
INTRODUCTION
The process of breast oncogenesis is believed to span up to several decades. Most (> 78 percent) of newly diagnosed breast cancer cases occur in women that are older than 50 years [1] and the median age at diagnosis is 61 years [2]. In most cases the tumors express the estrogen receptor (ER). ER+ tumors are exquisitely sensitive to anti-estrogen therapy. However, ER+ breast cancer is often metastatic at the time of diagnosis and metastatic ER+ tumors also frequently appear after many years of dormancy [3, 4]. In either case, the metastatic disease is generally incurable and even targeted therapies are generally only palliative. Therefore, it is necessary to understand more about deregulated molecular mechanisms that confer invasive properties on ER+ breast cancer cells. Clearly, both pre-menopausal and post-menopausal events that influence breast tumor invasiveness are clinically highly significant in breast tumor progression. Profound decreases in the levels of circulating estrogen and progesterone are a hallmark of post-menopausal physiology although, in post-menopausal women, breast, endometrial and adipose tissues contain much higher levels of estrogen and progesterone, compared to plasma levels of the hormones [5–9].
As the progesterone receptor (PR) gene is a target of estrogen, the PR expression status of ER+ breast tumors is believed to reflect the robustness of ER signaling and hence predict patient response to anti-estrogen therapy. Nevertheless, PR agonists do directly support invasiveness and metastatic potential in ER+/PR+ breast cancer cells as demonstrated using in vivo experimental models [10, 11]. The physiological relevance of these model systems is supported by the observation that in postmenopausal women, hormone replacement therapy with the combination of estrogen and progestin was associated with increased incidence of invasive breast cancer and breast cancer mortality compared with non-users [12] whereas estrogen monotherapy in women with prior hysterectomy was associated with a persistent decrease in the onset of invasive breast cancer [13]. However, in post-menopausal women who are not undergoing hormone replacement, the role of the endogenous hormones in the progression of ER+/PR+ breast tumors is unclear.
PR has two isoforms, A and B, that are expressed by alternative promoter usage from a single gene; PR-B is identical to PR-A except for the presence of an additional 164 amino acid amino-terminal segment that contains within it, an additional activation function, AF3 [14]. PR-B and PR-A exhibit both distinctive and overlapping patterns of agonist-induced gene activation or gene repression, depending on the variable contexts of the target promoters and the nature of the associated chromatin sites of PR binding [14–16]. In cells expressing equal amounts of PR-A and PR-B, a substantial proportion of the two proteins are sequestered by forming a heterodimer; the heterodimer regulates a smaller and unique set of genes compared to the homodimers [15, 17]. Clinical studies have shown that although in normal breast PR-A and PR-B are expressed at comparable levels, this balance is commonly altered during breast oncogenesis with a predominance of a high PR-A:PR-B ratio in early as well as progressed lesions [18]. An elevated PR-A:PR-B ratio, which is frequently due to overexpression of PR-A, is associated with a lower rate of disease free survival [19].
In vitro molecular studies have shown that when hormone-depleted breast cancer cells are treated with PR agonists, they induce invasiveness through several non-genomic and genomic signaling pathways of progestin [20–26]. Some of those studies have further reported that it is PR-B that mediates progestin-induced invasiveness in vitro [21, 27]. The progesterone doses that were used to demonstrate substantial PR-B dependent effects on invasiveness in vitro were relatively high, corresponding to the plasma range of the hormone levels associated with only the luteal phase of the menstrual cycle or with pregnancy. Horwitz and co-workers have also elegantly demonstrated in vitro that the mere overexpression of PR-A confers an inherently more aggressive phenotype in breast cancer cells, including adhesion to extracellular matrix, migratory capacity and survival, due to hormone-independent gene regulation by PR-A [28].
Most breast tumors are ER+ [29] and continue to retain ER expression even as they progress to hormone-independence [30, 31]. Estrogen supports the growth of ER+ breast tumors but it suppresses invasiveness of the tumor cells whether or not their growth is hormone-sensitive and also suppresses breast tumor progression [31–37]. However, in vitro studies of the role of PR in breast cancer cell invasiveness have generally been investigated mechanistically in the absence of estrogen signaling. The studies have either used ER+ cell line models in the absence of estrogen or they have relied on forced expression of PRs in ER-negative cells [21, 27, 38–40]. The relative contributions of PR ligands to invasiveness through opposing the suppressive effect of estrogen and the underlying mechanisms are still unclear in the literature.
Further, although the gene regulatory profile of ER has been shown to be estrogen dose-dependent [41], it is less clear whether PR has distinct mechanisms of action that depend on progesterone dose. The plasma levels of estrogen in pre-menopausal women is 1.4 nM–1.6 nM in the follicular phase and 3.6 nM–4.2 nM in the luteal phase [42]. Plasma levels of estrogen in post-menopausal women is 0.027 + 0.01 nM wheras breast tissue levels of estrogen in post-menopausal women is 1.4 + 0.7 nM [5, 6, 43]. The plasma level of progesterone ranges from 0.6 nM to 4 nM in the follicular phase and increases up to > 50 nM in the luteal phase [44] whereas post-menopausal women have a wide range of 0.047nM to 0.318nM (median 0.127 nM) [45]. The breast tissue level of progesterone in post-menopausal women is above an order of magnitude greater than its plasma levels [7]. Therefore, further investigation of the role of the individual PR isoforms on ER+ breast cancer cell invasiveness in the context of estrogen signaling and in the physiological range of breast tissue hormone levels was needed to more fully understand early events in hormonal regulation of breast cancer progression.
The ER+ model cell lines used in this study included T47D (ER+/PR+), ZR-75-1 (ER+/PR+) and BT474 (ER+/PR+/HER2+) cells. All three cell lines express both PR-A and PR-B. To dissect the actions of the individual PR isoforms, we also used recombinant T47D cells generated by Dr. Kathryn Horwitz and co-workers that virtually exclusively expressed PR-A or PR-B in addition to ER [46].
RESULTS
Estrogen dose dependence for inhibition of invasiveness
Estrogen (E2) is known to inhibit breast cancer cell invasiveness [47–51]. To relate the effect of E2 on invasiveness to physiological E2 levels, the E2 dose response for inhibition of invasiveness was determined in BT474, T47D and ZR-75-1 cells. E2 was able to inhibit invasiveness of the cells in the sub-nanomolar range with most of the inhibition occurring below 0.01 nM and virtually complete inhibition occurring at 0.1 nM in all three cell lines (Figure 1A, 1B, 1C). Thus the E2 dose that was required for substantial or virtually complete suppression of invasiveness in the three ER+ cell lines is at the low end of the literature consensus for both plasma and breast tissue levels of E2 in pre-menopausal (1.4nM–4.2nM) or post-menopausal (0.027 + 0.01 nM in plasma; 1.4nM + 0.7 in breast tisue) women [5, 6, 42, 43]. Invasiveness remained completely suppressed at higher concentrations of E2 (10nM and 20nM) (Supplemental Figure 1).
