
INTRODUCTION
Gallbladder cancer (GBC) is the most common type of biliary-tract carcinomas [1]. Most cases are diagnosed in advanced stages and patients cannot undergo curative surgery. Chemotherapy has been the main treatment option with agents such as gemcitabine, cisplatin, oxaliplatin, capecitabine, and 5-fluorouracil (5-FU) [2, 3]. A recent randomized phase III clinical trial (ABC-02 trial) established the cisplatin/gemcitabine (Gem/CDDP) combination as standard treatment regimen for patients with advanced biliary-tract cancer [4, 5]. However, the response to chemotherapy and radiotherapy is extremely limited, with modest impact in overall survival [4, 6–12].
The recent progress in identification and understanding of molecular alterations of GBC may improve the clinical management of patients through the application of more specific and effective therapies [11, 13, 14]. The phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathway has been proposed as an interesting therapeutic target in cancer with a pivotal role in cell cycle progression, cell proliferation and angiogenesis [15–30]. This pathway is frequently activated in different human cancers by a variety of genetic and epigenetic events [31]. Molecular therapies targeting mTOR, have being actively investigated and many of these have now progressed to clinical trials in many different types of cancer [16, 17, 32].
Previous studies by our group have shown the upregulation of the AKT/mTOR signaling pathway in advanced GBC. Through immunohistochemical assays, we have found that phospho-mTOR and phospho-P70S6K expression are significantly elevated in human gallbladder carcinoma compared to non-neoplastic tissues [33, 34]. Moreover, high phospho-mTOR expression was correlated with a worse prognosis in patients with advanced GBC [34]. In vitro analysis in GBC cell lines have demonstrated the efficacy of mTOR inhibitors on reducing cell growth, cell migration, and phospho-P70S6K expression [33, 34]. Preclinical studies also have confirmed the therapeutic effects of mTOR inhibitors. Wu et al., have reported that rapamycin can reduce the incidence of gallbladder carcinoma in BK5.erbB2 transgenic mouse models [35]. Zong et al., reported that the mTOR pathway inhibition attenuates the migration and invasion capacities of GBC using in vivo tumor metastasis mouse model [36]. These findings provide a rationale for the potential use of mTOR inhibitors as a therapeutic strategy for human gallbladder carcinoma.
Rapamycin is an mTOR inhibitor and an antifungal agent with immunosuppressive properties, which has an established effect on suppressing tumor growth in a number of solid tumors [37]. The efficiency of mTOR inhibitors has enabled the development of a number of rapalogs (rapamycin analogs). These rapalogs, which include sirolimus, temsirolimus, everolimus, and deforolimus [15], are allosteric inhibitors of mTORC1 that form a complex with the intracellular receptor FKBP12, which binds to mTOR and inhibits mTORC1 downstream signaling. Apart from rapalogs, there is now strong interest in small-molecule ATP-competitive inhibitors of mTOR kinase, which can either act selectively on mTORC1 and mTORC2 (AZD8055, WYE-354, PP30, PP242) or as dual PI3K- and mTOR inhibitors (BEZ235 and XL765) [18, 38].
In this study we evaluated the antitumor activity of the allosteric mTORC1 inhibitor, rapamycin and of the ATP-competitive mTOR inhibitor WYE-354 on preclinical xenograft GBC tumor models.
RESULTS
WYE-354 reduces cell viability and phosphorylation of AKT/mTOR downstream proteins in gallbladder cancer cells
We decided to study the in vitro effect of an ATP-competitive mTOR inhibitor, WYE-354, which inhibits the catalytic activity of mTORC1 and mTORC2, unlike rapamycin and its rapalogs, which are specific inhibitors of mTORC1 [21, 38]. In an earlier publication of our group, rapamycin also has been shown to significantly decrease cell viability in gallbladder cancer cells [33].
Cell viability was analyzed by MTS assay according to the manufacturer's protocol. Cells were treated with increasing concentrations of WYE-354 for 24, 48, and 72 hours. As shown in Figure 1A, WYE-354 significantly reduced cell viability starting at a 1 μM concentration after a 24 hours exposure, in both studied cell lines (P < 0.001). We did not observe a decrease in cell viability at a dose of 100 nM, except for the TGBC-2TKB cell line after 72 hours of treatment.
