUC1 was constructed to encode the entire MUC1 transgene with eight of the agonist epitopes previously described above [50, 51], including those in the C-terminus region. One potential pitfall in the use of an admixture of three vectors, each containing a different TAA, is that there may be “antigenic competition” and one or two TAAs would become dominant as to greatly inhibit or eliminate the expression of another TAA. Studies were thus designed to determine whether a mixture of Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, and Ad5 [E1-, E2b-]-brachyury (designated Tri-Ad5) has the ability to generate human T-cell responses in vitro, and murine T-cell responses in vivo, to each of the TAA transgenes.
RESULTS
Recombinant Ad5 [E1-, E2b-]-CEA was generated and characterized as previously described [6]. Recombinant Ad5 [E1-, E2b-]-MUC1 and Ad5 [E1-, E2b-]-brachyury were generated as described in the Methods section. As seen in Figure 1A, Western blot analysis using an anti-brachyury–specific monoclonal antibody (MAb 54–1) [57] revealed brachyury expression when human dendritic cells (DCs) were infected with Ad5 [E1-, E2b-]-brachyury. An Ad5 [E1-, E2b-] vector devoid of any transgene (Ad5 [E1-, E2b-]-null) was used as a negative control and SW620 human colon carcinoma cells that endogenously express brachyury were used as a positive control. An anti-MUC1–specific MAb was used to detect the expression of MUC1 in Ad5 [E1-, E2b-]-MUC1–infected human DCs (Figure 1B). SW620 cells, which also express MUC1 endogenously, were used as a positive control. The difference in molecular weights seen in the human DCs versus the SW620 human carcinoma cells is most likely due to the differential glycosylation of the MUC1 protein. As has been previously shown by others [58–61], it would appear that MUC1-C is being expressed in the human DCs predominantly as the unglycosylated 17 or 15 kDa form and not the 25–20 glycosylated species. A Western blot of Ad5 [E1-, E2b-]-CEA–infected human cells is shown in Supplemental Figure 1. Human DCs infected with Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1 and Ad5 [E1-, E2b-]-null were analyzed for evidence of DC maturation versus uninfected human DCs. There were no differences between the Ad5 [E1-, E2b-]-null and the recombinant Ad5 [E1-, E2b-] vectors expressing the TAAs in that each slightly upregulated surface CD80 and CD83 expression and strongly upregulated HLA-DR surface expression (Supplemental Table 1); it is thus apparent that any changes in DC maturation are due to the Ad5 vector alone and not any TAA transgene insertion.
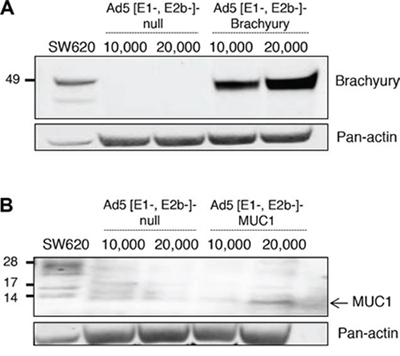
Figure 1: Expression of brachyury and MUC1 protein in human dendritic cells (DCs) infected with Ad5 [E1-, E2b-]- brachyury and Ad5 [E1-, E2b-]-MUC1. SW620 tumor cells were used as positive control. Actin was used as a loading control. A. Expression of brachyury was robust in DCs infected with Ad5 [E1-, E2b-]-brachyury. B. MUC1 expression was observed in human DCs infected with Ad5 [E1-, E2b-]-MUC1 vector as compared to DCs infected with Ad5 [E1-, E2b-]-null (no transgene).
We have previously reported on the generation of brachyury-, CEA-, and MUC1-specific human CD8+ T cells employing the corresponding peptide for each TAA [22, 50, 51, 62–64]. As shown in Table 1, Ad5 [E1-, E2b-]-null did not activate any of the T cells to produce IFN-γ. Ad5 [E1-, E2b-]-brachyury–infected DCs activated brachyury-specific T cells and not CEA-specific T cells (as a negative control). This demonstrates that the Ad5 [E1-, E2b-]-brachyury–infected DCs could process brachyury in a manner that generates brachyury–MHC Class I complexes capable of specific T-cell activation. Similarly, Ad5 [E1-, E2b-]-CEA–infected DCs specifically activated CEA-specific T cells but not MUC1-specific T-cell lines. Both Class I HLA-A2 and -A24 MUC1-specific T-cell lines have been previously generated [51] and the Ad5 [E1-, E2b-]-MUC1–infected DCs were capable of activating both of these T-cell lines but not the CEA-specific T-cell line (Table 1A). Human DCs were similarly infected with the Tri-Ad5 vector. As seen in Table 1B, T cells specific for CEA, MUC1, and brachyury were each activated to induce similar levels of IFN-γ as seen with the use of the individual Ad5 vectors.
Table 1A: Infection of human dendritic cells with recombinant adenovirus vectors encoding CEA, MUC1 or brachyury can activate antigen-specific T-cell lines
Dendritic cells (DCs) infected with |
Antigen-specific T-cell lines |
|||
CEA |
MUC1 |
MUC1 |
Brachyury |
|
Ad5 [E1-, E2b-]-null |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
Ad5 [E1-, E2b-]-brachyury |
<15.6 |
— |
— |
351.9 |
Ad5 [E1-, E2b-]-MUC1 |
<15.6 |
335.2 |
806.4 |
— |
Ad5 [E1-, E2b-]-CEA |
350.0 |
<15.6 |
<15.6 |
— |
Uninfected DCs |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
T cells only |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
Human DCs (6-day culture in IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) 2 × 104 cells/well in 0.5 ml of AIM-V) were infected with indicated adenovirus vectors at 20,000 multiplicity of infection (MOI). After 48 hours, DCs were washed and used for stimulation of human antigen-specific T cells. Results are expressed in pg/ml of IFN-γ per 1 × 105 T cells/ml. Numbers in bold indicate a significant enhancement of IFN-γ secretion compared to corresponding wells with uninfected DCs. [— indicates that the assay was not performed.]
Table 1B: Infection of human dendritic cells with Tri-Ad5 vectors encoding transgenes can activate antigen-specific T-cell lines to produce IFN-γ
Dendritic cells (DCs) infected with |
Antigen-specific T-cell lines |
|||
CEA |
MUC1 |
MUC1 |
Brachyury |
|
Tri-Ad5 |
480 |
236 |
763 |
496 |
Ad5 [E1, E2b]–null |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
Uninfected DCs |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
T cells only |
<15.6 |
<15.6 |
<15.6 |
<15.6 |
Human DCs (6-day culture in IL-4 and GM-CSF) from an HLA-A2 and -A24 donor were infected with Tri-Ad5 vector at 2 × 104/well (24-well plate) in 0.5 ml of AIM-V. Tri-Ad5 vectors were used at 20,000 MOI for 1 hour and then 1.5 ml of AIM-V were added to each well. Infected DCs were incubated for 48 hours and then washed and used for stimulation of human antigen-specific T cells. Results are expressed in pg of IFN-γ per 1 × 105 T cells/ml. Numbers in bold indicate a significant enhancement of IFN-γ secretion compared to corresponding wells with uninfected DCs.
Studies were then undertaken to determine whether simultaneous infection of human DCs with the CEA/MUC1/brachyury mixture of Tri-Ad5 could generate T-cell lines specific for all three TAAs. As seen in Table 2, when the T cells were activated by incubation with autologous B cells pulsed with the corresponding peptide, and not a control peptide, specific T-cell activation was observed. For example, the brachyury-specific T-cell line, generated by infecting human DCs with Tri-Ad5, was stimulated to produce IFN-γ when incubated with autologous DCs pulsed with brachyury peptide, but was not activated with the same autologous DCs pulsed with a CEA peptide. Similar results were seen with CEA and MUC1 T-cell lines generated with Tri-Ad5–infected DCs. These results indicate the lack of so-called “antigenic competition” in the in vitro use of Tri-Ad5.