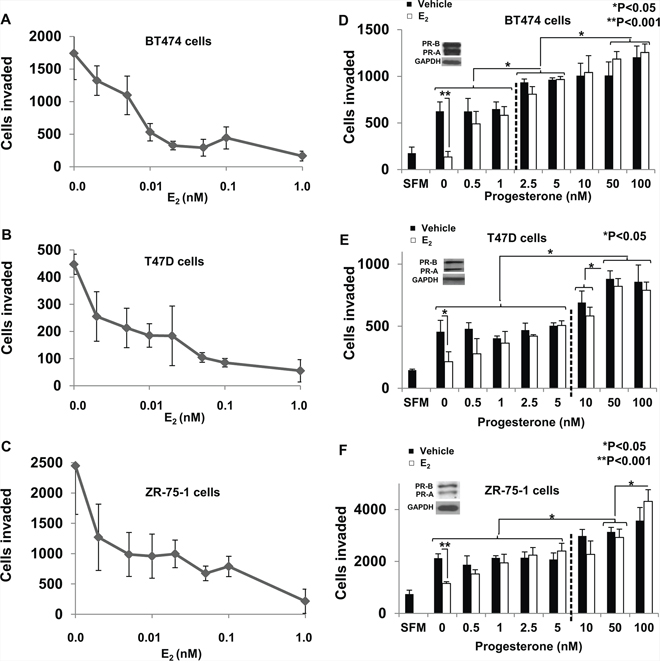
Figure 1: Dose response of regulation of breast cancer cell invasiveness by estrogen and progesterone. In panels A–C. hormone depleted BT474 cells (Panel A), T47D cells (Panel B) and ZR-75-1 cells (Panel C) at 30% confluence were treated with vehicle or the indicated concentrations of E2 for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentration of hormone present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. Data points in the plots in Panels A-C represent values for invasiveness represented as average number of cells invaded with the background (SFM) values subtracted. In panels D–F. Hormone depleted BT474 cells (Panel D), T47D cells (Panel E) and ZR-75-1 cells (Panel F) at 30% confluence were treated with vehicle or E2 (1 nM) and the indicated concentrations of progesterone for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentration of each hormone present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. Insets show western blots of whole cell lysates from BT474 cells (Panel D), T47D cells (Panel E) and ZR-75-1 cells (Panel F) probed for PR and for GAPDH. In panels A–F, values for invasiveness are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One-way ANOVA was performed on triplicate treatment sets and P values are indicated.
Dose-dependent dual regulation of invasiveness by natural and synthetic progestins
Plasma levels of progesterone are known to change throughout a woman's menstrual cycle ranging from 0.6 nM to 4 nM in the follicular phase and upwards to greater than 50 nM in the luteal phase [44]. Furthermore the median plasma concentration of progesterone in post-menopausal women is 0.127 nM [45] with breast tissue concentrations an order of magnitude greater than in the plasma [7]. To examine the effects on progesterone in the context of estrogen signaling BT474, T47D and ZR- 75-1 cells were treated at varying concentrations (0 nM– 100 nM) of progesterone either alone or in the presence of a fixed concentration (1 nM) of E2 (Figure 1D, 1E, 1F). In all the three cell lines E2 alone inhibited invasiveness. However, progesterone at 0.5 nM at least partially rescued invasiveness from the effects of E2 and showed virtually complete rescue in all cases at a concentration of 1 nM. It may be noted that progesterone alone (in the absence of E2) did not influence invasion below a concentration of 2.5 nM –5 nM but only rescued invasiveness from E2 regulation in the low concentration range (Figure 1D, 1E, 1F). At higher concentrations, progesterone progressively increased invasiveness of the cells independent of estrogen (Figure 1D, 1E, 1F). The hormones regulated invasiveness of the cells without affecting their migratory capacity (i.e., in the absence of matrigel in the transwells) (Supplemental Figure 2). Thus the data in Figure 1 (D–F) reveals two components of progesterone's effect on invasiveness in vitro in the three cell line models studied: (i) at low concentrations, progesterone rescues invasiveness from suppression by E2 and (ii) at higher concentrations, progesterone also induces invasiveness independent of E2.
As noted above, during the luteal phase of the menstrual cycle, the E2 level is elevated to about 4 nM along with an increase in the progesterone levels from about 4 nM to about 50 nM. Therefore, we tested the effect of 4 nM and 50 nM R5020 (a more stable synthetic progestin), in the presence of 4 nM E2 on invasiveness of T47D, ZR-75-1 and BT474 cells (Figure 2A–2C). In all the three cell lines, suppression of invasiveness by E2 was completely prevented by both concentrations of R5020 and at 50 nM R5020, there was a further increase in invasiveness (Figure 2A–2C).
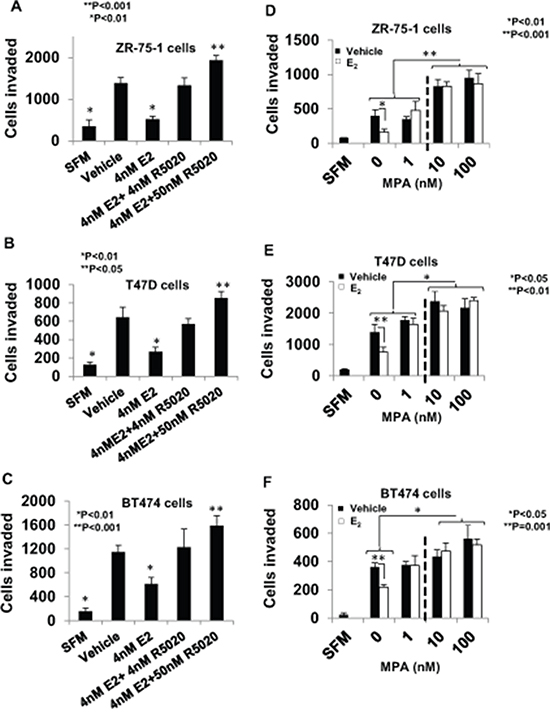
Figure 2: Regulation of breast cancer cell invasiveness by pre-menopausal concentrations of estrogen and progestin and dose-dependent effects of medroxyprogesterone acetate. In panels A–C. hormone depleted ZR-75-1 cells (Panel A), T47D cells (Panel B), and BT474 cells (Panel C) at 30% confluence were treated with vehicle or E2 (4 nM), alone or in combination with R5020 (5 nM or 50 nM) for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentration of E2 and/or R5020 present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. In panels D–F. hormone-depleted ZR-75-1 cells (Panel D), T47D cells (Panel E), and BT474 cells (Panel F) cells at 30% confluence were treated with vehicle or the indicated concetrations of MPA either with or without 1nM E2 for 48 h. Cells were trypsinized and subjected to the matrigel invasion assay with vehicle or the appropriate concentration of E2 and/or MPA present in the top and bottom chambers, as described under Materials and Methods. In panels A–F, values for invasiveness are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One-way ANOVA was performed on triplicate treatment sets and P values are indicated.
The dual effect of progesterone on invasiveness was recapitulated in all the three cell lines using the potent synthetic progestin medroxyprogesterone acetate (MPA). When MPA is used as a contraceptive (intramuscular route of administration) it has a mean plasma concentration of 2.58 nM [52] and has a 10–20 fold higher plasma concentration when administered orally during hormone replacement therapy [53]. At a concentration of 1 nM, MPA only reversed suppression of invasion by E2, but at higher concentrations (10 nM and 100 nM) MPA induced an increase in invasiveness well above the basal level whether or not E2 was present (Figure 2D–2F).
The distinctive role of each PR isoform in the regulation of invasiveness by progestins
The above observations led to the question of which PR-isoform(s) could mediate each of the two components of the regulation of invasiveness by progestins. To address this question, the effect of progestin dose on breast cancer cell invasiveness was tested in the absence or presence of 1nM E2 using recombinant T47D cells that exclusively express PR-A (T47D-A cells) or PR-B (T47D-B cells) (Figure 3A). The recombinant cells were a kind gift from Dr. Kathryn Horwitz who generated the cells as previously described [46]. Due to possible variance in absolute values of the number of cells invaded across experiments for a given cell line, we compared the invasive capacity of the T47D-A and T47D-B cells plated together under the same conditions at the same time. There was no difference in the invasive capacity of the two isogenic cell lines (Supplemental Figure 3). In T47D-A cells both R5020 and MPA only rescued invasiveness from E2 regulation at all concentrations tested (1nM, 10nM or 100 nM) but had no effect on invasiveness in the absence of E2 (Figure 3B and 3C). In contrast, in T47D-B cells, 1 nM of either R5020 or MPA was unable to rescue invasiveness from E2 regulation but at the higher concentrations (10 nM or 100 nM) they induced invasiveness well above the basal level and this was uninfluenced by the presence of E2 (Figure 3D and 3E).
