
INTRODUCTION
Medulloblastoma is the most common malignant brain tumor in children. Risk-adapted therapeutic protocols in non-infant patients encompass maximal surgical resection, cranio-spinal radiotherapy and chemotherapy. Treatment groups and intensity are defined by the presence (‘high-risk’ disease) or absence (‘standard-risk’) of clinical features associated with a poor prognosis; metastatic disease at diagnosis and/or significant post-operative tumor residues, and this stratification currently forms the basis of patient selection into clinical trials [1].
Recent advances in the biological sub-classification of medulloblastoma are leading to the conception of clinical trials aimed at more precise therapeutic stratification and improved outcomes [1]. Historical studies have identified biomarkers consistently associated with favorable (β-catenin nuclear immunopositivity as a marker of the WNT medulloblastoma molecular subgroup (MBWNT)) and poor (large-cell/anaplastic (LCA) pathology, MYC gene family amplification) prognosis [2–9], and their retrospective evaluation in the SIOP-UKCCSG-PNET3 clinical trial has validated their use alongside clinical factors for the improved definition of disease risk-stratification groups in disease-wide studies of non-infant medulloblastoma [10]. These stratification schemes will now form the basis of treatment selection in contemporary international clinical trials [1]. Additionally, further biomarkers with potential prognostic value, most notably the discovery of the four consensus medulloblastoma molecular subgroups (MBWNT, Sonic hedgehog (MBSHH), Group 3 (MBGroup3) and Group 4 (MBGroup4)), are emerging from recent research studies on retrospective cohorts of medulloblastoma patients [1, 11–14].
The validation of novel biomarkers and risk-stratification schemes in clinically-controlled cohorts is thus essential to their clinical application. Moreover, the specific therapeutic regimens used may potentially impact the prognostic significance of specific biomarkers, and validation of their relevance within the defined treatment groups used in current clinical trials (i.e. the clinical standard- or high-risk disease groups) is necessary. This will require large-scale and coordinated international studies.
Here, we report the first European prospective study of medulloblastoma biomarkers, undertaken as part of the multi-center HIT-SIOP-PNET4 trial (2001–2006), which enrolled 338 children from 120 centres, with clinically-defined non-metastatic, standard-risk medulloblastoma [15]. Sufficient FFPE tumor material was collected for prospective assay (2004–2010) of a selected panel of biomarkers of previously reported biological or prognostic significance (i.e. in ≥2 published series). The study aimed to (i) improve the early identification of the ~20% patients with standard-risk medulloblastoma which cannot be cured by current treatment concepts, and (ii) identify patients with a favorable prognosis who may qualify for a controlled reduction of adjuvant treatment schemes.
RESULTS
HIT-SIOP-PNET4: Clinical and treatment-related factors
338 patients, aged 4 to 21 at diagnosis, were enrolled and their clinical characteristics have been reported previously [15]. In summary, male patients predominated (211 male, 127 female) and the five-year EFS (all patients, including R+ disease) was 79 ± 2%. Features significantly associated with reduced EFS in univariate analysis in the clinical study were: (i) R+ disease (31/317 (9.8%); p = 0.020), and (ii) a delay to the start of radiotherapy (≥49 days after surgery (30/335 (9.0%); p = 0.050) or as a continuous variable (p = 0.025)). Patient gender and age at diagnosis were not associated with EFS [15]. Histopathological review was completed for 336/338 patients, and identified 273 CMB (81%), 47 DMB (14%) and 16 LCA (5%) tumors. There was no survival difference between CMB and DMB patients. The 16 patients with LCA subtype tumors enrolled on the study prior to amendment showed a higher frequency of relapses, but these did not reach significance [15]. The exclusion of further LCA patients, together with M+ patients, from the HIT-SIOP-PNET4 cohort, as well as sharpening of the definitions of DMB and LCA in the revised WHO classification of tumors of the CNS in 2007 [16] may account for any variation in the distribution of histopathological variants compared to previously-reported non-infant disease-wide trials (i.e. 71 CMB (61%), 22 DMB (19%) and 23 LCA (20%) in SJMB96 [3]; 174 CMB (84%), 14 DMB (7%) and 19 LCA (9%) in SIOP-UKCCSG-PNET3 [10]). Data are summarized in Table 1.
Table 1: Clinical, pathological and molecular characteristics of the HIT-SIOP-PNET4 cohort (n = 338; all patients with available data are shown), and univariate (Cox proportional hazards) analysis of their prognostic associations.