Table 2: Infection of human dendritic cells with Tri-Ad5 can generate antigen-specific T cells to brachyury, MUC1 and CEA and produce IFN-γ when stimulated with autologous B cells pulsed with the corresponding peptides
Antigen-specific T-cell lines |
Peptides (10 μg/ml) |
|||
CEA |
MUC1 (A2) |
MUC1 (A24) |
Brachyury |
|
T-brachyury |
<15.6 |
— |
— |
243 |
T-MUC1 (A2) |
<15.6 |
174 |
— |
— |
T-MUC1 (A24) |
<15.6 |
— |
206 |
— |
T-CEA |
211 |
<15.6 |
— |
— |
Human dendritic cells (DCs) from a prostate cancer patient (6-day culture in IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) 2 × 104 cells/well in 0.5 ml of AIM-V) were infected with Tri-Ad5 at 20,000 MOI. After 48 hours, infected DCs were washed and used to generate specific cytotoxic T lymphocytes (CTLs) using autologous peripheral blood mononuclear cells (PBMCs) as effectors. Following 3 cycles of in vitro stimulations, autologous peptide-pulsed B cells were used as antigen-presenting cells. Results are expressed in pg/ml of IFN-γ. [— indicates that the assay was not performed.]
We then investigated whether brachyury-, MUC1-, and CEA-specific human T cells generated using DCs infected with Tri-Ad5 could lyse human carcinoma cells that endogenously express these TAAs. SW620 human colon carcinoma cells express all three TAAs and possess the HLA-A2 and -A24 Class I alleles. ASPC-1 human pancreatic carcinoma cells were used as a negative control since they express the three TAAs but in the context of HLA-A1 and -A26 molecules. The results (Table 3) demonstrated that Tri-Ad5–infected human DCs can generate T cells capable of lysing, in an MHC-restricted manner, human tumor cells that endogenously express brachyury, CEA, and MUC1.
Table 3: Infection of human DCs with Tri-Ad5 can generate brachyury-, MUC1- and CEA-specific CTLs that efficiently lyse tumor cells expressing all three antigens
Antigen-specific T-cell lines |
SW620 Brachyury+ MUC1+ CEA+ (HLA-A2+/A24+) |
ASPC-1 Brachyury+ MUC1+ CEA+ (HLA-A1+/A26+) |
T-brachyury |
64.4 (3.6) |
8.3 (2.7) |
T-MUC1 (P93L) |
28.5 (1.3) |
2.0 (1.6) |
T-MUC1 (C6A) |
49.3 (3.3) |
5.0 (1.8) |
T-CEA |
42.4 (3.7) |
4.3 (1.9) |
Human dendritic cells (DCs) were infected with Tri-Ad5 at 20, 000 MOI. Infected DCs were used to generate specific cytotoxic T lymphocytes (CTLs) using autologous peripheral blood monoclonal cells (PBMCs). Autologous DCs were used as antigen-presenting cells for three in vitro stimulations (IVS). Autologous peptide-pulsed B cells were used to re-stimulate antigen-specific CTLs for two additional IVS. The effector-to-target ratio used was 30:1; CTLs were used at IVS 5. Results are expressed in % specific lysis (SD).
Studies were next undertaken to determine whether Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-MUC1, and Ad5 [E1-, E2b-]-CEA could each generate TAA-specific T-cell responses in vivo, and whether the Tri-Ad5 mixture could generate comparable responses. C57Bl/6 mice (n = 5 per group) were injected subcutaneously (s.c.) three times at 2-week intervals with 1010 viral particles (VP) of Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, Ad5 [E1-, E2b-]-brachyury, or Tri-Ad5 (1:1:1 mixture of 1010 VP each). An additional group of mice (n = 5) received 3 × 1010 VP of Ad5 [E1-, E2b-]-null (an empty vector control). Two weeks after the final vaccination, splenocytes from vaccinated mice were stimulated with corresponding brachyury, CEA, or MUC1 peptide pools and analyzed for IFN-γ and IL-2 secreting cells by the enzyme-linked immunospot (ELISPOT) assay. Mice vaccinated with singular constructs or with Tri-Ad5 responded to brachyury, CEA, and MUC1 peptides, respectively, with significant increases in IFN-γ and IL-2 spot forming cells (SFCs) as compared to control mice (Figure 2A and 2B). There was no significant difference in the average number of IFN-γ SFCs in mice vaccinated with Ad5 [E1-, E2b-]-brachyury or Ad5 [E1-, E2b-]-CEA individually as compared with the Tri-Ad5 vaccine. There was a significant decrease in IFN-γ SFCs in mice treated with the Tri-Ad5 vaccine as compared to Ad5 [E1-, E2b-]-MUC1 alone, although the MUC1–specific immune response induced by Tri-Ad5 remained significantly elevated over control mice (p < 0.0001) (Figure 2A). IL-2 responses were similar in mice treated with Tri-Ad5 versus single vaccine constructs; moreover, there was a significant increase (p = 0.004) in CEA-specific IL-2 SFCs when mice were vaccinated with the Tri-Ad5 vaccine versus the Ad5 [E1-, E2b-]-CEA vaccine alone (Figure 2B). Splenocytes from mice vaccinated with empty vector did not respond to brachyury, CEA, or MUC1 peptide pools. In addition, there was no reactivity to control peptide pools (simian immunodeficiency virus (SIV)–Nef and SIV-Vif) in splenocytes from any of the vaccinated groups (data not shown). Taken together, these data indicate that combining Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-CEA, and Ad5 [E1-, E2b-]-MUC1 in a Tri-Ad5 vaccine admixture has the effect of generating antigen-specific IFN-γ– and IL-2–producing cells similar to that achieved when using each vaccine alone.
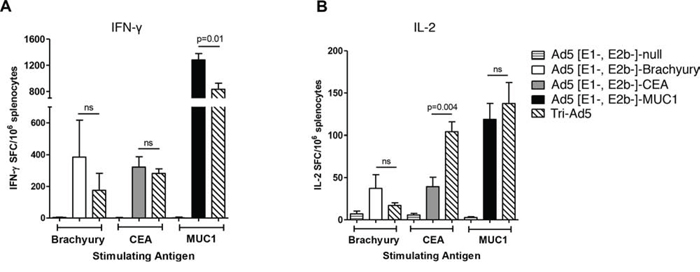
Figure 2: Analysis of IFN-γ− and IL-2−expressing splenocytes following vaccination of mice with Ad5 [E1-, E2b-]- brachyury, Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, Tri-Ad5, or Ad5 [E1-, E2b-]-null. C57Bl/6 mice (n = 5/group) were vaccinated three times at 2-week intervals with 1010 VP (viral particle) of Ad5 [E1-, E2b-]-brachyury (white bar), Ad5 [E1-, E2b-]-CEA (grey bar), Ad5 [E1-, E2b-]-MUC1 (black bar) or Tri-Ad5 (1:1:1 mixture of 1010 VP each of Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1) (diagonal hatched bar). Controls received 3 × 1010 VP of Ad5 [E1-, E2b-]-null (horizontal striped bar). Splenocytes were collected 14 days after the final vaccination and assessed for IFN-γ−secreting cells A. or IL-2-secreting cells B. by ELISPOT assay. For positive controls, splenocytes were exposed to Concanavalin A (Con A) (data not shown). Data reported as the number of spot forming cells (SFCs) per 106 splenocytes. The error bars depict the SEM. Significant differences (p < 0.05) between columns are reported in p-values, not significant = ns.
Intracellular accumulation of IFN-γ and TNF-α in CD8+ and CD4+ lymphocyte populations was also evaluated by flow cytometry using splenocytes from mice vaccinated with the adenovirus vectors and stimulated with overlapping pools of the respective synthetic peptides (Figure 3). No significant differences were observed between the IFN-γ production observed with CD8+ splenic lymphocytes isolated from mice vaccinated with Ad5 [E1-, E2b-]-brachyury compared with those isolated from mice vaccinated with Tri-Ad5 (Figure 3A). We did observe significant reductions between the CEA-specific and MUC1-specific IFN-γ accumulation in CD8+ splenocytes isolated from mice vaccinated with Tri-Ad5 as compared to single construct vaccinated mice, although the relative number of SFCs remained significantly elevated over controls (p < 0.0001) (Figure 3A). However, we found no significant differences in IFN-γ accumulation between CD4+ splenocytes isolated from each single construct vaccinated mice or Tri-Ad5 vaccinated mice (Figure 3B).