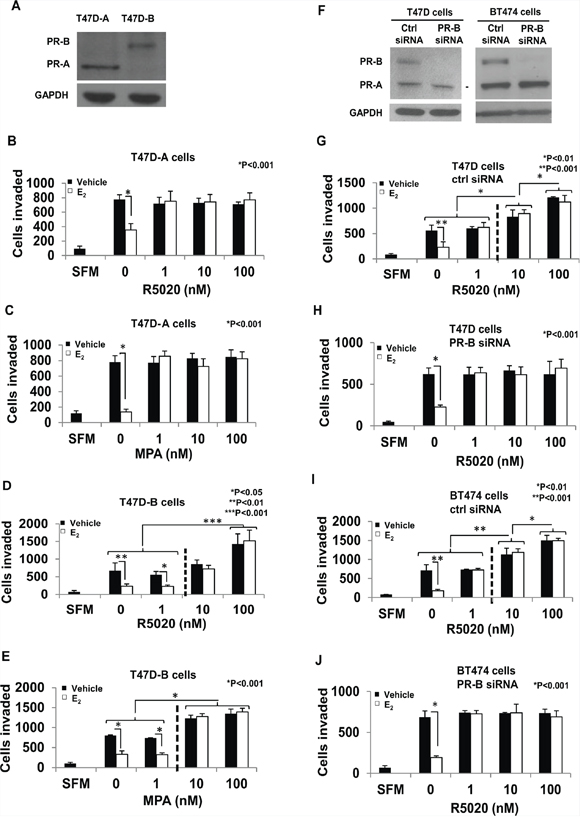
Figure 3: PR-A vs. PR-B mediated effects of progestins on invasiveness of breast cancer cells. Panel A shows a western blot of cell lysates from T47D-A and T47D-B cells probed for PR or GAPDH (loading control); the position of the PR band is used to identify the PR isoform. In panels A–E. hormone-depleted T47D-A cells (Panels B and C) and T47D-B cells (Panels D and E) at 30% confluence were treated with vehicle or 1 nM E2 in combination with the indicated concentrations of R5020 (Panels B and D) or MPA (Panels C and E) for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentrations of E2/R5020/MPA present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. In panels F–J. hormone depleted cells were transfected with either siRNA directed against PR-B or non-targeted control siRNA and incubated for 48 h. In Panel F cell lysates from the transfected cells were prepared 96 h post-transfection and analyzed by western blot for PR or GAPDH (loading control); the PR isoforms are identified by their positions on the western blots. T47D cells transfected with control siRNA (Panel G) or PR-B targeted siRNA (Panel H) and also BT474 cells transfected with control siRNA (Panel I) or PR-B targeted siRNA (Panel J) were treated with vehicle or 1 nM E2 in combination with the indicated concentrations of R5020 for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentrations of E2 and R5020 present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. In panels B–E and G–J, values for invasiveness are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One-way ANOVA was performed on triplicate treatment sets and P values are indicated.
As a complementary approach, we utilized siRNA knockdown of PR-B in parental T47D cells and in BT474 cells that express equal amounts of PR-A and PR-B. Both cell lines were transfected with siRNA directed against a target site in the unique 5′ segment of the PR-B mRNA or with control non-silencing siRNA using Lipofectamine. Knockdown of PR-B (western blots in Figure 3F) resulted in a loss of E2-independent induction of invasiveness at the higher concentrations (10 nM and 100 nM) of R5020 in both T47D cells and BT474 cells (Figure 3H and 3J) in contrast to the control siRNA transfected cells (Figure 3G and 3I). However the selective depletion of PR-B did not alter R5020′s ability to rescue invasiveness in the presence of E2, at all concentrations (1nM, 10nM, and 100nM) of R5020 (Figure 3G–3J). This result is consistent with those observed above using T47D-A cells.
The above results demonstrate that PR-A exclusively mediates the role of low dose progestins in opposing suppression of invasiveness by E2, whereas PR-B exclusively mediates E2-independent induction of invasiveness at high doses of progestins.
Effect of RU486 on PR-A mediated induction of invasiveness by progestins
RU486 is a synthetic antagonist of progesterone that is PR isoform selective in specific target gene contexts. Therefore it was of interest to test the effect of RU486 on the PR-A dependent actions of progesterone on breast cancer cell invasiveness. The cell lines T47D-A, T47D-B and BT474 were treated with either E2, the progestin R5020, or the anti-progestin RU486, each at a concentration of 1 nM in the various combinations indicated in Figure 4. In T47D-A cells, RU486 disrupted the ability of R5020 to rescue invasiveness from E2 suppression but did not have any effect by itself on invasiveness, in either the presence or absence of E2 (Figure 4A). On the other hand, in T47D-B cells, RU486 had no effect on invasiveness under any of the conditions tested when each of the ligands was used at a concentration of 1 nM (Figure 4B). In BT474 cells, which express equal amounts of both PR isoforms, the effect of RU486 was similar to that observed in the T47D-A cells, demonstrating that agonist or antagonist actions that modulate the effect of PR-A on invasiveness are functionally independent of PR-B expression (Figure 5C).
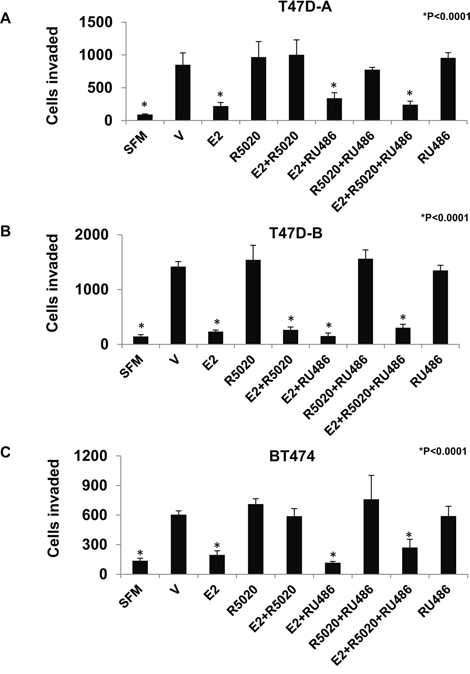
Figure 4: Effect of RU486 on regulation of breast cancer cell invasiveness by R5020 in relation to estrogen, PR-A and PR-B. Hormone depleted T47D-A Panel A., T47D-B Panel B. and BT474 Panel C. cells at 30% confluence were treated with vehicle or the indicated combinations of E2, R5020 and RU486, each at a concentration of 1nM for 48 h. Cells were trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentration of E2, R5020 or RU486 present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. Values are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One-way ANOVA was performed on triplicate treatment sets and P values are indicated.
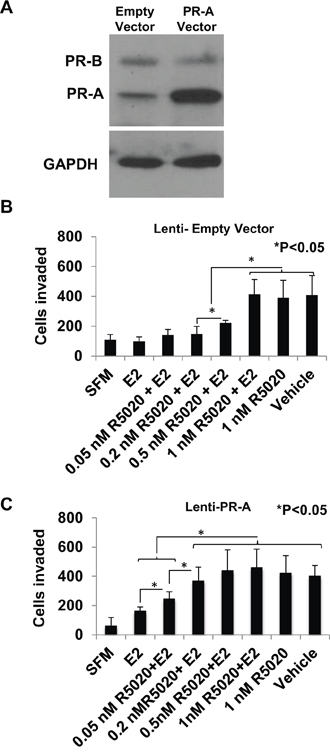
Figure 5: Effect of overexpressing of PR-A on the progestin dose response for rescue of invasiveness from estrogen regulation. PR-A was ectopically overexpressed in hormone-depleted T47D cells by lentiviral transduction, as described under Materials and Methods. Whole cell lysates from cells transduced with either the PR-A expression vector or the control empty vector were probed for PR and for GAPDH Panel A. Cells transduced with the control empty vector Panel B. or PR-A expression vector Panel C. at 30% confluence were treated with vehicle or the indicated concentrations of R5020 in the absence or in the presence of E2 (1 nM) for 48 h. Cells were then trypsinized and subjected to the matrigel transwell invasion assay with vehicle or the appropriate concentration of E2 or R5020 present in the top and bottom chambers, as described under Materials and Methods. In the negative control, serum free media (SFM) was used instead of the FBS chemoattractant. Values are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One-way ANOVA was performed on triplicate treatment sets and P values are indicated.