Variable |
Categories |
n |
Five-year pEFS ±SE |
Univariate Hazard Ratio (± CI) |
p-value |
+Gender |
Male (M) |
211 |
0.79 ± 0.03 |
1.0 |
0.533 |
Age* |
Median (years) |
9.0 |
− |
0.99 (0.93–1.05) |
0.754 |
+Pathology** |
All others |
320 |
0.80 ± 0.02 |
1.0 |
0.262 |
+Residual tumor |
≤1.5 cm2 |
286 |
0.82 ± 0.02 |
1.0 |
0.020 |
+Time from diagnosis to radiotherapy |
<49 days |
305 |
0.81 ± 0.02 |
1.0 |
0.050 |
Time from diagnosis to radiotherapy* |
Median (days) |
35 |
− |
1.03 (1.00–1.05) |
0.025 |
β-catenin nuclear accumulation |
No |
196 |
0.75 ± 0.03 |
1.0 |
0.019 |
CTNNB1 mutation |
No |
164 |
0.75 ± 0.04 |
1.0 |
0.058 |
MYC/MYCN amplification (PCR) |
No |
160 |
0.79 ± 0.03 |
1.0 |
0.606 |
MYC amplification (iFISH) |
No |
157 |
0.81 ± 0.03 |
1.0 |
0.542 |
MYCN amplification (iFISH) |
No |
147 |
0.82 ± 0.03 |
1.0 |
0.588 |
17p loss and/or 17q gain (diploid(cen)) (iFISH) |
No |
127 |
0.85 ± 0.03 |
1.0 |
0.007 |
Polyploid |
No |
72 |
0.78 ± 0.05 |
1.0 |
0.572 |
PTCH1 (9q22) loss |
No |
138 |
0.80 ± 0.03 |
1.0 |
0.781 |
*Assessed as a continuous variable.
**Patients with LCA tumors recruited prior to study amendment in November 2003. pEFS, event-free survival probability; SE, standard error; CI, 95% confidence interval. Significant prognostic associations are marked in bold type.
+Previously reported data shown for information [15].
MBWNT subgroup
22.8% (58/254) of assessable tumors were MBWNT positive by β-catenin IHC [10] (Figure 1A). 15.9% of tumors (31/195) harbored CTNNB1 activating mutations (Supplementary Figure 1); all except one were observed in tumors with strong nuclear protein accumulation (n = 30; p < 0.001; Figure 1B). A further 12 tumors displayed β-catenin positivity (28.6% (12/42)) in the absence of CTNNB1 exon 3 mutation.
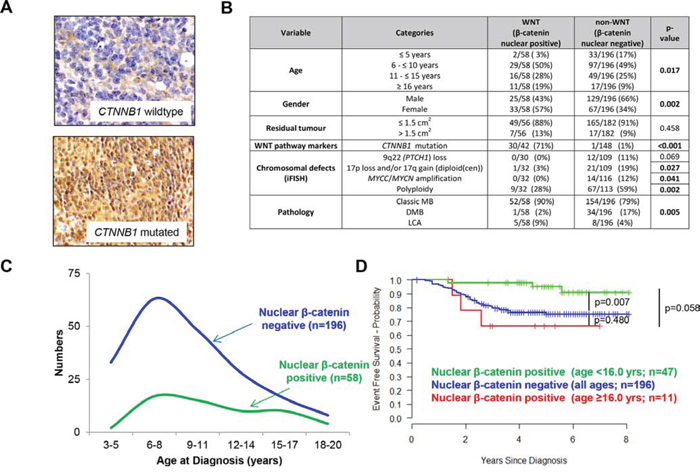
Figure 1: MBWNT subgroup tumors: clinical, pathological and molecular correlates. A. Examples of tumors scored negative and positive for β-catenin nuclear accumulation, with CTNNB1 status also indicated. B. Distribution of clinical, histopathological and molecular markers between the MBWNT (nuclear β-catenin accumulation) and non-MBWNT medulloblastoma subgroups. C. Age distributions of MBWNT and non-MBWNT patients. D. Kaplan-Meier plots and associated ‘p’ values (log-rank test) shown for MBWNT patients with age at diagnosis younger than 16 years, older than 16 years versus non-MBWNT cases.
MBWNT subgroup tumors were clinically, pathologically and molecularly distinct [11]: All except one displayed CMB or LCA histology (p = 0.005), and the group displayed gender parity in contrast to the male predominance in non-MBWNT patients (p = 0.002). All copy number aberrations (CNAs) tested were infrequent or absent in MBWNT and polyploidy was less frequent (p = 0.002; Figure 1B). MBWNT patients showed a broader age distribution than non-MBWNT, with 11/58 (19.0%) MBWNT patients ≥16.0 years old at diagnosis, suggesting a secondary peak in adolescents/young adults (Figure 1B, 1C).
Favorable outcomes for patients with MBWNT tumors were confirmed in univariate analysis, (Table 1, Figure 2 (p = 0.019, Cox proportional hazards test; p = 0.003, log-rank test)). CTNNB1 mutation did not reach significance (Table 1, Figure 2A, 2B). 6/58 (10.3%) MBWNT tumors relapsed; all had β-catenin nuclear accumulation in >50% of cells and 3/4 assessed tumors harbored a CTNNB1 mutation (Supplementary Figure 2A). Of note, the EFS rate in MBWNT patients aged ≥16.0 years at diagnosis appeared lower than in MBWNT patients <16.0 years (p = 0.058; Figure 1D). Direct comparison of patients aged below 16.0 years from the HIT-SIOP-PNET4 and SIOP-UKCCSG-PNET3 trials showed equivalent five-year survival rates (±SE) for MBWNT patients (0.951 (±0.034) vs. 0.877 (±0.058); p = 0.434), but significantly better outcomes (0.754 (±0.034) vs. 0.671 (±0.036); p = 0.033) in non-MBWNT patients, and reduced frequencies of clinical high-risk features (M+ and/or R+ disease), in the HIT-SIOP-PNET4 cohort (Supplementary Figure 3).