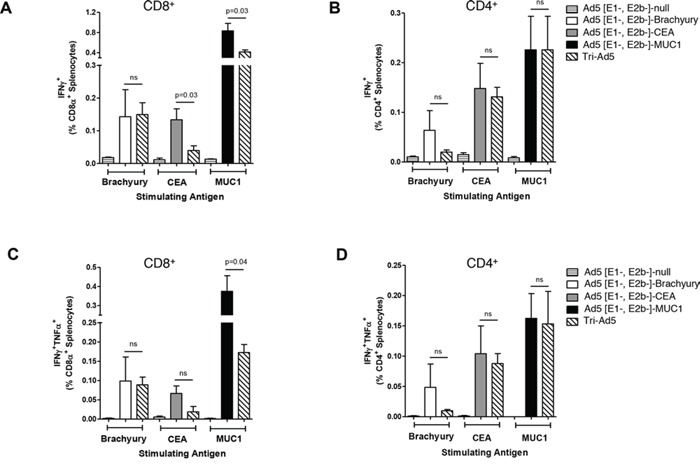
Figure 3: Analysis of CD8+ and CD4+ and multifunctional cellular populations following vaccination with Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, Tri-Ad5, or Ad5 [E1-, E2b-]-null. C57Bl/6 mice (n = 5/group) were vaccinated three times at 2-week intervals with 1010 VP (viral particle) of Ad5 [E1-, E2b-]-brachyury (white bar), Ad5 [E1-, E2b-]-CEA (grey bar), Ad5 [E1-, E2b-]-MUC1 (black bar) or Tri-Ad5 (1:1:1 mixture of 1010 VP (viral particle) each of Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1) (diagonal hatched bar). Controls received 3 × 1010 VP of Ad5 [E1-, E2b-]-null (horizontal striped bar). Splenocytes were collected 14 days after the final vaccination and were assessed by FACS for CD8α+ A. and CD4+ B. IFN-γ–secreting cells, or for CD8α+ C. and CD4+ D. cells secreting IFN-γ and TNF-α. For positive controls, splenocytes were exposed to Concanavalin A (Con A) (data not shown). The error bars depict the SEM. Significant differences (p < 0.05) between columns are reported in p-values, not significant = ns.
Peptide-stimulated splenocytes were also assessed by flow cytometry for the intracellular accumulation of both IFN-γ and TNF-α. We detected antigen-specific multifunctional CD8+ and CD4+ splenocytes in mice vaccinated with each single-antigen vector as well as with Tri-Ad5. When directly comparing the frequencies of dual-functional CD8+ and CD4+ splenocytes isolated from mice vaccinated with a single vector versus those from a mouse vaccinated with Tri-Ad5, we found very few differences (Figure 3C and 3D). We detected no significant differences between the dual-functional CD8+ splenocytes isolated from mice vaccinated with Ad5 [E1-, E2b-]-brachyury or Ad5 [E1-, E2b-]-CEA against the respective antigen as compared with those isolated from mice vaccinated with Tri-Ad5 (Figure 3C). We did observe a significant reduction in dual-functional CD8+ splenocytes from mice vaccinated with Ad5 [E1-, E2b-]-MUC1 compared with Tri-Ad5 (p = 0.04); this reduced frequency, however, was significantly elevated as compared to controls (p < 0.001). We found no significant differences in the frequencies of multifunctional CD4+ splenocytes isolated from each single construct or Tri-Ad5 vaccinated mice (Figure 3C).
To assess whether humoral responses were induced by Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, Ad5 [E1-, E2b-]-brachyury, or Tri-Ad5 vaccines, antigen-specific quantitative enzyme-linked immunosorbent assays (ELISAs) were employed. Significant and comparable antibody responses were detected against CEA in sera from mice vaccinated with Ad5 [E1-, E2b-]-CEA or Tri-Ad5 (Figure 4A). Antibodies against CEA were not detected in mice vaccinated with control vector (Figure 4A), or mice vaccinated with Ad5 [E1-, E2b-]-brachyury, or Ad5 [E1-, E2b-]-MUC1 (data not shown). Antigen-specific antibodies to brachyury or MUC1 were not detected in sera of mice vaccinated with Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-MUC1, or Tri-Ad5, respectively.
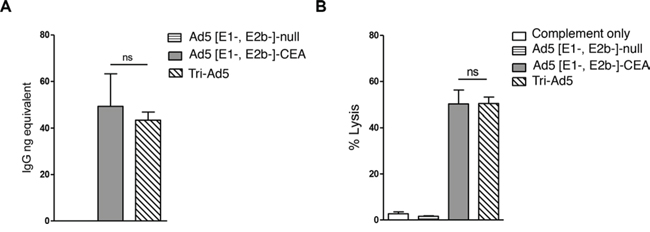
Figure 4: CEA antibody activity from sera from mice vaccinated with Ad5 [E1-, E2b-]-CEA or Tri-Ad5. CEA IgG levels in mice vaccinated three times with 1010 VP (viral particle) of Ad5 [E1-, E2b-]-CEA (grey bar), Tri-Ad5 (diagonal hatched bar) or 3 × 1010 VP of Ad5 [E1-, E2b-]-null (horizontal striped bar) were determined by ELISA A. Complement-dependent cytotoxicity (CDC) against MC38-CEA2 cells was performed B. The error bars depict the SEM. Significant differences (p < 0.05) between columns are reported in p-values, not significant = ns.
To determine the propensity of the CEA antibodies in the sera of Ad5 [E1-, E2b-]-CEA or Tri-Ad5 vaccinated mice to lyse tumor cells expressing CEA, we utilized a complement-dependent cytotoxicity (CDC) assay. Heat-inactivated sera from vaccinated mice were incubated with MC38-CEA2 tumor cells (murine CEA colon carcinoma cells transfected with human CEA), followed by rabbit sera as a source of complement. Lysis was determined by the release of lactate dehydrogenase (LDH) from MC38-CEA2 cells. There was significant lysis of MC38-CEA2 cells in sera from mice vaccinated with Tri-Ad5 or Ad5 [E1-, E2b-]-CEA, and this effect was similar between the two groups (Figure 4B).
Studies were then undertaken to determine whether the Tri-Ad5 vaccine regimen was as effective as the use of a single recombinant adenovirus construct in eliciting an anti-tumor effect. C57BL/6 mice (n = 7/group) were implanted s.c. with 1 × 106 MC38 cells expressing MUC1 (MC38-MUC1) in the left flank. Mice were vaccinated weekly with s.c. injections in the opposite flank using 1010 VP of Ad5 [E1-, E2b-]-MUC1 or Tri-Ad5, respectively. A control group of mice received 3 × 1010 VP of Ad5 [E1-, E2b-]-null (no transgene). Mice vaccinated with Ad5 [E1-, E2b-]-MUC1 or Tri-Ad5 had significantly smaller tumors than control mice on days 25 (p < 0.01) and 29 (p < 0.05) (Figure 5). There was no significant difference (p > 0.1) in anti-tumor effect for the groups of mice vaccinated with Ad5 [E1-, E2b-]-MUC1 or Tri-Ad5 at all time points.
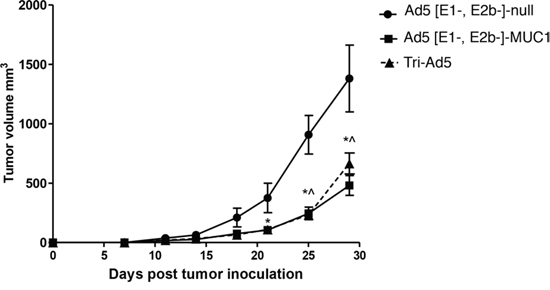
Figure 5: Comparison of immunotherapy of MUC1-expressing tumors using Ad5 [E1-, E2b-]-MUC1 vs. Tri-Ad5. C57Bl/6 mice (n = 7/group) were inoculated with 106 MC-38-MUC1 cells subcutaneously in the left flank. Mice were administered 1010 VP (viral particle) of Ad5 [E1-, E2b-]-MUC1 or Tri-Ad5 (1:1:1 mixture of 1010 VP each of Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, and Ad5 [E1-, E2b-]-brachyury, 3 × 1010 VP total). A control group of mice received 3 × 1010 VP of Ad5 [E1-, E2b-]-null (no transgene). Tumor growth was monitored and volumes calculated. (*) indicates days when Ad5 [E1-, E2b-]-MUC1 treated mice had significantly smaller (p < 0.05) tumors than control mice and (^) indicates days when Tri-Ad5–treated mice had significantly smaller (p < 0.05) tumors than control mice. There was no significant difference (p > 0.1) between Ad5 [E1-, E2b-]-MUC1 vs. Tri-Ad5–treated mice at any time point. Error bars represent the SEM.