Hypersensitization of PR-A to progestin through overexpression of the receptor
As noted above, PR-A is frequently overexpressed in invasive clinical breast tumors. It was therefore of interest to examine the possibility that overexpression of PR-A in the tumor cells may sensitize PR-A mediated regulation of invasiveness to post-menopausal breast tissue levels of progesterone.
T47D cells express comparable amounts of PR-A and PR-B protein as observed on a western blot probed with an antibody against a common carboxyl-terminal peptide of the two receptor isoforms (Figure 5A). Lentiviral transduction of a PR-A expression plasmid increased the level of PR-A by approximately 3.7-fold, without altering the expression of PR-B (Figure 5A). The R5020 dose-dependence for rescue of invasiveness from E2 regulation was compared between the PR-A overexpressing cells and the control cells transduced with the empty vector. Overexpression of PR-A clearly conferred hypersensitivity to R5020 as the progestin partially rescued invasiveness even at a concentration of 0.05 nM and fully rescued invasiveness at a concentration of 0.2 nM in the PR-A overexpressing cells (Figure 5C); in comparison, in the control cells a concentration of 0.5 nM –1.0 nM R5020 was required to observe similar effects (Figure 5B).
PR isoform A-dependent regulation of E2 target genes by progestin and their functional role
E2 acts through its receptor ER to repress expression of genes known to be involved in breast tumor invasion, EMT, and metastasis [49, 50, 54–56]. The ability of progestins to oppose E2 regulation of invasiveness did not involve a decrease in ER expression as evident from a western blot of T47D-A cells treated with R5020 (Figure 6A).
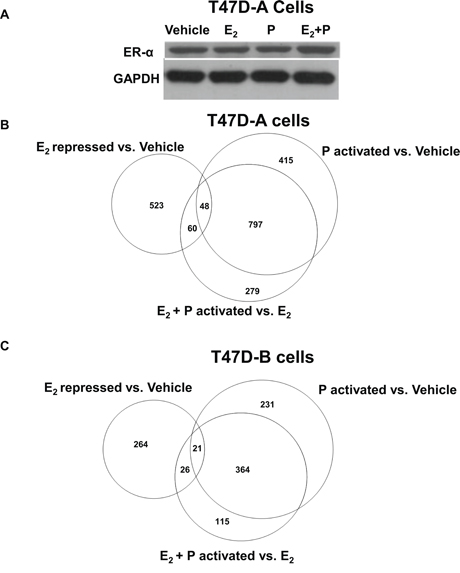
Figure 6: Effect of low dose progestin on the gene repression profile of estrogen in relation to PR-A and PR-B. Hormone depleted T47D-A or T47D-B cells at 30% confluence were treated with vehicle, 1 nM E2, 1 nM R5020 (P) or 1nM E2 plus1nM R5020 (E2 + P) for 48 h. In Panel A. whole cell lysates from the treated T47D-A cells were probed by western blot for ERα and for GAPDH. In parallel, total RNA was extracted from the treated T47D-A and T47D-B cells and subjected to mRNA expression profiling as described under Materials and Methods. The mRNA profiling data is represented in the Venn diagrams in Panel B. (for T47DA cells) and in Panel C. (for T47D-B cells).Panels B and C show comparisons among the gene set repressed by E2 (E2 repressed vs. Vehicle), the gene set activated by R5020 in the absence of E2 (P activated vs. Vehicle) and the gene set activated by R5020 in the presence of E2 (E2 + P activated vs. E2). The data represents results from experimental triplicates.
Next, we undertook to examine PR isoform-specific effects on transcriptional signaling by E2 using T47D-A or T47D-B cells. A concentration of 1 nM R5020 was chosen because at this concentration the progestin completely rescued invasiveness from E2 regulation (through PR-A) but did not exert E2-independent effects on invasiveness (through PR-B) (please refer back to Figure 3B and 3D). The cells were treated with vehicle, 1 nM E2, 1 nM R5020 or 1 nM R5020 + 1 nM E2 for an extended duration of 48 hours to examine expression of both direct and indirect target genes of the hormones. mRNA expression profiles were examined by DNA microarray analysis using the Illumina platform and an arbitrary cut off value of 1.5-fold was applied to identify patterns of changes in mRNA expression. In T47D-A cells, among 631 genes that were repressed by E2 (Supplemental Table 1) (Figure 6B), R5020 opposed the repression of 108 genes (Supplemental Table 2) (Figure 6B) including 48 genes that were activated by R5020, independent of E2 (Supplemental Table 3) (Figure 6B). In T47D-B cells, among 311 genes that were repressed by E2 (Supplemental Table 4) (Figure 6C), R5020 opposed the repression of 47 genes (Supplemental Table 5) (Figure 6C) including 21 genes that were activated by R5020, independent of E2 (Supplemental Table 6) (Figure 6C). Inspection of these gene lists revealed that of the 108 E2 repressed genes whose expression was rescued by R5020 in T47D-A cells, only 9 genes were also rescued by R5020 in T47D-B cells. The E2 repressed genes that were activated by progesterone alone were also cell-type specific, with only 8 exceptions. Thus, repression of 99 genes by E2 was opposed by R5020 in an exclusively PR isoform A-dependent manner. We next searched the literature to identify all the genes in this group that had suggested or established roles in breast tumor biology. A total of 19 genes were clearly known to be associated with breast tumor biology and they predominantly supported breast tumor progression, including invasiveness and metastasis (Supplemental Table 7). The DNA microarray data is validated for 4 representative genes (HES1, PRKCH, ELF5 and TM4SF1) by quantitative real time RT-PCR in Figure 7A and 7B using T47D-A and T47D-B cells. We also confirmed that these four genes were regulated in T47D (parental), BT474, and ZR-75-1 cells in the same pattern as that observed in T47D-A cells (Figure 7C–7E).
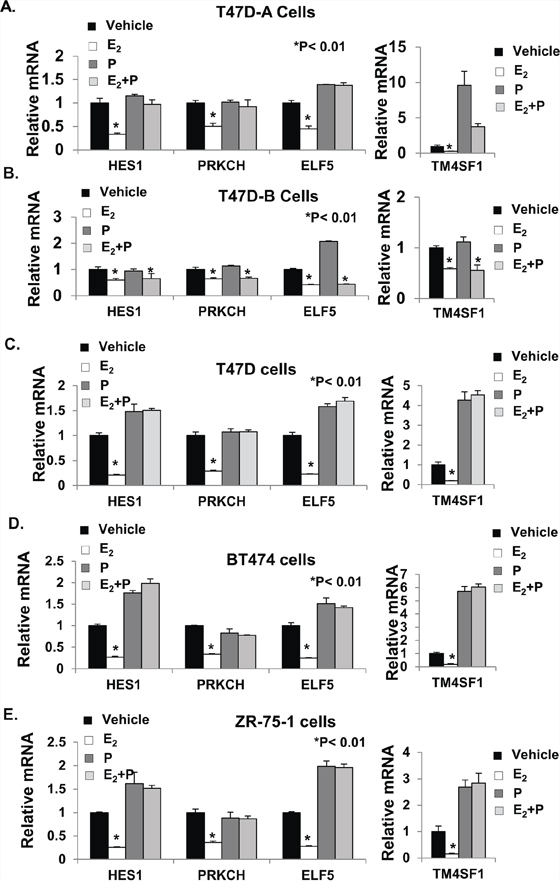
Figure 7: Validation of gene expression profiling. The total RNA samples used for mRNA expression profiling in Figure 6 were used for validation of the mRNA profiling data for selected genes in T47D-A cells Panel A. and T47D-B cells Panel B. Validation of estrogen and progestin regulation of these genes in the PR-A+ cells was also extended to T47D (parental) cells Panel C. BT474 cells Panel D. and ZR-75-1 cells Panel E. RNA purified from the treated cells was reverse transcribed and the cDNA was analyzed by real-time PCR using TaqMan Probes, as described under Materials and Methods. Relative mRNA levels were measured in the samples for HES1, PRKCH, ELF5, and TM4SF1 genes. All CT Values were normalized to GAPDH and represented as fold change in comparison to vehicle treated controls. The mRNA values are the average (+/− standard deviation) from triplicate assays performed for each one of the triplicate treatment sets.