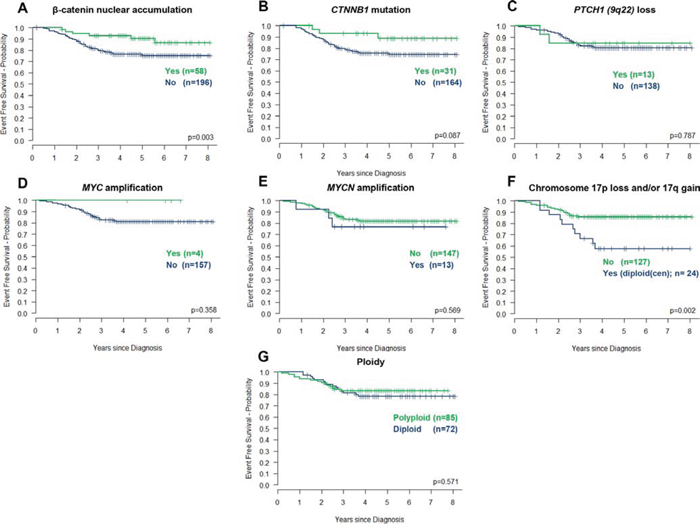
Figure 2: Prognostic significance of molecular disease features in the HIT-SIOP-PNET4 cohort. Kaplan-Meier plots and associated ‘p’ values (log-rank test) are shown for each feature. Chromosome 17 losses and gains observed against a diploid centromeric reference score (diploid(cen)) are represented.
Copy number aberrations
MYC (2.5% (4/161) tumors tested) and MYCN (8.1% (13/160)) amplifications detected by iFISH were mutually exclusive and were detected in non-MBWNT disease (Figure 1). Neither was associated with previously reported high-risk clinical (residual tumor) or pathological (LCA tumors included in this study) [1] disease features, or an adverse prognosis (Table 1, Figure 2D, 2E). iFISH and qPCR estimations of MYC/MYCN copy number were concordant in 90% of tumors analyzed by both methods (n = 131). Polyploidy (54% (85/157)) and PTCH1 (9q22) losses (10.6% (13/151)) were not associated with prognosis (Table 1, Figure 2C, 2G).
Chromosome 17 imbalances (q-arm gains and/or p-arm losses) were frequent (45.7% (69/151) tumors assessed). Imbalances were molecularly heterogeneous (Figure 3), and occurred against diploid (2 signals) and polyploid (>2 signals) centromeric reference backgrounds (Figure 3A, 3C). Strikingly, this heterogeneity was clinically significant. Tumors with chromosome 17 imbalances/diploid background (ch17(im)/diploid(cen); 16% (24/151)) were significantly associated with a poor outcome (p < 0.007; Table 1, Figure 2F), while tumors with imbalances/polyploid background (ch17(im)/polyploid(cen); 30% (45/151)) were not, and behaved equivalently to balanced tumors (Figure 3B). Ch17(im)/diploid(cen) (n = 24) most commonly involved p-arm loss (to a single copy) in conjunction with q-arm gain (17/24), consistent with isochromosome (17q), but isolated p-arm losses (4/24) and q-arm gains (3/24) also contributed. This tumor group peaked in children 6–10 years at diagnosis and all but one tumor was found in non-MBWNT disease, but the group was not associated with other clinico-pathological factors (Figure 3D). Notably, the prognostic significance of ch17(im)/diploid(cen) was gender-specific and 8/9 relapses in this group occurred in male patients (Supplementary Figure 4).
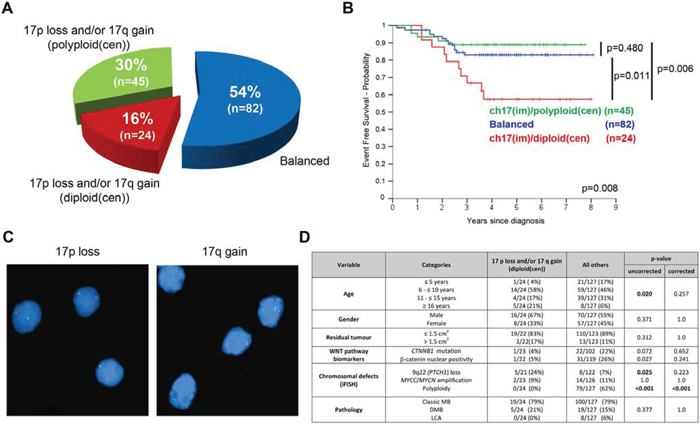
Figure 3: Chromosome 17 defects in HIT-SIOP-PNET4 cohort tumors. Patterns (A, C) and prognostic significance (B.; ‘p’, log-rank tests) of chromosome 17 defects detected by iFISH. C. iFISH analysis showing (i) 17p loss (single green signals) and (ii) 17q gain (three green signals) against a diploid centromeric background (two red signals). Nuclei are counterstained blue. D. Relationship of ch17(im)/diploid(cen) defects to clinico-pathological and molecular disease features assessed (‘p’, Fisher's exact or χ2 tests; corrected and uncorrected values are shown). Abbreviations: ch, chromosome; im, imbalance (p-gain and/or q-loss); diploid(cen), diploid centromeric signal; polyploidy(cen), polyploidy centromeric signal.