DISCUSSION
Recent clinical studies have brought cancer immunotherapy into the area of the management of several tumor types. The U.S. Food and Drug Administration (FDA) approval of the checkpoint inhibitor anti-CTLA4 and the Provenge prostate cancer vaccine has been followed by recent FDA approvals of anti-PD-L1/PD-1 immune checkpoint inhibitors [65–67]. The phenomenon of tumor heterogeneity, including diversity of TAA expression, is well established. The previously described [16, 22–25, 31, 32, 52–56, 68–71] wide level expression of CEA, brachyury, and MUC1 in a range of human carcinomas, along with their diverse activity in human tumors, renders the simultaneous targeting of these three TAAs of potential clinical benefit. We thus set out to determine whether a mixture of three recombinant adenovirus-TAA vaccines would be as effective as the use of each one individually.
The generation and use in a preclinical model of the recombinant Ad5 [E1-, E2b-]-CEA vaccine have been described previously [6]. A clinical trial of the Ad5 [E1-, E2b-]-CEA (ETBX-011) vaccine in patients with metastatic colorectal cancer demonstrated the ability to administer multiple vaccinations to immunocompromised patients safely, and provided a favorable survival profile [1, 10]. The potential advantages of targeting brachyury and MUC1 (including the C-terminus of MUC1) have also been described previously [24, 34, 72], as has the use of vaccines that also contain enhancer agonist epitopes of these TAAs [50, 51, 62–64].
In the studies reported here, we demonstrate that multi-TAA targeted immunotherapy (Tri-Ad5), which consists of a mixture of three Ad5 vectors expressing different TAAs, is as efficient in the activation of human T cells as the use of each of the adenovirus vectors alone, with only minor differences. We analyzed nine different in vivo parameters via vaccinating mice with each of the Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, and Ad5 [E1-, E2b-]-brachyury vectors individually versus vaccination with Tri-Ad5 (Figures 2–5). Of the 21 assays performed, the only statistical differences observed were (a) an enhanced number of MUC1-specific splenocytes and CD8+ IFN-producing and multifunctional CD8+ T cells, and (b) more CEA-specific CD8+ IFN-producing T cells, in the mice vaccinated with one vector than in the Tri-Ad5 vaccinated mice. On the other hand, the Tri-Ad5 vaccinated mice produced more CEA-specific IL-2–producing cells than the Ad5 [E1-, E2b-]-CEA vaccinated mice. In the other 16/21 assays, however, there were no statistical differences in the results between the use of the individual vector versus the use of the Tri-Ad5 in terms of antigen-specific activation of (a) splenocytes for IFN-γ and IL-2 production, (b) CD8+ T cells for IFN-γ production, (c) CD4+ T cells for IFN-γ production, (d) multifunctional CD8+ T cells for IFN-γ and TNF-α production, (e) multifunctional CD4+ T cells for IFN-γ and TNF-α production, and (f) production of antigen-specific antibodies (Figures 2–5). There was also no difference in anti-tumor activity using the single vector Ad5 [E1-, E2b-]- MUC1 versus the Tri-Ad5 vaccine; while both vaccines did not eliminate the tumor, both vaccines reduced the tumor growth rate in a similar manner. It should be noted that the reduction of tumor growth rate has been seen with several forms of immunotherapy in clinical studies [66, 73, 74]. It should also be pointed out that while Tri-Ad5 was not as efficient in T-cell activation in some assays, we believe that the potential ability of the Tri-Ad5 platform to overcome the TAA heterogeneity that exists in human solid tumors far outweighs the relatively minor differences in potency of T-cell activation of Tri-Ad5 vs. individual vectors in some assays.
CEA, MUC1 and brachyury are all human TAAs and are not expressed in murine solid tumors. Moreover, human solid tumors are very heterogeneous with respect to expression of different TAAs. It would be extremely difficult to transfect a murine tumor cell line with all three transgenes to define the effect of vaccination of Tri-Ad5 vs. each vector alone. We chose to use the targeting of a murine tumor expressing MUC1 because the single Ad5 [E1-, E2b-]-MUC1 vector was more potent in some of the murine T-cell assays (Figures 2 and 3) compared to Tri-Ad5 than the CEA and brachyury vectors compared to Tri-Ad5. Thus this appeared to be the most stringent model to compare the Tri-Ad5 platform to a single vector platform. The studies reported herein were designed to provide the rationale for potential clinical studies as a vaccine immunotherapy, or use in combination with other therapeutics, using this novel adenovirus vaccine delivery platform (Ad5 [E1-, E2b-]) targeting a diverse range of TAA transgenes in the Tri-Ad5 regimen.
Several other vaccine platforms are currently being evaluated in clinical studies that target CEA, MUC1, or brachyury. We have previously shown [75] in preclinical studies that two diverse vaccine platforms targeting the same TAA can and will induce quite different T-cell populations, including diverse epitope specificity, cytokine production, and avidity. It is thus speculated that different vaccine platforms targeting the same TAAs can be used in tandem in clinical studies to obtain a more diverse T-cell population, resulting in enhanced anti-tumor activity.
While the checkpoint inhibitor antibodies have shown evidence of clinical activity in melanoma and squamous non-small cell lung cancer, clinical benefit in other cancer types has been observed in a minority of patients. For some tumor types, such as colorectal cancer and prostate cancer, the anti-PD-L1/PD-1 checkpoint inhibitors have shown little clinical activity. One hypothesis that has been put forth for the lack of PD-L1/PD-1 therapeutic activity in some patients is the lack of T-cell infiltrates in tumors. Consequently, if a vaccine targeting TAAs in the tumor would result in the presence of antigen-specific T cells in the tumor microenvironment, then a checkpoint inhibitor employed in combination or following vaccination would be able to “release the brakes” of the tumor-infiltrating anergized T cells leading to clinical effect.
MATERIALS AND METHODS
Viral construction
Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]- CEA and Ad5 [E1-, E2b-]-MUC1 were constructed and produced as previously described [6, 12]. Briefly, the transgenes were subcloned into the E1 region of the Ad5 [E1-, E2b-] vector using a homologous recombination-based approach. The replication deficient virus was propagated in the E.C7 packaging cell line, CsCl2 purified, and titered as previously described [12]. Viral infectious titer was determined as plaque-forming units (PFUs) on an E.C7 cell monolayer. The VP concentration was determined by sodium dodecyl sulfate (SDS) disruption and spectrophotometry at 260 nm and 280 nm (ViraQuest, North Liberty, IA). The CEA transgene also contains a modified CEA containing the highly immunogenic epitope CAP1–6D [63, 64].
The sequence encoding for the human brachyury protein (T, NM_003181.3) was modified by introducing the enhancer T-cell HLA-A2 epitope (WLLPGTSTV) [62] and removal of a 25 amino acid fragment involved in DNA binding. The resulting construct was subsequently subcloned into the Ad5 vector to generate the Ad5 [E1-, E2b-]-brachyury construct.
The MUC1 molecule consists of two regions: the N-terminus (MUC1-N), which is the large extracellular domain of MUC1, and the C-terminus (MUC1-C), which has three regions: a small extracellular domain, a single transmembrane domain, and a cytoplasmic tail [76]. The cytoplasmic tail contains sites for interaction with signaling proteins and has been shown to act as an oncogene and a driver of cancer motility, invasiveness and metastasis [56, 77]. For construction of the Ad5 [E1-, E2b-]-MUC1, the entire MUC1 transgene, including eight agonist epitopes previously described [50, 51], was subcloned into the Ad5 vector. The agonist epitopes included in the Ad5 [E1-, E2b-]-MUC1 vector bind to HLA-A2 (epitope P93L in the N-terminus, V1A and V2A in the VNTR region, and C1A, C2A and C3A in the C-terminus), HLA-A3 (epitope C5A), and HLA-A24 (epitope C6A in the C-terminus) [50, 51]. The Tri-Ad5 vaccine was produced by combining 1010 VP of Ad5 [E1-, E2b-]-brachyury, Ad5 [E1-, E2b-]-CEA and Ad5 [E1-, E2b-]-MUC1 at a ratio of 1:1:1 (3 × 1010 VP total). The vaccines used in this study are available to qualified researchers under a material transfer agreement.