The four genes (HES1, PRKCH, ELF5 and TM4SF1) validated above have all been associated with cancer progression. To directly test whether regulation of these genes by E2 mediated the hormonal effects of E2 on invasiveness in ER+ breast cancer cells, we used a loss-of-function approach. T47D and BT474 cells were transfected with siRNAs against the four genes either individually (Figure 8A and 8E) or together (Figure 8B and 8F); in all cases, the siRNAs effectively knocked down the genes, as observed by their mRNA levels compared to the cells transfected with control non-targeted siRNA (Figure 8A, 8B, 8E and 8F). For the protein products of the genes for which high quality antibodies were available (i.e., ELF5 and HES1), the knockdown was also confirmed by western blot (Supplemental Figure 4). Individually knocking down the genes decreased invasiveness of the cells to different degrees (Figure 8C and 8G) and the combined knockdown completely suppressed invasiveness (Figure 8D and 8H). The results indicate the functional relevance of genes whose regulation by E2 was found in this study to be opposed by low dose progesterone acting through PR-A. Clearly the subset of E2 repressed genes that are counter-regulated by progesterone/PR-A include genes that mediate hormonal regulation of invasiveness in breast cancer cells.
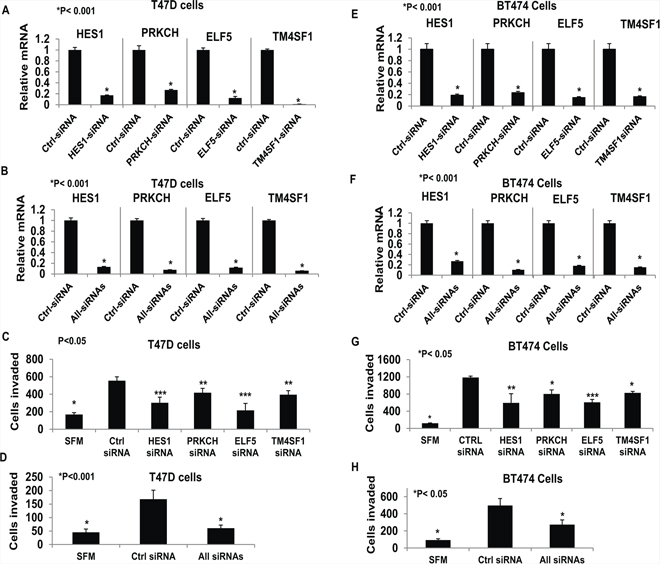
Figure 8: Functional testing of selected tumor progression genes. Hormone-depleted T47D cells Panels A–D. and BT474 Panels E–H. were transfected with control siRNA, TM4SF1 siRNA, HES1 siRNA, ELF5 siRNA and PRKCH siRNA independently (Panel A, C, E, and G) or all four targeted siRNAs in combination (Panels B, D, F, H). After 72 hours cell were subjected to the transwell matrigel invasion assay (Panels C, D, G, and H) as described under Materials and Methods. In the negative controls, serum free medium (SFM) was used instead of the FBS chemoattractant. Values are represented as average number of cells invaded from triplicate treatment sets and the error bars represent standard deviation. One way ANOVA was performed on triplicate treatment sets and P values are indicated.
A similar analysis was then conducted for E2 activated genes in T47D-A (Figure 9A and Supplemental Tables 8–10) and T47D-B (Figure 9B and Supplemental Tables 11–13) cells. We found that activation of 112 genes by E2 was opposed by R5020 in an exclusively PR-A isoform dependent manner. Within this group, the small number of genes with better known functions in breast tumor biology tended to support growth and inhibit invasiveness.
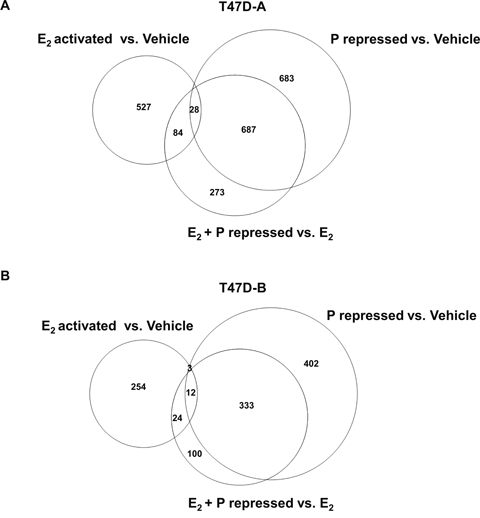
Figure 9: Effect of low dose progestin on the gene activation profile of estrogen in relation to PR-A and PR-B. Hormone depleted T47D-A or T47D-B cells at 30% confluence were treated with vehicle, 1 nM E2, 1 nM R5020 (P) or 1nM E2 plus1nM R5020 (E2 + P) for 48 h. Total RNA was extracted from the treated T47D-A and T47D-B cells and subjected to mRNA expression profiling as described under Materials and Methods. The mRNA profiling data is represented in the Venn diagrams in Panel A. (for T47DA cells) and in Panel B. (for T47D-B cells). Panels A and B show comparisons among the gene set activated by E2 (E2 activated vs. Vehicle), the gene set repressed by R5020 in the absence of E2 (P repressed vs. Vehicle) and the gene set repressed by R5020 in the presence of E2 (E2 + P repressed vs. E2). The data represents results from experimental triplicates.
DISCUSSION
The results of this study reveal that the positive effect of progestins on invasiveness of ER+ breast cancer cells has two components: 1. rescue of invasiveness from estrogen repression at relatively low progestin concentrations that is mediated exclusively by PR isoform A and 2. estrogen-independent induction of invasiveness at high progestin concentrations that is mediated exclusively by PR isoform B. Moreover, PR-A was sensitized to even lower levels of progestin when this receptor isoform was overexpressed relative to PR-B. Similar to the observations here on PR isoforms, other steroid receptors are also known to induce distinct genotropic and phenotypic effects at different hormone doses as well as hypersensitization to hormone by a few fold overexpression of the receptor [41, 57].
The relevance of the above findings to the physiological hormone status prior to and after menopause is apparent. The estrogen dose that was required for substantial or virtually complete suppression of invasiveness in ER+ cells is well under the plasma levels of estrogen in pre-menopausal women. It is also within the range of plasma and breast tissue levels of estrogen in post-menopausal women. The full effect of PR-A on the invasiveness of the various breast cancer cell lines occurred at < 1 nM progesterone and the dose requirement was reduced to < 0.2 nM when the expression level of PR-A was elevated. Thus, dysregulated PR-A has the potential to rescue invasiveness of breast cancer cells from estrogen regulation in response to post-menopausal plasma/breast tissue progesterone levels. This is in contrast to PR-B, which only induced invasiveness progressively with progesterone dose in the range of 5 nM to 50 nM. Thus, isoform A of PR plays the predominant hormone-dependent role in increasing invasiveness of ER+ breast cancer cells at progesterone concentrations that include the entire range of follicular phase, luteal phase and post-menopausal hormone levels, particularly when the cells overexpress PR-A. The findings on the role of PR isoforms also extend to plasma progestin levels associated with the use of MPA, either in contraception or in hormone replacement therapy. Therefore in luminal breast cancer, prior to diagnosis or after cessation of treatment, PR-A may have a greater mechanistic role in promoting invasiveness than PR-B.