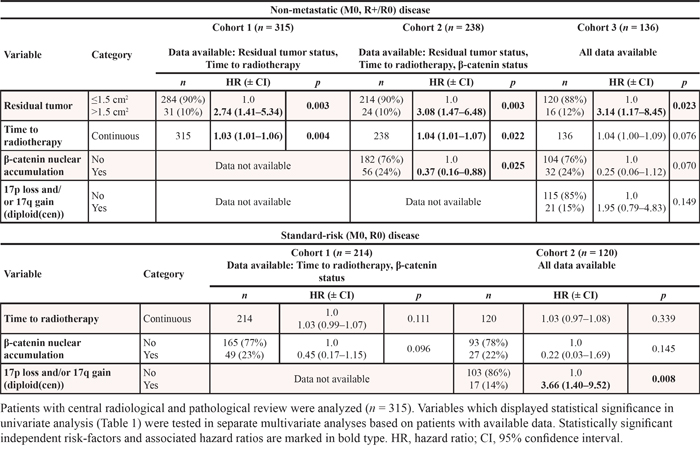
Clinical and biological prognostic factors: Multivariate analysis
Multivariate analyses were performed separately on the clinically-defined non-metastatic (i.e. M0) and standard-risk (i.e. M0/R0) patient cohorts within the HIT-SIOP-PNET4 study; these analyses were based on all features showing significance in univariate analysis (p < 0.05; Table 1, Figure 2). To account for all features not having been assessed in all patients, we performed this multivariate analysis on different patient groups, based on the data available (Table 2). In the non-metastatic patient cohort, R+ disease was independently significant in all analyses, while MBWNT and time to radiotherapy were either significant or marginally significant, consistent with findings from the univariate analysis. In the standard-risk cohort (i.e. following removal of R+ patients), ch17(im)/diploid(cen) was the only feature independently and significantly predictive of a poor outcome (Table 2).
Table 2: Multivariate analysis of independent risk factors in the HIT-SIOP-PNET4 cohort, shown for patients within the clinically-defined non-metastatic (M0, R+/R0) disease and standard-risk (M0, R0) patient groups
Risk stratification models for standard-risk medulloblastoma
The standard-risk patient group, defined by M0/R0 disease, forms the basis of current clinical trials [1]. We therefore next used our data to develop risk-stratification models for this patient group. First, we generated Cox models from all variables (listed in Table 1) using 90% of patients in our cohort, and selected the most significant model to predict survival for the remaining 10% of patients, using a 10-fold cohort re-selection strategy for cross-validation. Ch17(im)/diploid(cen) was selected as the sole prognostic feature in every fold, thus forming a model for the prediction of poor outcome within the standard-risk disease group (61 ± 13% vs. 89 ± 3% survival at five-years, p = 0.009) (Figure 4). In each fold, the model was not improved by the addition of any other covariate. We next compared model performance in our standard-risk cohort, against the current clinico-biological stratification scheme for the SIOP-PNET5 trial [1], which was defined previously based on disease-wide non-infant cohorts [1, 10, 14] (Figure 4A). Survival prediction at 5-years using the new cross-validated model improved performance and increased the area-under-curve (AUC) from 0.609 to 0.630 in ROC curve analysis (Figure 4D), and allowed 86% (102/118) of patients to be classified into a favorable-risk group, compared to 20% (24/118) in the established model. In view of the established favorable prognosis of MBWNT tumors [1, 3, 10], confirmed in our cohort, we assessed the impact of MBWNT status within this new model (Figure 4C). Inclusion was not detrimental to model performance (Figure 4D), however five-year survival for the favorable-risk MBWNT (96 ± 4%) and non-MBWNT ch17(im)/diploid(cen) negative (87 ± 4%) patient groups were not significantly different (p = 0.189). Finally, findings were equivalent when patients aged up to 16.0 years at diagnosis were considered in isolation (Supplementary Figure 5).