Generation of human DCs from PBMCs
Dendritic cells were generated from the peripheral blood mononuclear cells (PBMCs) of a prostate cancer patient (HLA-A2+ and -A24+) enrolled in a clinical trial employing a PSA-TRICOM vaccine in combination with ipilimumab [35], using the method previously described [78]; using PBMCs from this patient post-vaccination, we were able to establish individual T-cell lines specific for CEA, MUC1, and brachyury. An Institutional Review Board of the National Institutes of Health (NIH) Clinical Center approved the procedures, and informed consent was obtained in accordance with the Declaration of Helsinki. Briefly, PBMCs were isolated using lymphocyte separation medium gradient (ICN Biochemicals, Aurora, VA), resuspended in AIM-V medium (Invitrogen, Carlsbad, CA) (2 × 107 cells) and allowed to adhere in a 6-well plate for 2 hours. Adherent cells were cultured for 5 days in AIM-V medium containing 100 ng/ml of recombinant human (rh) GM-CSF and 20 ng/ml of rhIL-4. The culture medium was replenished every 3 days.
Infection of human DCs with adenovirus vectors
Dendritic cells (2 × 105) in 1 ml of AIM-V medium were infected with adenovirus vectors (Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-MUC1, Ad5 [E1-, E2b-]-brachyury, and Ad5 [E1-, E2b-]-null at indicated multiplicity of infection (MOI of 10,000 or 20,000) for 1 hour in 6-well plates. AIM-V medium (4 ml) was then added to each well and incubated for an additional 2 days. To analyze the efficacy of transgene expression, DCs were harvested and analyzed using flow cytometry and Western blot. For phenotypic analysis, DCs were stained for the expression of CD80, CD83, CD86, CEA, and HLA-DR using BV421-conjugated anti-CD80, PerCP Cy5.5-conjugated anti-CD83, APC-Cy7-conjugated anti-HLA-DR, PE-conjugated anti-CD86, and FITC-conjugated anti-CEA. Antibodies for flow cytometry were purchased from BD Bioscience (San Jose, CA).
Generation of T-cell lines using adenovirus-infected DCs
A modification of the method described by Tsang et al. [79] was used to generate CEA-, MUC1- and brachyury-specific cytotoxic T lymphocytes (CTLs). Dendritic cells (1–2 × 105/well in 1 ml of AIM-V) were infected with 20,000 MOI of Tri-Ad5, as described above. Infected DCs were used as APCs for stimulation of autologous nonadherent cells at an effector-APC ratio of 10:1. Cultures were incubated for 3 days at 37oC in a humidified atmosphere containing 5% CO2. The cultures were then supplemented with rhIL-2 for 7 days; IL-2 containing medium was replenished every 3 days. The 10-day stimulation constituted one in vitro stimulation (IVS) cycle. Autologous vector-infected DCs were used as APCs for three IVS. Autologous peptide-pulsed B cells were used to restimulate antigen-specific CTLs after three IVS. T-cell lines were maintained in medium containing IL-7 and IL-15 (10 ng/ml; PeproTech, Rocky Hill, NJ).
Cytotoxic assay
A modification of the protocol described by Tsang et al. [80] was used for CTL analysis. In brief, target cells were labeled with 50 μCi of 111In oxide (GE Health Care, Vienna, VA) at 37°C for 20 min and used at 3,000 cells/well in 96-well round-bottom culture plates. T cells were added at different ratios and incubated at 37°C for 16 hours. Supernatants were harvested for gamma counting. Determinations were carried out in triplicate and SDs were calculated. Spontaneous release was determined by incubating target cells with medium alone and complete lysis was determined by incubating with 0.25% Triton X-100. Specific lysis was calculated with the use of the following formula: Lysis (%) = [observed release (CPM)-spontaneous release (CPM)] / [Complete release (CPM)-spontaneous release (CPM)] × 100.
Tumor cell culture
Human colon carcinoma SW620 (HLA-A2+, HLA-A24+, brachyury+, MUC1+, CEA+) and pancreatic carcinoma ASPC-1 (HLA-A1+, HLA-A26+, MUC1+, brachyury+, CEA+) cell lines were obtained from American Type Culture Collection (Manassas, VA). Cell cultures were free of mycoplasma and maintained in complete medium (RPMI-1640 supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine) (Mediatech, Herndon, VA).
Detection of cytokines
Supernatants of T cells stimulated for 24 hours with DCs infected with adenovirus vectors or peptide-pulsed DCs in IL-2–free medium were evaluated for secretion of IFN-γ using an ELISA kit (Invitrogen, Frederick, MD). The antigen-specific T-cell lines used in this analysis have been reported previously: (a) an HLA-A2 CEA-specific CTL [81], (b) an HLA-A2 MUC1-specific CTL [50], (c) an HLA-A24 MUC1-specific CTL [51], and (d) an HLA-A2 brachyury-specific CTL [62].
Peptides
The following HLA-A2 and HLA-A24 binding peptides were used in this study: (a) the HLA-A2 binding CEA agonist peptide CAP1–6D (YLSGADLNL) [64], (b) the HLA-A2 MUC1 agonist peptide P93L (ALWGQDVTSV) [50], (c) the HLA-A24 binding MUC1 agonist peptide C6A (KYHPMSEYAL) [51], and (d) the HLA-A2 binding brachyury agonist peptide (WLLPGTSTV) [62]. All peptides were greater than 96% pure and manufactured by American Peptide Company, Inc. (Sunnyvale, CA).
Mice
Specific pathogen-free, female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) of ages 8−10 weeks were housed in animal facilities at the Infectious Disease Research Institute (IDRI) (Seattle, WA, USA). All procedures were conducted according to Institutional Animal Care and Usage Committee (IACUC) approved protocols.
Vaccination and splenocyte preparation
Female C57BL/6 mice (n = 5) were injected s.c. with 1010 VP of Ad5 [E1-, E2b-]-brachyury or Ad5 [E1-, E2b-]-CEA or Ad5 [E1-, E2b-]-MUC1 or a combination of 1010 VP of all three viruses at a ratio of 1:1:1 (Tri-Ad5). Control mice were injected with 3 × 1010 VP of Ad5 [E1-, E2b-]-null (no transgene insert). Doses were administered in 25 μ1 of injection buffer (20 mM HEPES with 3% sucrose) and mice were vaccinated three times at 14-day intervals. Fourteen days after the final injection spleens and sera were collected. Sera were frozen at −20°C. Splenocyte suspensions were generated by gently crushing the spleens through a 70 μM nylon cell strainer (BD Falcon, San Jose, CA). Red cells were removed by the addition of red cell lysis buffer (Sigma-Aldrich, St. Louis, MO) and the splenocytes were washed twice and resuspended in R10 (RPMI 1640 supplemented with L-glutamine (2 mM), HEPES (20 mM) (Corning, Corning, NY), penicillin 100 U/ml and streptomycin 100 μg/ml (Hyclone, GE Healthcare Life Sciences, Logan, UT), and 10% fetal bovine serum (Hyclone). Splenocytes were assayed for cytokine production by ELISPOT and flow cytometry.
ELISPOT assay
Brachyury-, CEA- and MUC1-specific IFN-γ– or IL-2–secreting T cells were determined by ELISPOT assay from freshly isolated mouse splenocytes, as described above. The ELISPOT assay was performed according to the manufacturer's specifications (Affymetrix Bioscience, San Diego, CA). Briefly, 2 × 105 splenocytes were stimulated with 0.2 μg/well of overlapping 15-mer peptides in a single pool derived from brachyury or CEA (JPT Peptide Technologies, Berlin, Germany) or MUC1. Cells were stimulated with Concanavalin A (Con A) at a concentration of 0.0625 μg/per well as a positive control and overlapping 15-mer complete peptides pools derived from SIV-Nef and SIV-Vif (AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH)) were used as irrelevant peptide controls. The numbers of SFCs were determined using an Immunospot ELISPOT plate reader (Cellular Technology, Shaker Heights, OH) and results were reported as the number of SFCs per 106 splenocytes.