The unique ability of only isoform A of PR to oppose regulation of invasiveness by estrogen at low progesterone concentrations is clearly reflected in the differential abilities of PR-A and PR-B to mediate cross-talk between progesterone and estrogen with respect to patterns of gene regulation. Gene expression analysis using isogenic recombinant (T47D) cells that exclusively expressed either the A or the B isoform of PR revealed that the cross-talk between estrogen and low dose progesterone affected the expression of estrogen target genes with diverse functions. However, among these genes, the subsets that were regulated by progesterone through PR-A vs. PR-B were largely non-overlapping. The genes whose regulation by estrogen was opposed by progesterone in an exclusively PR-A dependent manner included both estrogen-activated and estrogen-repressed genes. The estrogen-repressed genes were more noteworthy in the context of this study as they included genes with established roles in progression of breast cancer. Moreover, we demonstrated that selected genes from this subset (HES1, PRKCH, ELF5 and TM4SF1) did support invasiveness in ER+ breast cancer cells.
In response to the binding of progesterone, several mechanisms could conceivably enable PR-A to oppose estrogen's action on a subset of estrogen target genes. The ligand-dependent activity of PR-A did not result in any change in ER expression. Rather, the exact mechanism of PR-A isoform dependent cross-talk between progesterone and estrogen signaling could depend on the target gene context. For example, (i) PR-A could compete with ER to bind to tethering proteins at repressive sites in the chromatin, either simply blocking repression by estrogen/ER or activating the target gene; (ii) PR-A could bind at chromatin sites that are different from the repressive sites of ER binding and compete with ER for interaction with the pre-initiation complex of the target genes; (iii) PR-A could indirectly oppose gene regulation by estrogen by regulating transcription of other regulatory proteins or microRNAs. The amino-terminal truncation in PR-A could expose protein binding motifs that are unexposed in PR-B enabling unique or higher affinity interactions of agonist bound PR-A with other regulatory proteins in the chromatin. Similar chromatin interactions of PR-B may therefore require higher doses of progestins. More extensive studies including ChIP-seq analyses should help to establish specific mechanisms by which PR-A may de- regulate estrogen target genes.
In mice, selective ablation of PR-B revealed that PR-B was not required for the normal physiology ofthe uterus or the ovary but was necessary for pregnancy-associated mammary gland morphogenesis [58]. That study demonstrated that the ability of progesterone to suppress estrogen-induced endometrial proliferation was due to PR-A. In contrast, when PR-A was selectively ablated, progesterone not only failed to inhibit estrogen-induced cell proliferation in the endometrium but actually further increased proliferation of the uterine epithelium, an effect mediated by PR-B [59]. Therefore, given the necessary role of PR-A in endometrial physiology, selectively disrupting its actions in breast cancer cells vs. endometrial tissue will require a better understanding of tissue-specific molecular pathways by which PR-A opposes estrogen signaling in breast cancer. Identifying and narrowly targeting a critical cross-talk pathway between PR-A and ER may enable suppression of tumor progression without disrupting the protective role of PR-A in the endometrium or the adverse effects of a broader PR antagonist. Such an intervention may also be useful in combination hormone replacement therapy. A molecular signature of hyperactive PR-A may also more effectively predict tumor progression.
MATERIALS AND METHODS
Chemicals and reagents
Dulbecco's modified Eagles medium (DMEM) and phenol red-free DMEM, glutamine, penicillin, streptomycin, Fetal Bovine Serum (FBS), charcoal stripped FBS and TaqMan probes were purchased (Life Technologies, Carlsbad, CA). 17β-estradiol (E2), R5020, RU486, progesterone and medroxyprogesterone acetate (MPA) were purchased from Sigma Aldrich (Saint Louis, MO). Growth factor reduced matrigel (Cat# 356231) and calcein AM fluorescent dye (Cat# 354216) were purchased from BD Biosciences (San Jose, CA). PR-B directed siRNAs [60, 61] and control non-silencing siRNA (Cat# SIC001) were ordered from Sigma Aldrich (St. Louis, MO). siRNAs targeting TM4SF1 (Cat# S8367), HES1 (Cat# S6920), PRKCH (CAT#S1107), and ELF5 (CAT# S4629) were purchase from Life Technologies (Carlsbad, CA).
Cell culture and treatment
BT474, T47D and ZR-75-1 breast cancer cells (American Type Culture Collection) were cultured in DMEM supplemented with FBS (10%) penicillin (100 unit/ml) streptomycin (100ug/ml) and L-glutamine (2 mM). T47D-A and T47D-B cells were a generous gift from Dr. Katherine Horowitz (University of Colorado, Denver, CO) and were cultured as previously described [16]. The cell lines were all cultured at 37°C with 5% CO2. Before hormone treatment, cells were plated in 6-well plates at 30% confluence in phenol red-free media supplemented with charcoal-stripped FBS and incubated at 37°C with 5% CO2 for 48 h. Cells were then treated with vehicle, progesterone, MPA, R5020, RU486 and E2 alone or in various combinations at concentrations as indicated for each individual experiment for a duration of 48 h. The cells were then harvested for mRNA analysis, western blot analysis or cell invasion assays.
Western blot analysis
Cells were lysed using RIPA Buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1% SDS, and 50 mM Tris pH 8.0) containing protease inhibitor cocktail (Pierce Biotechnology, Rockford IL). The lysates were chilled on ice and agitated by vortex every ten minutes for one hour. Total protein concentration was measured by Bradford assay (Bio-Rad laboratories, Hercules, CA). A total amount of 10–40 μg protein per sample was resolved by electrophoresis on a 8% SDS-polyacrylamide gel and transferred to a PVDF membrane (Millipore Corporation, Bedford MA). Membranes were probed with primary polyclonal rabbit anti-PR antibody (sc-539, Santa Cruz biotechnologies, CA), polyclonal rabbit anti-ERα antibody (sc-543, Santa Cruz Biotechnologies, CA), mouse monoclonal anti-GAPDH antibody (sc-4472, Santa Cruz Biotechnologies, CA), rabbit polyclonal anti-HES1 (sc-25392, Santa Cruz Biotechnologies, CA) or mouse monoclonal anti-ELF-5(sc-376737, Santa Cruz Biotechnologies, CA). The blots were then probed with appropriate horseradish peroxidase conjugated secondary antibody (Vector Laboratories, MD). The protein bands were visualized using enhanced chemiluminescence reagent Hyglo Quick spray (Denville Scientific, South Plainfield, NJ) per the manufacturer's suggested protocol. Relative protein expression was determined by ImageJ (National Institutes of Health, USA).
RNA isolation, reverse transcription PCR and real time PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen, MD). Reverse transcription PCR reactions were performed using high capacity complementary DNA archive kit (Life Technologies Corporation, Carlsbad, CA) according to manufacturer's protocol. cDNA was measured by quantitative real time PCR using the StepOne Plus Real time PCR system (Life technologies Corporation, Carlsbad, CA). All mRNA measurements were performed in biological triplicates, and all CT values were normalized to intra-sample GAPDH. mRNA values were represented as fold difference, which is calculated using the formula = 2−ΔΔCT, where ΔΔCT = ΔCT sample − ΔCT calibrator (ΔCT = CT of gene of interest- CT of GAPDH).
Boyden chamber transwell invasion assay
Cells (1 × 105) were re-suspended in the appropriate culture media devoid of serum and phenol red and added to the top chamber of the flouroblok inserts (Cat# 351152, 8 μM pore membrane: BD biosciences, Bedford, MA) coated with growth factor reduced matrigel (0.2 mg/ml). The chemoattractant comprised phenol red-free media supplemented with FBS (20%). The appropriate hormone treatment was included in both the top and bottom chambers. Each treatment was replicated in three wells and the entire experiment was replicated at least three times. Cells were allowed to invade for 24 h at 37°C with 5% CO2. Cells that invaded to the bottom surface were stained with calcein AM (4ug/ml) in serum free media in the dark for 1 h at 37°C with 5% CO2. Images were captured in an identical manner from each well in 5 non-overlapping fields (the middle of the well and surrounding fields) using a 4x objective. Images were analyzed using ImageJ software (National Institutes of Health, USA) and the number of cells invaded was quantified by brightness and pixel size.