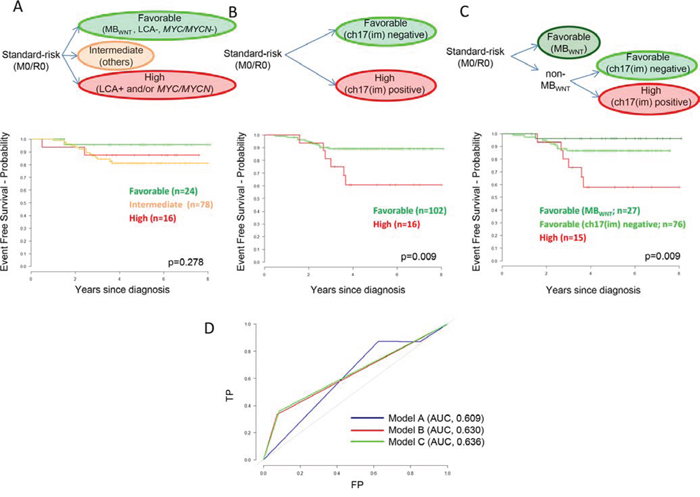
Figure 4: Biomarker-driven risk-stratification models for standard-risk (M0/R0) medulloblastoma based on patients from the HIT-SIOP-PNET4 cohort with data available for all parameters (n = 118). A. Established disease-wide survival model for non-infant medulloblastoma [1, 10, 14] (LCA pathology and/or MYC/MYCN amplified, high-risk; MBWNT and no high-risk features, favorable-risk; others, intermediate-risk). B. Empirically-derived survival model for non-infant, standard-risk medulloblastoma. C. Illustrative survival model for non-infant, standard-risk medulloblastoma, incorporating the distinction of MBWNT patients into the empirically-derived model. Kaplan-Meier plots and associated ‘p’ values (log-rank tests) show EFS. M0, non-metastatic; R0, no significant post-surgical tumor residuum; ch17(im), ch17(im)/diploid(cen) tumors. D. Time-dependent receiver operator characteristic (ROC) curves showing predictive performance of the three models for survival at five-years, determined as the area-under-curve (AUC). TP, true positive; FP, false positive. Chromosome 9 defects were not assessed in survival modelling due to missing data points.
DISCUSSION
The prospective assessment within HIT-SIOP-PNET4 of disease-relevant biomarkers, with reported significance in ≥2 previous retrospective series, alongside clinical and pathological factors, has provided important new insights to biomarker-driven risk stratification in clinically-defined standard-risk medulloblastomas. Post-operative residual tumor, delayed radiotherapy and MBWNT were validated as independent prognostic factors. Importantly, in the standard-risk (M0/R0) disease group, which forms the basis of current clinical trials [1], the use of distinct biomarkers (chromosome 17 status determined at the cellular copy-number level) and novel survival models allows the improved stratification of disease-risk. Moreover, we have established first mechanisms for prospective pan-European biological studies as a basis for the assessment of biomarker-driven therapies, and future therapeutic advances.
Assessment of MBWNT subgroup patients in the HIT-SIOP-PNET4 cohort revealed significant new insights to their clinical behavior, and comparison to other trials-based studies, to support future trial design. Their favorable prognosis was validated, supporting their consideration for individualized risk-stratified therapies; notably, despite equivalent EFS rates in the MBWNT groups, a significantly higher EFS was observed for the non-MBWNT group in our non-metastatic HIT-SIOP-PNET4 cohort (75% five-year EFS) compared to previous studies of the SIOP-UKCCSG-PNET3 trials cohort which included high-risk patients [2, 3]. MBWNT relapses were observed at higher frequency in patients with delayed radiotherapy or aged >16.0 years at diagnosis (Figure 1D; Supplementary Figure 2). Patients >16.0 years were not ascertained in our previous analyses of SIOP-UKCCSG-PNET3 which defined the favorable prognosis of the MBWNT group [2, 10], and the present data are consistent with the bi-modal age distribution and worse prognosis reported for adults compared to children within MBWNT in retrospective series [4, 17, 18]. Although the incidence of such cases is too low to draw firm conclusions, these observations thus indicate patient age >16.0 years and delayed radiotherapy negatively influence survival in MBWNT, and indicate radiotherapy is important component of its multi-modal treatment. Similarly, only 1 event was observed in MBWNT tumors also displaying previously reported high-risk disease features (MBWNT/R+ (n = 7), MBWNT/LCA (n = 5)) supporting their favorable prognosis following standard-risk therapy. CTNNB1 mutation rates in MBWNT tumors, defined by β-catenin IHC, were equivalent to SIOP-UKCCSG-PNET3 (30/42 (71.4%) and 20/31 (64.5%), respectively [10]) and our data did not support reduced survival rates in mutation-negative MBWNT (Supplementary Figure 2A).
Study design, and the prospective collection of clinical material and biological data, was undertaken in 2000–2010. This limited our ability to assess in the PNET4 cohort the other non-MBWNT molecular subgroups, on which consensus emerged in 2012 [11]. Insufficient FFPE-derived tumor material remained following our planned prospective analysis (see Materials and Methods; typically <200 ng double-stranded (ds)DNA [measured by picogreen fluorometric quantitation] and ≤1 4 μm section remaining), to enable subgroup assessment using established methods (approximately 1 μg FFPE-extracted dsDNA required for DNA methylation array analysis [14, 19, 20]; >2 sections for IHC-based assignment to MBSHH subgroup [21]).