Intracellular cytokine stimulation
Splenocytes were prepared as indicated above. Stimulation assays were performed using 1 × 106 live splenocytes per well in 96-well U-bottom plates. Pools of overlapping peptides spanning the entire coding sequences of brachyury, CEA and MUC1 were synthesized as 15-mers with 11-amino acid overlaps (JPT GmbH) and lyophilized peptide pools were dissolved in Dimethyl sulfoxide (DMSO). Similarly constructed peptide pools corresponding to SIV-Vif and SIV-Nef served as off-target controls. Splenocytes in R10 media (RPMI 1640, 10% fetal bovine serum, and antibiotics) were stimulated by the addition of peptide pools at 2 μg/mL/peptide for 6 h at 37°C and 5% CO2, with protein transport inhibitor (GolgiStop, BD) added 2 hours into the incubation. Stimulated splenocytes were then stained for lymphocyte surface markers CD8α and CD4, fixed, permeabilized, and then stained for the intracellular accumulation of IFN-γ and TNF-α. Antibodies against mouse CD8α (clone 53–6.7), CD4 (clone RM4–5), IFN-γ (clone XMG1.2), and TNF-α (clone MP6-XT22) were purchased from BD and staining was performed in the presence of anti-CD16/CD32 (clone 2.4G2). Flow cytometry was performed using an Accuri C6 Flow Cytometer (BD) and analyzed in BD Accuri C6 Software.
ELISA to detect antibodies against CEA
ELISA plates (Nunc Maxisorp, Sigma-Aldrich, st Louis, mo) were coated with 100 ng of human CEA in 0.05M carbonate-bicarbonate buffer pH 9.6 and incubated overnight at room temperature. Plates were washed three times with phosphate buffered saline containing 1% Tween-20 (PBS-T) and then blocked with PBS containing 1% BSA for 60 min at room temperature. After an additional three washes, sera diluted 1/50 in PBS-T were added to the wells and the plates were incubated for 1 hour at room temperature. Peroxidase labeled goat anti-mouse immunoglobulin (Ig) G (γ-chain specific) (Sigma-Aldrich) antibody at a 1:5000 dilution was added to the wells after washings and plates were incubated for 1 hour. Plates were washed three times and 1,2-phenylene-diamine substrate solution (Thermo-Fisher Scientific, Waltham, MA) was added to each well. The reaction was stopped by adding 10% phosphoric acid. Absorbance was measured at 492 nm on a SpectraMax 190 ELISA reader (Molecular Devices, Sunnyvale, CA). The nanogram equivalents of IgG bound to CEA per well were obtained by reference to a standard curve generated using purified mouse IgG and developed at the same time as the CEA ELISA (Sigma-Aldrich) as previously described [5]. The results were analyzed and quantitated using SoftMax Pro 6.3 software (Molecular Devices).
Complement-dependent cytotoxicity assay (CDC)
MC38-CEA2 tumor cells were cultured overnight at a density of 2 × 104 cells per well in 96-well tissue culture microplates. Pooled heat inactivated mouse sera were added at a 1:50 dilution and incubated at 37°C for 1 hour. Rabbit serum was then added at a 1:50 dilution as a source of complement and cells were incubated an additional 2.5 hours at 37°C. Cell culture supernatants were assayed using Promega Cytotox 96 non-radioactive cytotoxicity assay (Promega, Madison, WI), according to the manufacturer's instructions. Percent lysis of MC38-CEA2 cells was calculated by the formula % lysis = (experimental – target spontaneous) / (target maximum – target spontaneous) × 100%.
Tumor immunotherapy
For in vivo tumor treatment studies, female C57BL/6 mice, 8–10 weeks old, were implanted with 106 MC38-MUC1 cells s.c. in the left flank. Mice were treated three times at a 7-day interval with 1010 VP Ad5 [E1-, E2b-]-MUC1 or Tri-Ad5. Control mice were injected with 3 × 1010 VP of Ad5 [E1-, E2b-]-null. Tumor growth was assessed by measuring two opposing dimensions (a, b) and the volume calculated as previously described [82] according to the formula V = (axb)2/2 where the shorter dimension was “a”. Tumor studies were terminated when tumors reached 1500 m3 or became severely ulcerated.
Supplemental materials
Supplemental data for this manuscript are available online at the publisher's website.
Abbreviations
Ad5, Adenovirus serotype-5; Ad5 [E1-], Adenovirus serotype-5 (Ad5)-based vector platforms with deletions in the early 1 (E1) gene and early 3 (E3) gene regions; Ad5 [E1-, E2b-], Ad5 [E1-] with additional deletions in the early 2 (E2) gene region and early (E3) gene region; APC, antigen presenting cell; CEA, carcinoembryonic antigen; CDC, complement-dependent cytotoxicity; Con A, Concanavalin A; CTL, cytotoxic T lymphocyte; DC, dendritic cell; DMSO, dimethyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; ELISPOT, enzyme-linked immunospot; EMT, epithelialmesenchymal transition; FDA, U.S. Food and Drug Administration; GM-CSF, granulocyte-macrophage colony-stimulating factor; HIV, human immunodeficiency virus; IFN, interferon; Ig, immunoglobulin; IVS, in vitro stimulation; LDH, lactate dehydrogenase; MAb, monoclonal antibody; mCRC, metastatic colorectal cancer; MHC, major histocompatibility complex; MOI, multiplicity of infection; MUC1, cancer-associated mucin; PBMC, peripheral blood mononuclear cells; PFU, plaque-forming unit; PSA, prostate-specific antigen; s.c., subcutaneously; SDS, sodium dodecyl sulfate; SFC, spot forming cells; SIV, simian immunodeficiency virus; TAA, tumor-associated antigen; Tri-Ad5, recombinant adenovirus admixture consisting of Ad5 [E1-, E2b-]-CEA, Ad5 [E1-, E2b-]-brachyury and Ad5 [E1-, E2b-]-MUC1; VP, viral particles
ACKNOWLEDGMENTS
We thank Debra Weingarten for her assistance in the preparation of this manuscript.
FUNDING
Laboratory of Tumor Immunology and Biology, NCI:
Studies conducted in the Laboratory of Tumor Immunology and Biology were funded by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health.
Etubics Corporation:
This study was funded in part by the National Cancer Institute Small Business Innovative Research (SBIR) Grants 2R44CA134063–02, 1R43CA186357–01A1; HHSN261201100097C.
CONFLICTS OF INTEREST
Laboratory of Tumor Immunology and Biology, NCI:
No potential conflicts of interest were disclosed.
Etubics Corporation:
Elizabeth S. Gabitzsch is a shareholder and employee of Etubics and has stock options in the Company. Adrian Rice is an employee of Etubics and has stock options in the Company. Yvette Latchman is an employee of Etubics and has stock options in the Company. Joseph P. Balint is a shareholder and employee of Etubics and has stock options in the Company. Frank R. Jones is a shareholder and employee of Etubics and has stock options in the Company.
REFERENCES
1. Balint JP, Gabitzsch ES, Rice A, Latchman Y, Xu Y, Messerschmidt GL, Chaudhry A, Morse MA, Jones FR. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol Immunother. 2015; 64:977–987.
2. Gabitzsch E, Jones F. New recombinant Ad5 vector overcomes Ad5 immunity allowing for multiple safe, homologous immunizations. J Clin Cell Immunol. 2011; S4:001.
3. Gabitzsch ES, Balint-Junior JP, Xu Y, Balcaitis S, Sanders-Beer B, Karl J, Weinhold KJ, Paessler S, Jones FR. Control of SIV infection and subsequent induction of pandemic H1N1 immunity in rhesus macaques using an Ad5 [E1-, E2b-] vector platform. Vaccine. 2012; 30:7265–7270.
4. Gabitzsch ES, Xu Y, Balcaitis S, Balint JP Jr., Jones FR. An Ad5[E1-, E2b-]-HER2/neu vector induces immune responses and inhibits HER2/neu expressing tumor progression in Ad5 immune mice. Cancer Gene Ther. 2011; 18:326–335.