Migration Assay
Pre-treatment and preparation of cells and the experimental protocol were identical to those described above for the Boyden chamber transwell invasion assay with the exception that the transwells were devoid of matrigel.
Lentiviral transduction
293FT cells were used to generate lentiviral particles by transfection using lipofectamine 2000 (Life Technologies Corporation, Carlsbad, CA). Packaging plasmids pMD2G, PMDLg/RRE, and pRSV/Rev were cotransfected with pCDH PR-A expression plasmid, or pCDH empty vector plasmid. Lentivirus containing supernatant was harvested at 48 h and 72 h after transfection. T47D cells were plated in phenol red-free DMEM supplemented with heat-inactivated charcoal-stripped FBS (10%) and 2 mM L-Glutamine two days before infection. For infection, T47D cells were transduced with either pCDH empty vector lentivirus or pCDH PR-A lentivirus with polybrene (8 μg/ml) for 5 h. A second transduction was performed similarly for another 5 h. The cells were then incubated in phenol red-free DMEM supplemented with charcoal-stripped serum (10%) and L-Glutamine (2 mM) for 48 h. Following infection, cells were harvested for western blots and cell invasion assays as described previously.
siRNA Transfection
Cells were plated to 30% confluence without antibiotic in phenol-red free DMEM medium supplemented with 10%charcoal-stripped FBS. 24 hours later cells were transfected with siRNA directed against specific gene targets or non-silencing siRNA using lipofectamine (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol.
mRNA expression profiling
T47D-A and T47D-B cells were depleted of hormone for 48 h as described above. Cells were then either treated with vehicle, 1nM E2, 1nM R5020, or 1nM E2+ 1nM R5020 for 48 h. Total RNA was isolated using the RNeasy mini kit (Qiagen, MD). Sample identities were randomized for blinded analysis. The samples were analyzed at the Wayne State University School of Medicine Applied Genomics Center (AGTC) using the HumanHT-12 v4 Expression BeadChip with the Illumina HiScan System (Illumina, San Diego, CA). A total of 47,000 probes were used to analyze the transcriptome expression for each treatment group. Data was analyzed using Partek V6.6 software (St. Louis, MO), and processed using genome Studio (Illumina, San Diego, CA). Expression values were normalized using quantile-normalization, with background subtraction. Log transformation to the base of 2, followed by one way ANOVA was used to determine error. The differentially expressed genes were identified by comparing E2 treatment with vehicle treatment, R5020 treatment with vehicle treatment and E2 treatment with E2+ R5020 treatment (repressed or activated with a fold difference of 1.5 and a p value < 0.05). Genes that had activated expression in E2+ R5020 treatment but were repressed by E2 treatment were identified. Genes that were activated in E2 treatment but repressed by E2+ R5020 were also identified. Gene ontology analysis was performed by literature mining by searching the MEDLINE database (National institutes of Health, USA) with a query of “name of gene” followed by the term “AND Cancer” or “ AND Breast Cancer”. All articles under the specified query were examined to determine gene function in breast cancer. Validation of Microarray Data was performed by real-time RT-PCR as described above using TaqMan probes.
Statistical analysis
Experimental values were presented as mean +/− standard deviation using triplicate treatment sets. The statistical difference between values was determined by using one way ANOVA followed by post hoc paired t-test. The significant P values are noted in the figures. Concordant results were obtained from at least three repetitions of the experiments conducted on different days.
ACKNOWLEDGMENTS
This study utilized the Wayne State University School of Medicine Applied Genomics Core. We thank Yanfang Huang for technical support. We are grateful to Dr. Kathryn Horwitz for sharing the recombinant T47D cells.
CONFLICTS OF INTEREST
The authors have no conflicts to declare.
Authors contributions
All of the authors made intellectual contributions to make this study possible and also participated in discussions of the results and in the manuscript preparation. All authors approve the final manuscript. T.M. was responsible for the design and execution of most of the experiments. M.P. performed trouble shooting, provided guidance to T.M. and also participated in experimental design. R.R. designed and conducted some of the experiments. M.R. conceptualized and directed the entire project including the manuscript preparation.
GRANT SUPPORT
This work was supported by NIH R01-grant CA 140690 to M. R. and WSUSOM Cancer Biology Program NRSA-T32 Training Grant fellowships (T32-CA009531) to T.M. and R.R. The Wayne State University School of Medicine Applied Genomics Core is supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne state University
REFERENCES
1. American Cancer Society. Cancer facts & figures. The Society; 2008.
2. Ries L, Eisner M, Kosary C, Hankey B, Miller B, Clegg L, Mariotto A, Feuer E, Edwards B. SEER Cancer Statistics Review 1975–2004. National Cancer Institute; Bethesda, MD: Available Seer Cancer Govcsr 1975–2001; 2007.
3. Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol Off J Eur Soc Med Oncol ESMO. 2012 Oct; 23Suppl 7:vii11–9.
4. Zhang XH-F, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis Dormancy in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res. 2013 Dec 1; 19:6389–97.
5. Geisler J. Breast cancer tissue estrogens and their manipulation with aromatase inhibitors and inactivators. J Steroid Biochem Mol Biol. 2003; 86:245–53.
6. Pasqualini J, Chetrite G, Blacker C, Feinstein M, Delalonde L, Talbi M, Maloche C. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre-and postmenopausal breast cancer patients. J Clin Endocrinol Metab. 1996; 81:1460–4.
7. Newton CJ, Samuel DL, James VH. Aromatase activity and concentrations of cortisol, progesterone and testosterone in breast and abdominal adipose tissue. J Steroid Biochem. 1986; May 24:1033–9.
8. Åkerlund M, Batra S, Helm G. Comparison of plasma and myometrial tissue concentrations of estradiol-17β and progesterone in nonpregnant women. Contraception. 1981; 23:447–55.
9. Kinoshita T, Honma S, Shibata Y, Yamashita K, Watanabe Y, Maekubo H, Okuyama M, Takashima A, Takeshita N. An innovative LC-MS/MS-based method for determining CYP 17 and CYP 19 activity in the adipose tissue of pre-and postmenopausal and ovariectomized women using 13C-labeled steroid substrates. J Clin Endocrinol Metab. 2014; 99:1339–47.
10. Liang Y, Besch-Williford C, Brekken RA, Hyder SM. Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res. 2007; 15; 67:9929–36.
11. Liang Y, Benakanakere I, Besch-Williford C, Hyder RS, Ellersieck MR, Hyder SM. Synthetic progestins induce growth and metastasis of BT-474 human breast cancer xenografts in nude mice. Menopause N Y N. 2010; 17:1040–7.
12. Chlebowski RT, Manson JE, Anderson GL, Cauley JA, Aragaki AK, Stefanick ML, Lane DS, Johnson KC, Wactawski-Wende J, Chen C. Estrogen plus progestin and breast cancer incidence and mortality in the Women’s Health Initiative Observational Study. J Natl Cancer Inst. 2013; 105:526–35.
13. LaCroix AZ, Chlebowski RT, Manson JE, Aragaki AK, Johnson KC, Martin L, Margolis KL, Stefanick ML, Brzyski R, Curb JD. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. Jama. 2011; 305:1305–14.
14. Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012; 357:18–29.
15. Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002; 277:5209–18.
16. Shatnawi A, Tran T, Ratnam M. R5020 and RU486 act as progesterone receptor agonists to enhance Sp1/Sp4-dependent gene transcription by an indirect mechanism. Mol Endocrinol. 2007; 21:635–50.
17. Graham JD, Yager ML, Hill HD, Byth K, O’Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol Baltim Md. 2005; 19:2713–35.
18. Mote P, Bartow S, Tran N, Clarke C. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002; 72:163–72.
19. Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004; 10:2751–60.