From our findings, high-risk biomarkers previously validated in disease-wide studies appear to have different prognostic relevance in the standard-risk clinical disease group, from which tumors with high-risk clinical features (e.g. M+ disease) have been excluded. In this clinical context, MYC/MYCN amplification and LCA were not associated with each other, with clinical high-risk factors (one MYCN/R+ and one MYCN/LCA tumor were observed) or with poor prognosis when observed in isolation as risk-factors in the clinically-defined non-metastatic/standard-risk group reported. These findings are consistent with observations based on SIOP-UKCCSG-PNET3 (which included high-risk patients), where MYC/MYCN amplification were associated with LCA pathology and were prognostic in high-risk (i.e. when observed in tumors from patients with other high-risk disease features) but not standard-risk disease [7, 10]. In most recent genomics studies, MYC-amplified tumors were most commonly observed in MBGroup3, while MYCN-amplified tumors were associated with MBSHH (where they were also associated with TP53 mutation and LCA) and MBGroup4, but were only prognostic in MBSHH [12, 14, 22–24]. Our findings may thus reflect (i) the numbers of MYC/MYCN amplified tumors observed in this cohort, or (ii) biological heterogeneity and/or subgroup-dependency, including the trial amendment to cease recruitment of patients with LCA tumors (see methods), potentially limiting ascertainment of tumors with interactions between these factors. Evaluation/outcome monitoring of further patients defined by these features will now be required, prior to any refinements of their prognostic relevance used in the design of future clinical studies.
Our findings fully support the continued consideration of R+ patients as high-risk, and their exclusion from the standard-risk disease group [1]. Most notably, ch17(im)/diploid(cen) was the strongest independent biomarker risk-factor in M0/R0 standard-risk disease, characterizing a patient group with <60% five-year EFS; however, imbalances against a polyploid background were not significant. This previously undisclosed and clinically-significant heterogeneity observed between chromosome 17 imbalanced tumors is important, and these data suggest the biological impact of different patterns of chromosome 17 imbalance is equivalently heterogeneous. Variable prognostic associations have been reported for chromosome 17 imbalances in previous large studies [6, 10]; however, the complex patterns of imbalance revealed by iFISH analysis of individual tumor cells in this study were either not investigated or not detectable using the whole-biopsy copy number methodologies (e.g. array-CGH, SNP array) employed in many previous studies [9, 24–28]. Chromosome 17 imbalances are predominantly observed in MBGroup3 and MBGroup4 [24] and, in whole-biopsy SNP-array based investigations, Shih et al. [12] recently reported that the association between iso (17q) and a poor prognosis was restricted to MBGroup3 patients. Concerted studies are now required to reconcile these findings and establish the relationship between tumor subgroup, cellular patterns of chromosome 17 imbalance, and prognosis.
Following designation of ch17 (im)/diploid (cen) tumors as high-risk in cross-validated survival models of standard-risk patients within the HIT-SIOP-PNET4 cohort, the EFS of remaining patients did not differ significantly from the MBWNT group, allowing the classification of >80% of patients into a favorable-risk category. This model outperforms established prognostication schemes in our standard-risk cohort.
Alongside methods developed for testing chromosome 17 imbalances at the cellular level in routinely-collected tumor material, these findings provide a straightforward scheme for risk-stratification in the clinically homogeneous group of children with standard-risk medulloblastoma, and a strong basis for their validation and further investigation in future clinical trials of this group. Future study concepts must ensure collection of sufficient FFPE alongside high-quality biological material (e.g. snap-frozen, histologically-controlled tumor tissue), from large patient numbers, to support further biomarker discovery and validation, including understanding their behavior in the context of the consensus medulloblastoma expression / DNA methylation subgroups.
MATERIALS AND METHODS
Patient cohort, pathological review, material collection & processing
338 patients with non-metastatic (M0 [29]) medulloblastoma, treated in 120 European centers and 11 countries, were enrolled on HIT-SIOP-PNET4 over a 6 year period (2001–2006) [15]. Patients were randomized to receive post-operative treatment with either hyper-fractionated (HFRT) or conventionally fractionated/standard (STRT) radiotherapy and were followed up for a median of 4.8 years; all patients received the same chemotherapy. Clinical features of the cohort have been reported [15] (summarized in Table 1). The two treatment arms showed no significant difference in 5-year event-free survival (EFS) [15], and were considered together for biological analysis.
Post-operative radiological review was undertaken for 317/338 (93.7%) patients, the remainder were reviewed locally. Patients without significant post-surgical tumor residuum (≤1.5 cm2; R0) were defined as standard-risk [1]. A histopathological diagnosis of medulloblastoma was confirmed by five neuropathologists (including DF-B and TP), who performed central reference review of all patients. Tumors were classified using WHO criteria [30], and assigned to the classic (CMB), desmoplastic/nodular (DMB) or large-cell/anaplastic (LCA) sub-entities [16]. LCA were defined by a predominant component of tumor cells with either characteristic large-cell or severe anaplastic cytology, or both [8, 16, 31, 32]. DMB showed a significant tumor component with reticulin fiber-free islands (nodules) surrounded by reticulin fiber-rich tumor areas. Reactive fiber induction (‘desmoplastic reaction’, e.g. due to leptomeningeal growth) did not qualify as DMB [16, 33]. A study amendment was made in November 2003 to not enroll further LCA tumors, on the basis of their reported poor prognosis [8, 15, 31, 32].
During sample processing for reference pathology, excess tumor material was collected for biological studies where available. Two slides (4 μm tissue sections) were prepared for β-catenin immunohistochemistry (IHC). In addition, two tubes with 4 × 20 μm sections were collected, one to isolate nuclei for interphase fluorescence in situ hybridization (iFISH) analysis, the other for genomic DNA extraction (CTNNB1 mutation, MYC/MYCN qPCR analysis (see below)). DNA was extracted using the QIAamp DNA Mini Tissue Kit (Qiagen GmbH, Düsseldorf, Germany) according to the manufacturer's instructions.