5. Gabitzsch ES, Xu Y, Balint JP Jr., Balcaitis S, Sanders-Beer B, Jones FR. Induction and comparison of SIV immunity in Ad5 naive and Ad5 immune non-human primates using an Ad5 [E1-, E2b-] based vaccine. Vaccine. 2011; 29:8101–8107.
6. Gabitzsch ES, Xu Y, Balint JP Jr., Hartman ZC, Lyerly HK, Jones FR. Anti-tumor immunotherapy despite immunity to adenovirus using a novel adenoviral vector Ad5 [E1-, E2b-]- CEA. Cancer Immunol Immunother. 2010; 59:1131–1135.
7. Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Amalfitano A, Jones FR. Novel Adenovirus type 5 vaccine platform induces cellular immunity against HIV-1 Gag, Pol, Nef despite the presence of Ad5 immunity. Vaccine. 2009; 27:6394–6398.
8. Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Gayle RB, Amalfitano A, Jones FR. A preliminary and comparative evaluation of a novel Ad5 [E1-, E2b-] recombinant-based vaccine used to induce cell mediated immune responses. Immunol Lett. 2009; 122:44–51.
9. Jones FR, Gabitzsch ES, Xu Y, Balint JP, Borisevich V, Smith J, Smith J, Peng BH, Walker A, Salazar M, Paessler S. Prevention of influenza virus shedding and protection from lethal H1N1 challenge using a consensus 2009 H1N1 HA, and NA adenovirus vector vaccine. Vaccine. 2011; 29:7020–7026.
10. Morse MA, Chaudhry A, Gabitzsch ES, Hobeika AC, Osada T, Clay TM, Amalfitano A, Burnett BK, Devi GR, Hsu DS, Xu Y, Balcaitis S, Dua R, et al. Novel adenoviral vector induces T-cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol Immunother. 2013; 62:1293–1301.
11. Amalfitano A, Chamberlain JS. Isolation and characterization of packaging cell lines that coexpress the adenovirus E1, DNA polymerase, and preterminal proteins: implications for gene therapy. Gene Ther. 1997; 4:258–263.
12. Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J Virol. 1998; 72:926–933.
13. Everett RS, Hodges BL, Ding EY, Xu F, Serra D, Amalfitano A. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum Gene Ther. 2003; 14:1715–1726.
14. Hu H, Serra D, Amalfitano A. Persistence of an [E1-, polymerase-] adenovirus vector despite transduction of a neoantigen into immune-competent mice. Hum Gene Ther. 1999; 10:355–364.
15. Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002; 20:2197–2207.
16. Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999; 9:67–81.
17. Greiner JW, Hand PH, Noguchi P, Fisher PB, Pestka S, Schlom J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984; 44:3208–3214.
18. Hand PH, Nuti M, Colcher D, Schlom J. Definition of antigenic heterogeneity and modulation among human mammary carcinoma cell populations using monoclonal antibodies to tumor-associated antigens. Cancer Res. 1983; 43:728–735.
19. Horan Hand P, Colcher D, Salomon D, Ridge J, Noguchi P, Schlom J. Influence of spatial configuration of carcinoma cell populations on the expression of a tumor-associated glycoprotein. Cancer Res. 1985; 45:833–840.
20. Lottich SC, Szpak CA, Johnston WW, Thor A, Schlom J. Phenotypic heterogeneity of a tumor-associated antigen in adenocarcinomas of the colon and their metastases as demonstrated by monoclonal antibody B72.3. Cancer Invest. 1986; 4:387–395.
21. Szpak CA, Johnston WW, Lottich SC, Kufe D, Thor A, Schlom J. Patterns of reactivity of four novel monoclonal antibodies (B72.3, DF3, B1.1 and B6.2) with cells in human malignant and benign effusions. Acta Cytol. 1984; 28:356–367.
22. Palena C, Polev DE, Tsang KY, Fernando RI, Litzinger M, Krukovskaya LL, Baranova AV, Kozlov AP, Schlom J. The human T-box mesodermal transcription factor Brachyury is a candidate target for T-cell-mediated cancer immunotherapy. Clin Cancer Res. 2007; 13:2471–2478.
23. Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest. 2010; 120:533–544.
24. Hamilton DH, Litzinger MT, Fernando RI, Huang B, Palena C. Cancer vaccines targeting the epithelial-mesenchymal transition: tissue distribution of brachyury and other drivers of the mesenchymal-like phenotype of carcinomas. Semin Oncol. 2012; 39:358–366.
25. Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011; 71:5296–5306.
26. Kilic N, Feldhaus S, Kilic E, Tennstedt P, Wicklein D, Wasielewski R, Viebahn C, Kreipe H, Schumacher U. Brachyury expression predicts poor prognosis at early stages of colorectal cancer. Eur J Cancer. 2011; 47:1080–1085.
27. Haro A, Yano T, Kohno M, Yoshida T, Koga T, Okamoto T, Takenoyama M, Maehara Y. Expression of Brachyury Gene Is a Significant Prognostic Factor for Primary Lung Carcinoma. Ann Surg Oncol. 2013; 3:S509–16.
28. Pinto F, Pertega-Gomes N, Pereira MS, Vizcaino JR, Monteiro P, Henrique RM, Baltazar F, Andrade RP, Reis RM. T-box transcription factor brachyury is associated with prostate cancer progression and aggressiveness. Clin Cancer Res. 2014; 20:4949–4961.
29. Du R, Wu S, Lv X, Fang H, Wu S, Kang J. Overexpression of brachyury contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. J Exp Clin Cancer Res. 2014; 33:105.
30. Palena C, Roselli M, Litzinger MT, Ferroni P, Costarelli L, Spila A, Cavaliere F, Huang B, Fernando RI, Hamilton DH, Jochems C, Tsang KY, Cheng Q, et al. Overexpression of the EMT driver brachyury in breast carcinomas: association with poor prognosis. J Natl Cancer Inst. 2014; 106:pii: dju054.
31. Huang B, Cohen JR, Fernando RI, Hamilton DH, Litzinger MT, Hodge JW, Palena C. The embryonic transcription factor Brachyury blocks cell cycle progression and mediates tumor resistance to conventional antitumor therapies. Cell Death Dis. 2013; 4:e682.
32. Larocca C, Cohen JR, Fernando RI, Huang B, Hamilton DH, Palena C. An autocrine loop between TGF-beta1 and the transcription factor brachyury controls the transition of human carcinoma cells into a mesenchymal phenotype. Mol Cancer Ther. 2013; 12:1805–1815.
33. Roselli M, Fernando RI, Guadagni F, Spila A, Alessandroni J, Palmirotta R, Costarelli L, Litzinger M, Hamilton D, Huang B, Tucker J, Tsang KY, Schlom J, et al. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res. 2012; 18:3868–3879.
34. Palena C, Fernando RI, Hamilton DH. An immunotherapeutic intervention against tumor progression: Targeting a driver of the epithelial-to-mesenchymal transition. Oncoimmunology. 2014; 3:e27220.
35. Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, Dahut WL, Schlom J, Gulley JL. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13:501–508.
36. Bilusic M, Heery CR, Arlen PM, Rauckhorst M, Apelian D, Tsang KY, Tucker JA, Jochems C, Schlom J, Gulley JL, Madan RA. Phase I trial of a recombinant yeast-CEA vaccine (GI-6207) in adults with metastatic CEA-expressing carcinoma. Cancer Immunol Immunother. 2014; 63:225–234.
37. Heery CR, Singh BH, Rauckhorst M, Marte JL, Donahue RN, Grenga I, Rodell TC, Dahut W, Arlen PM, Madan R, Schlom J, Gulley JL. Phase I trial of a yeast-based therapeutic cancer vaccine (GI-6301) targeting the transcription factor brachyury. Cancer Immunol Res. 2015 Jun 30. [Epub ahead of print].
38. Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004; 4:45–60.
39. Kawano T, Ito M, Raina D, Wu Z, Rosenblatt J, Avigan D, Stone R, Kufe D. MUC1 oncoprotein regulates Bcr-Abl stability and pathogenesis in chronic myelogenous leukemia cells. Cancer Res. 2007; 67:11576–11584.
40. Yin L, Ahmad R, Kosugi M, Kufe T, Vasir B, Avigan D, Kharbanda S, Kufe D. Survival of human multiple myeloma cells is dependent on MUC1 C-terminal transmembrane subunit oncoprotein function. Mol Pharmacol. 2010; 78:166–174.