20. Diaz J, Aranda E, Henriquez S, Quezada M, Espinoza E, Bravo ML, Oliva B, Lange S, Villalon M, Jones M. Progesterone promotes focal adhesion formation and migration in breast cancer cells through induction of protease-activated receptor-1. J Endocrinol. 2012; 214:165–75.
21. Ibrahim YH, Byron SA, Cui X, Lee AV, Yee D. Progesterone Receptor-B Regulation of Insulin-Like Growth Factor-Stimulated Cell Migration in Breast Cancer Cells via Insulin Receptor Substrate-2. Mol Cancer Res. 2008; 6:1491–8.
22. Fu X-D, Giretti MS, Baldacci C, Garibaldi S, Flamini M, Sanchez AM, Gadducci A, Genazzani AR, Simoncini T. Extra-nuclear signaling of progesterone receptor to breast cancer cell movement and invasion through the actin cytoskeleton. PLoS One. 2008; 3:e2790.
23. Fu X-D, Goglia L, Sanchez AM, Flamini M, Giretti MS, Tosi V, Genazzani AR, Simoncini T. Progesterone receptor enhances breast cancer cell motility and invasion via extranuclear activation of focal adhesion kinase. Endocr Relat Cancer. 2010; 17:431–43.
24. Holley AK, Kiningham KK, Spitz DR, Edwards DP, Jenkins JT, Moore MR. Progestin stimulation of manganese superoxide dismutase and invasive properties in T47D human breast cancer cells. J Steroid Biochem Mol Biol. 2009; 117:23–30.
25. Henriquez S, Calderon C, Quezada M, Oliva B, Bravo ML, Aranda E, Kato S, Cuello MA, Gutiérrez J, Quest AF. Progesterone utilizes distinct membrane pools of tissue factor to increase coagulation and invasion and these effects are inhibited by TFPI. J Cell Physiol. 2011; 226:3278–85.
26. Carnevale RP, Proietti CJ, Salatino M, Urtreger A, Peluffo G, Edwards DP, Boonyaratanakornkit V, Charreau EH, de Kier Joffé EB, Schillaci R. Progestin effects on breast cancer cell proliferation, proteases activation, and in vivo development of metastatic phenotype all depend on progesterone receptor capacity to activate cytoplasmic signaling pathways. Mol Endocrinol. 2007; 21:1335–58.
27. Kariagina A, Xie J, Langohr IM, Opreanu RC, Basson MD, Haslam SZ. Progesterone Decreases Levels of the Adhesion Protein E-Cadherin and Promotes Invasiveness of Steroid Receptor Positive Breast Cancers. Horm Cancer. 2013; 4:371–80.
28. Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol. 2005; 19:574–87.
29. Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007; 9:R6.
30. Gomez-Fernandez C, Daneshbod Y, Nassiri M, Milikowski C, Alvarez C, Nadji M. Immunohistochemically determined estrogen receptor phenotype remains stable in recurrent and metastatic breast cancer. Am J Clin Pathol. 2008; 130:879–82.
31. Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001; 344:276–85.
32. Lin K-H, Wang W-J, Wu Y-H, Cheng S-Y. Activation of antimetastatic Nm23-H1 gene expression by estrogen and its α-receptor. Endocrinology. 2002; 143:467–75.
33. Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004; 51:55–67.
34. Beauchemin D, Lacombe C, Van Themsche C. PAX2 is activated by estradiol in breast cancer cells of the luminal subgroup selectively, to confer a low invasive phenotype. Mol Cancer. 2011; 10:148.
35. Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S, Garcia M. Estrogen receptor mediated inhibition of cancer cell invasion and motility: an overview. J Steroid Biochem Mol Biol. 1998; 65:163–8.
36. Guttilla IK, Adams BD, White BA. ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab, TEM. 2012; 23:73–82.
37. Sisci D, Maris P, Cesario MG, Anselmo W, Coroniti R, Trombino GE, Romeo F, Ferraro A, Lanzino M, Aquila S, Maggiolini M, Mauro L, Morelli C, Andò S. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle Georget Tex. 2013; 1;12:3405–20.
38. Lin VC, Ng EH, Aw SE, Tan MG, Ng EH, Chan VS, Ho GH. Progestins inhibit the growth of MDA-MB-231 cells transfected with progesterone receptor complementary DNA. Clin Cancer Res. 1999; 5:395–403.
39. Lin VC, Eng AS, Hen NE, Ng EH, Chowdhury SH. Effect of progesterone on the invasive properties and tumor growth of progesterone receptor-transfected breast cancer cells MDA-MB-231. Clin Cancer Res. 2001; 7:2880–6.
40. Sumida T, Itahana Y, Hamakawa H. Reduction of human metastatic breast cancer cell aggressiveness on introduction of either form A or B of the progesterone receptor and then treatment with progestins. Cancer Res. 2004; 64:7886–92.
41. Chandrasekharan S, Kandasamy KK, Dayalan P, Ramamurthy V. Estrogen induced concentration dependent differential gene expression in human breast cancer (MCF7) cells: role of transcription factors. Biochem Biophys Res Commun. 2013; 2;437:475–81.
42. Barnett JB, Woods MN, Lamon-Fava S, Schaefer EJ, McNamara JR, Spiegelman D, Hertzmark E, Goldin B, Longcope C, Gorbach SL. Plasma lipid and lipoprotein levels during the follicular and luteal phases of the menstrual cycle. J Clin Endocrinol Metab. 2004; 89:776–82.
43. Hankinson SE, Manson JE, Spiegelman D, Willett WC, Longcope C, Speizer FE. Reproducibility of plasma hormone levels in postmenopausal women over a 2–3-year period. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 1995; 4:649–54.
44. Test Guide Historical Reference Ranges. Progesterone. NIH Clinical Center. 2014;. [Internet].
45. Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004; 15;96:1856–65.
46. Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, Horwitz KB. New T47D breast cancer cell lines for the independent study of progesterone B-and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994; 54:3868–77.
47. Wilson BJ, Giguère V. Meta-analysis of human cancer microarrays reveals GATA3 is integral to the estrogen receptor alpha pathway. Mol Cancer. 2008; 7:49.
48. Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem. 2010; 285:14042–51.
49. Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-α. Mol Endocrinol. 2007; 21:2907–18.
50. Ye Y, Xiao Y, Wang W, Yearsley K, Gao J, Shetuni B, Barsky S. ERα signaling through slug regulates E-cadherin and EMT. Oncogene. 2010; 29:1451–62.
51. Guttilla IK, Adams BD, White BA. ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab. 2012; Feb 23:73–82.
52. Mishell DR. Pharmacokinetics of depot medroxyprogesterone acetate contraception. J Reprod Med. 1996; 41:381–90.
53. Osborne CK. Endocrine Therapies in Breast and Prostate Cancer. Springer US; 1988. Available from: https://books.google.com/books?id=gW0eBAAAQBAJ.
54. Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010; 15:169–90.
55. Patki M, Trumbly R, Ratnam M. Differential effects of estrogen-dependent transactivation vs. transrepression by the estrogen receptor on invasiveness of HER2 overexpressing breast cancer cells. Biochem Biophys Res Commun. 2015.
56. Wang X, Belguise K, O’neill CF, Sánchez-Morgan N, Romagnoli M, Eddy SF, Mineva ND, Yu Z, Min C, Trinkaus-Randall V. RelB NF-κB represses estrogen receptor α expression via induction of the zinc finger protein Blimp1. Mol Cell Biol. 2009; 29:3832–44.
57. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10:33–9.
58. Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci. 2003; 100:9744–9.
59. Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000; 289:1751–4.
60. Hardy DB, Janowski BA, Corey DR, Mendelson CR. Progesterone receptor plays a major antiinflammatory role in human myometrial cells by antagonism of nuclear factor-κB activation of cyclooxygenase 2 expression. Mol Endocrinol. 2006; 20:2724–33.
61. Wu Y, Shi X. The knockdown of progesterone receptor isoform B (PR-B) promotes proliferation in immortalized endometrial stromal cells. Fertil Steril. 2008; 90:1320–3.