Assessment of copy number aberrations
Tumor nuclei were isolated and CNAs of reported biological or prognostic significance assessed by iFISH using probes specific for MYCN (2p24) and MYC (8q24) (amplification previously associated with poor prognosis and LCA pathology [5–7, 10, 21]), PTCH1 (9q22; loss associated with the sonic hedgehog medulloblastoma molecular subgroup (MBSHH) and DMB [5, 6, 11, 17, 21]), and the p- and q-arms of chromosome 17 (imbalances associated with a poor prognosis [5, 6]), alongside reference probes to the chromosome 2, 8, 9 and 17 centromeric regions, as previously described [5, 10]. Signals in >200 non-overlapping nuclei were scored to give region of interest: centromere signal ratios for individual cells. For chromosome gains and losses, the modal score was considered representative of genetic status. MYC or MYCN gene amplification was defined by double-minute patterns or homogeneously staining regions in ≥5% of nuclei [5, 7, 10]. Tumor ploidy was determined as the modal status of the four centromeric probes (>2 signals at ≥2 probes, polyploid) assessed. Quantitative PCR (qPCR) was used to estimate MYC and MYCN gene copy numbers, as described [34, 35].
MBWNT subgroup status
Assessment of β-catenin nuclear accumulation by IHC has been widely studied as an MBWNT biomarker [9, 14, 36–38], and was performed as described using the monoclonal anti-β-catenin antibody 14 (Transduction laboratories) [2, 10, 39]. Tumors with >10% positive nuclei were scored positive (nuclear accumulation); the same cut-off as used in the published SIOP-UKCCSG-PNET3 cohort [10]. The few tumors showing nuclear accumulation in single cells (typically <5%) were classified negative. For CTNNB1 mutation analysis, exon 3 was PCR-amplified from tumor DNA using the primers 5′-GATTTGATGGAGTTGGACATGG-3′/5′-TGTTCTTGAGTGAAGGACTGAG-3′, and sequenced using standard methods [39].
Prospective biological studies: molecular biomarker assessment
Overall, biological data were collected prospectively from FFPE tumor material for 269/338 (79.5%) patients. Data collection rates varied according to the individual assays and the amount of tissue available. Notably, the success rate of sampling for β-catenin IHC did not differ between centers recruiting low (≤2; 77% success) and high (>7; 80%) patient numbers. Cases with available biological data from each assay were distributed randomly across the major disease demographics were thus considered representative of the whole cohort for further analysis (Supplementary Table 1).
Statistical analysis
EFS was defined as time from diagnosis to recurrence, progression or death during remission (of any reason). Patients not experiencing an event were censored at last follow-up. The database and biological data collection for this analysis was closed July 1st 2010. Kaplan–Meier curves, log-rank tests and unadjusted Cox proportional hazards models were used to test univariate EFS markers. Adjusted Cox proportional hazards models were used to test for independent disease-risk markers. For univariate and multivariate survival analyses which included ‘time to radiotherapy’ (as a continuous variable), EFS times were landmarked to the commencement of radiotherapy. Risk stratification models for standard-risk medulloblastoma were developed in the cohort of M0/R0 patients with data available for all prognostic parameters (n = 118). This cohort was demographically representative of the entire standard-risk cohort within HIT-SIOP-PNET4 (n = 286; Supplementary Table 2). Associations between clinico-pathologic and/or molecular variables were examined using Fisher's exact or X2 tests, as appropriate. P-values were corrected for multiplicity using the Bonferroni method where indicated. Analysis was performed using SPSS (SPSS, Chicago, U.S.A.) and R [40] software. Data proportions presented in Tables and Figures are based on patients with available data and may not add to 100% due to rounding.
ACKNOWLEDGMENTS
The contributions to this study of the SIOP-Europe PNET working group, and national study group members, are gratefully acknowledged.
CONFLICTS OF INTEREST
No author had any potential conflicts of interest to disclose.
GRANT SUPPORT
Supported by grants from Cancer Research UK, Swedish Childhood Cancer Foundation, French Ministry of Health/French National Cancer Institute and German Children's Cancer Foundation.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Pizer BL, Clifford SC. The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg. 2009; 23:364–375.
2. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005; 23:7951–7957.
3. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006; 7:813–820.
4. Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, Sturm D, Wittmann A, Schottler A, Felsberg J, Reifenberger G, Rutkowski S, Scheurlen W, Kulozik AE, von Deimling A, Lichter P, et al. Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol. 2010; 28:3054–3060.
5. Lamont JM, McManamy CS, Pearson AD, Clifford SC, Ellison DW. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004; 10:5482–5493.
6. Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Gerber NU, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler OD, Radlwimmer B, Scheurlen W, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009; 27:1627–1636.
7. Ryan SL, Schwalbe EC, Cole M, Lu Y, Lusher ME, Megahed H, O’Toole K, Nicholson SL, Bognar L, Garami M, Hauser P, Korshunov A, Pfister SM, Williamson D, Taylor RE, Ellison DW, et al. MYC family amplification and clinical risk-factors interact to predict an extremely poor prognosis in childhood medulloblastoma. Acta Neuropathol. 2012; 123:501–513.