41. Yin L, Kufe D. MUC1-C Oncoprotein Blocks Terminal Differentiation of Chronic Myelogenous Leukemia Cells by a ROS-Mediated Mechanism. Genes Cancer. 2011; 2:56–64.
42. Gendler SJ, Lancaster, Carole A, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemherton L, Lalani E-N, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. 1990; 265:15286–15293.
43. Kufe D, Inghirami G, Abe M, Hayes D, Justi-Wheeler H, Schlom J. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma. 1984; 3:223–232.
44. Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, Remondo C, Cereda V, Jones JL, Pazdur MP, Higgins JP, Hodge JW, Steinberg SM, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008; 14:3060–3069.
45. Limacher JM, Quoix E. TG4010: A therapeutic vaccine against MUC1 expressing tumors. Oncoimmunology. 2012; 1:791–792.
46. Mohebtash M, Tsang KY, Madan RA, Huen NY, Poole DJ, Jochems C, Jones J, Ferrara T, Heery CR, Arlen PM, Steinberg SM, Pazdur M, Rauckhorst M, et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011; 17:7164–7173.
47. Morse MA, Niedzwiecki D, Marshall JL, Garrett C, Chang DZ, Aklilu M, Crocenzi TS, Cole DJ, Dessureault S, Hobeika AC, Osada T, Onaitis M, Clary BM, et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann Surg. 2013; 258:879–886.
48. Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Levy E, Krzakowski M, Hess D, Tartour E, Chenard MP, Limacher JM, Bizouarne N, Acres B, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol. 2008; 3:735–744.
49. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009; 9:874–885.
50. Tsang KY, Palena C, Gulley J, Arlen P, Schlom J. A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1. Clin Cancer Res. 2004; 10:2139–2149.
51. Jochems C, Tucker JA, Vergati M, Boyerinas B, Gulley JL, Schlom J, Tsang KY. Identification and characterization of agonist epitopes of the MUC1-C oncoprotein. Cancer Immunol Immunother. 2014; 63:161–174.
52. Hu XF, Yang E, Li J, Xing PX. MUC1 cytoplasmic tail: a potential therapeutic target for ovarian carcinoma. Expert Rev Anticancer Ther. 2006; 6:1261–1271.
53. Khodarev NN, Pitroda SP, Beckett MA, MacDermed DM, Huang L, Kufe DW, Weichselbaum RR. MUC1-induced transcriptional programs associated with tumorigenesis predict outcome in breast and lung cancer. Cancer Res. 2009; 69:2833–2837.
54. Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003; 22:6107–6110.
55. Ren J, Agata N, Chen D, Li Y, Yu WH, Huang L, Raina D, Chen W, Kharbanda S, Kufe D. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell. 2004; 5:163–175.
56. Wei X, Xu H, Kufe D. Human mucin 1 oncoprotein represses transcription of the p53 tumor suppressor gene. Cancer Res. 2007; 67:1853–1858.
57. Hamilton DH, Fernando RI, Schlom J, Palena C. Aberrant expression of the embryonic transcription factor brachyury in human tumors detected with a novel rabbit monoclonal antibody. Oncotarget. 2014; 6:4853–4862.
58. Poland PA, Kinlough CL, Rokaw MD, Magarian-Blander J, Finn OJ, Hughey RP. Differential glycosylation of MUC1 in tumors and transfected epithelial and lymphoblastoid cell lines. Glycoconj J. 1997; 14:89–96.
59. Abe M, Kufe DW. Identification of a family of high molecular weight tumor-associated glycoproteins. J Immunol. 1987; 139:257–261.
60. Friedman EL, Hayes DF, Kufe DW. Reactivity of monoclonal antibody DF3 with a high molecular weight antigen expressed in human ovarian carcinomas. Cancer Res. 1986; 46:5189–5194.
61. Ramasamy S, Duraisamy S, Barbashov S, Kawano T, Kharbanda S, Kufe D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007; 27:992–1004.
62. Tucker JA, Jochems C, Boyerinas B, Fallon J, Greiner JW, Palena C, Rodell TC, Schlom J, Tsang KY. Identification and characterization of a cytotoxic T-lymphocyte agonist epitope of brachyury, a transcription factor involved in epithelial to mesenchymal transition and metastasis. Cancer Immunol Immunother. 2014; 63:1307–1317.
63. Salazar E, Zaremba S, Arlen PM, Tsang KY, Schlom J. Agonist peptide from a cytotoxic t-lymphocyte epitope of human carcinoembryonic antigen stimulates production of tc1-type cytokines and increases tyrosine phosphorylation more efficiently than cognate peptide. Int J Cancer. 2000; 85:829–838.
64. Zaremba S, Barzaga E, Zhu M, Soares N, Tsang KY, Schlom J. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997; 57:4570–4577.
65. Adachi K, Tamada K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015; 106:945–950.
66. Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst. 2012; 104:599–613.
67. Webster RM. The immune checkpoint inhibitors: where are we now? Nat Rev Drug Discov. 2014; 13:883–884.
68. Konstantopoulos K, Thomas SN. Cancer cells in transit: the vascular interactions of tumor cells. Annu Rev Biomed Eng. 2009; 11:177–202.
69. Thomas SN, Tong Z, Stebe KJ, Konstantopoulos K. Identification, characterization and utilization of tumor cell selectin ligands in the design of colon cancer diagnostics. Biorheology. 2009; 46:207–225.
70. Thomas SN, Zhu F, Schnaar RL, Alves CS, Konstantopoulos K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J Biol Chem. 2008; 283:15647–15655.
71. von Kleist S, Migule I, Halla B. Possible function of CEA as cell-contact inhibitory molecule. Anticancer Res. 1995; 15:1889–1894.
72. Hamilton DH, Litzinger MT, Jales A, Huang B, Fernando RI, Hodge JW, Ardiani A, Apelian D, Schlom J, Palena C. Immunological targeting of tumor cells undergoing an epithelial-mesenchymal transition via a recombinant brachyury-yeast vaccine. Oncotarget. 2013; 4:1777–1790.
73. Gulley JL, Madan RA, Tsang KY, Jochems C, Marte JL, Farsaci B, Tucker JA, Hodge JW, Liewehr DJ, Steinberg SM, Heery CR, Schlom J. Immune impact induced by PROSTVAC (PSA-TRICOM), a therapeutic vaccine for prostate cancer. Cancer Immunol Res. 2014; 2:133–141.
74. Stein WD, Gulley JL, Schlom J, Madan RA, Dahut W, Figg WD, Ning YM, Arlen PM, Price D, Bates SE, Fojo T. Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res. 2011; 17:907–917.
75. Boehm AL, Higgins J, Franzusoff A, Schlom J, Hodge JW. Concurrent vaccination with two distinct vaccine platforms targeting the same antigen generates phenotypically and functionally distinct T-cell populations. Cancer Immunol Immunother. 2010; 59:397–408.
76. Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA. Cloning and sequencing of a human pancreatic tumor mucin cDNA. J Biol Chem. 1990; 265:15294–15299.
77. Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, Carraway KL 3rd, Kufe D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J Biol Chem. 2001; 276:35239–35242.
78. Cereda V, Vergati M, Huen NY, di Bari MG, Jochems C, Intrivici C, Gulley JL, Apelian D, Schlom J, Tsang KY. Maturation of human dendritic cells with Saccharomyces cerevisiae (yeast) reduces the number and function of regulatory T cells and enhances the ratio of antigen-specific effectors to regulatory T cells. Vaccine. 2011; 29:4992–4999.
79. Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995; 87:982–990.
80. Tsang KY, Zhu M, Even J, Gulley J, Arlen P, Schlom J. The infection of human dendritic cells with recombinant avipox vectors expressing a costimulatory molecule transgene (CD80) to enhance the activation of antigen-specific cytolytic T cells. Cancer Res. 2001; 61:7568–7576.
81. Palena C, Arlen P, Zeytin H, Greiner JW, Schlom J, Tsang KY. Enhanced expression of lymphotactin by CD8+ T cells is selectively induced by enhancer agonist peptides of tumor-associated antigens. Cytokine. 2003; 24:128–142.
82. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989; 24:148–154.