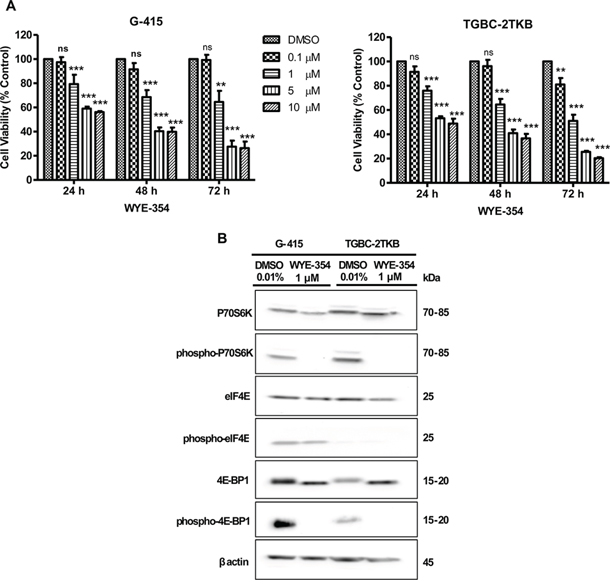
Figure 1: In vitro effects of WYE-354 on cell growth and mTOR signaling pathway in two gallbladder cancer cell lines. A. G-415 and TGBC-2TKB cells were treated with increasing concentrations of WYE-354. Cell viability was determined after 24, 48, and 72 hours of treatment. Data are shown as mean ± SD of at least three independent experiments in quintuplicate (**P < 0.01; ***P < 0.001; ns: not significant). B. G-415 and TGBC-2TKB cells were treated with WYE-354 (1 μM), for 18 hours. Control cells received an equivalent amount of solvent (0.01% dimethylsulfoxide). Western blot analysis was carried out using antibodies against the total and phosphorylated portion of P70S6K, 4E-BP1, and eIF4E proteins. Protein loading was normalized using an antibody recognizing β-actin.
To further investigate the in vitro effects of WYE-354 on mTOR signaling, we evaluated the phosphorylation status of mTOR effectors by immunoblotting. Cells were exposed to WYE-354 (1 μM) and 0.01% dimethylsulfoxide (as control) for 18 hours and were lysed and analyzed by Western blot using commercial antibodies. As shown in Figure 1B, the phosphorylation of the downstream effectors of mTOR, 4E-BP1 and P70S6K, were strongly inhibited in vitro by WYE-354 treatment. No significant changes were observed in phospho-eIF4E and in total P70S6K, 4E-BP1 and eIF4E protein expression under the treatment conditions assayed.
Exposure to mTOR inhibitors decreases in vitro cell migration and invasion in gallbladder cancer cells
In order to establish the effect on cell migration and cell invasion of WYE-354 and rapamycin, G-415 and TGBC-2TKB were exposed to 0.01% dimethylsulfoxide (as control), WYE-354 (1 μM), or rapamycin (50 nM) for 12 hours. After 24 hours, the migration and invasion rates were significantly lower in treated cells compare with untreated cells (P < 0.001; P < 0.01, respectively). Relative migration rates observed in G-415 were 36.7% (rapamycin) and 32.8% (WYE-354), while TGBC-2TKB showed a migration rate of 21.0% (rapamycin) and 28.9% (WYE-354) (Figure 2A). Relative invasion rates in G-415 cells were 51.6% (rapamycin) and 41.5% (WYE-354), while TGBC-2TKB exhibited 41.0% (rapamycin) and 38.1% (WYE-354) (Figure 2B).
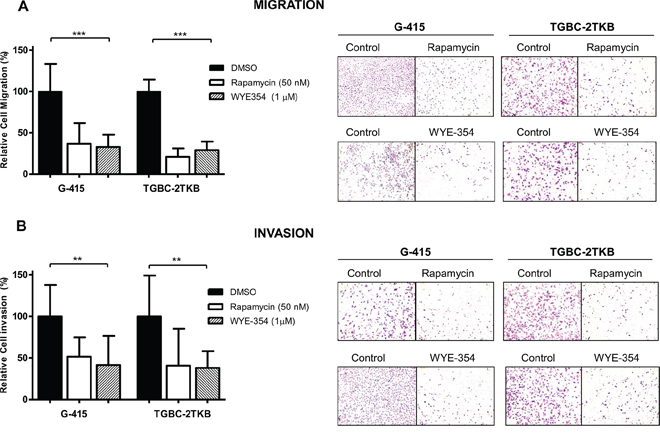
Figure 2: Effect of WYE-354 and rapamycin on cell migration and invasion. A. Migration and B. invasion assays were performed using 24-well transwell plates containing polycarbonate filters with an 8 μm pore size. Before seeding, G-415 and TGBC-2TKB cells were exposed to WYE-354 (1 μM), rapamycin (50 nM) or dimethylsulfoxide (0.01%) (control) for 12 hours at 37°C. Cells were counted in six randomly selected fields after 24 hours. Results are expressed as mean ± SD (**P < 0.001; ***P < 0.001).