8. Brown HG, Kepner JL, Perlman EJ, Friedman HS, Strother DR, Duffner PK, Kun LE, Goldthwaite PT, Burger PC. “Large cell/anaplastic“ medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol. 2000; 59:857–865.
9. Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Ellison DW. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006; 5:2666–2670.
10. Ellison DW, Kocak M, Dalton J, Megahed H, Lusher ME, Ryan SL, Zhao W, Nicholson SL, Taylor RE, Bailey S, Clifford SC. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011; 29:1400–1407.
11. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012; 123:465–472.
12. Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M, Luu B, Yao Y, Wang X, Dubuc AM, Garzia L, Peacock J, Mack SC, Wu X, Rolider A, Morrissy AS, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014; 32:886–896.
13. Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, Pfister SM. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012; 12:818–834.
14. Schwalbe EC, Williamson D, Lindsey JC, Hamilton D, Ryan SL, Megahed H, Garami M, Hauser P, Dembowska-Baginska B, Perek D, Northcott PA, Taylor MD, Taylor RE, Ellison DW, Bailey S, Clifford SC. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol. 2013; 125:359–371.
15. Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, Massimino M, Reddingius R, Benesch M, Carrie C, Taylor R, Gandola L, Bjork-Eriksson T, Giralt J, Oldenburger F, Pietsch T, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012; 30:3187–3193.
16. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system. Lyon: IARC Press; 2007.
17. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, Clifford SC, Pietsch T, von Bueren AO, Rutkowski S, McCabe M, Collins VP, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta neuropathologica. 2012; 123:473–484.
18. Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A, Benner A, von Deimling A, Scheurlen W, Perry A, Croul S, Kulozik AE, Lichter P, Taylor MD, Pfister SM, Korshunov A. Adult Medulloblastoma Comprises Three Major Molecular Variants. J Clin Oncol. 2011; 29:2717–23.
19. Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R, Cavalli FM, Ramaswamy V, Zapatka M, Reifenberger G, Rutkowski S, Schick M, Bewerunge-Hudler M, Korshunov A, Lichter P, Taylor MD, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013; 125:913–916.
20. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, Ryan SL, Poon E, Robinson SP, Ruddle R, Raynaud FI, Howell L, et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 2015; 27:72–84.
21. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, Gilbertson RJ. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta neuropathologica. 2011; 121:381–396.
22. Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, Sturm D, Pfaff E, Stark S, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014; 25:393–405.
23. Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, von Bueren AO, Jones DT, Northcott PA, Kool M, Sturm D, Pugh TJ, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013; 31:2927–2935.
24. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012; 488:49–56.
25. Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PloS one. 2008; 3:e3088.
26. Nicholson J, Wickramasinghe C, Ross F, Crolla J, Ellison D. Imbalances of chromosome 17 in medulloblastomas determined by comparative genomic hybridisation and fluorescence in situ hybridisation. Mol Pathol. 2000; 53:313–319.
27. Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009; 41:465–472.
28. Reardon DA, Michalkiewicz E, Boyett JM, Sublett JE, Entrekin RE, Ragsdale ST, Valentine MB, Behm FG, Li H, Heideman RL, Kun LE, Shapiro DN, Look AT. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997; 57:4042–4047.
29. Chang CH, Housepian EM, Herbert C, Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969; 93:1351–1359.
30. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61:215–225. discussion 226–219.
31. Eberhart CG, Kepner JL, Goldthwaite PT, Kun LE, Duffner PK, Friedman HS, Strother DR, Burger PC. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002; 94:552–560.
32. McManamy CS, Lamont JM, Taylor RE, Cole M, Pearson AD, Clifford SC, Ellison DW. Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. Journal of neuropathology and experimental neurology. 2003; 62:627–632.
33. McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE, Grundy RG, Clifford SC, Ellison DW. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007; 17:151–164.
34. Rutkowski S, von Bueren A, von Hoff K, Hartmann W, Shalaby T, Deinlein F, Warmuth-Metz M, Soerensen N, Emser A, Bode U, Mittler U, Urban C, Benesch M, Kortmann RD, Schlegel PG, Kuehl J, et al. Prognostic relevance of clinical and biological risk factors in childhood medulloblastoma: results of patients treated in the prospective multicenter trial HIT’91. Clin Cancer Res. 2007; 13:2651–2657.
35. Waha A, Watzka M, Koch A, Pietsch T, Przkora R, Peters N, Wiestler OD, von Deimling A. A rapid and sensitive protocol for competitive reverse transcriptase (cRT) PCR analysis of cellular genes. Brain Pathol. 1998; 8:13–18.
36. Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S, Manie E, Raquin MA, Bours D, Carpentier S, Barillot E, Grill J, Doz F, Puget S, Janoueix-Lerosey I, Delattre O. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009; 218:86–94.
37. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006; 24:1924–1931.
38. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011; 29:1408–1414.
39. Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer research. 1999; 59:269–273.
40. RDevelopmentCoreTeam. R: a language and environment for statistical computing. In R Foundation for Statistical Computing. Vienna: 2011.