Rapamycin and WYE-354 inhibit tumor growth on xenograft gallbladder cancer model
Based on the above information and previous work, we decided to study further whether mTOR inhibitors can be therapeutically effective in vivo on subcutaneously established human gallbladder tumors. The effect of rapamycin and WYE-354 on tumor growth was evaluated in xenograft GBC tumor models. 2 × 106 or 5 × 106 cells of G-415 or TGBC2TKB, respectively, were xenotransplanted into NOD-SCID mice subcutaneously. When tumors reached an average volume of 100 mm3, the mice were treated either with rapamycin or WYE354. Rapamycin was administered i.p. at a concentration of 10 mg/kg, daily for 5 days per week for 3 weeks, while WYE-354 was administrated at a daily i.p. dose of 50 mg/kg for 5 days. Mice were sacrificed 30 days after the initiation of the treatments and an autopsy was performed that included removal of the entire tumor area. Mice bearing G-415 or TGBC-2TKB tumors and treated with rapamycin exhibited 92.7% and 97.1% reduction in average tumor size (P < 0.001; P < 0.5), as well as 84.3% and 88.7% in tumor weight (P < 0.001; ns) compared to the control, respectively (Figures 3A, 3B and 3C). While mice treated with WYE-354 exhibited 68.6% and 52.4% reduction in average tumor size (P < 0.01; P < 0.01), as well as 82.9% and 45.5% (P < 0.01; ns) reduction in tumor weight, respectively. (Figures 4A, 4B and 4C). Macroscopic analysis of the tumors revealed decreased vascularization in treated groups compared to the control groups. This effect was stronger in mice treated with rapamycin. Inhibition of mTOR signaling by rapamycin and WYE-354 in tumor tissues obtained at the end of the in vivo studies was assessed by Western blot analysis. As shown in Figures 3D and 4D, treatment with either mTOR inhibitor partially decreased the phosphorylated status of P70S6K in both G-415 and TGBC-2TBK tumors. With the WYE-354 regimen, only half of the analyzed tumors, showed a significant but marginally decrease in phosphorylation status of P70S6K, compared to control mice. By contrast, all the tumors from animals treated with rapamycin showed a significant but marginally reduction in phospho-P70S6K. As shown in Supplementary Figure S1 and Supplementary Figure S2 (Figure S1 and Figure S2), an immunohistochemical analysis of phospho-4E-BP1 was performed on the xenograft tumors tissues. We observed that treatments with either mTOR inhibitor partially decreased the phosphorylated status of 4E-BP1 in both G-415 and TGBC-2TBK tumors. Mice treated with rapamycin showed lower expression of phospho-4E-BP1 in both cell lines, G-415 and TGBC-2TKB (Supplementary Figure S1). Mice treated with WYE-354 display regions with lower staining expression and smaller area of phospho-4E-BP1 compared with controls in both cell lines (Supplementary Figure S2). Altogether, the qualitative analysis of the staining of phospho-4E-BP1 showed a decrease in its expression in mice treated with each of the inhibitors in both cell lines. For WYE-354, the effect appears to be more noticeable for the cell line G-415, which is related to the greater cytotoxic effect observed in this cell line compared to TGBC-2TKB (Figure 2. Tumor weight reduction 82.9% vs 45.5%).
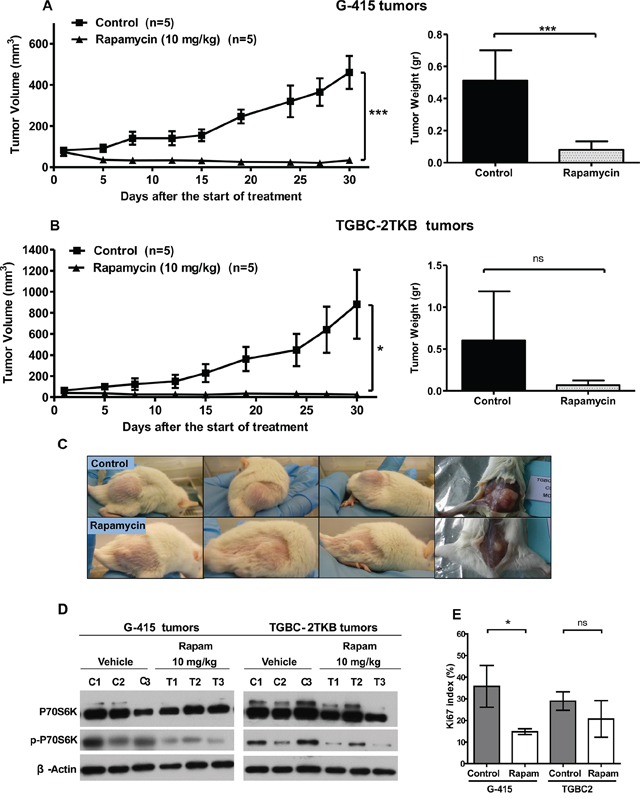
Figure 3: In vivo efficacy of rapamycin on human gallbladder cancer xenografts. A. and B. Tumor growth of mice harboring G-415 or TGBC2TKB tumors. G-415 or TGBC2TKB cells were injected subcutaneously into NOD-SCID mice. When tumors reached a volume of approximately 100 mm3, they were treated with rapamycin or vehicle, as described in the Materials and Methods section. Animals were sacrificed when tumors reached approximately 1500 mm3 or at day 30. Data are expressed as mean ± SD. Rapamycin exerted a statistically significant antitumor effect (compared with the groups treated with the vehicle (*P < 0.05; ***P < 0.001 at day 30)). C. Representative photographs of mice treated with vehicle or rapamycin. D. Western blot analysis of total P70S6K and phospho-P70S6K of tumor tissue from mice treated with vehicle or rapamycin. E. Ki67 proliferation index was assessed by immunohistochemistry in G-415 and TGBC-2TKB xenografts collected 30 days after starting the treatment. Ki67 positive cells were counted to calculate the Ki67 proliferation index. Data are expressed as mean ± SD. (*P < 0.05; ns: not significant).
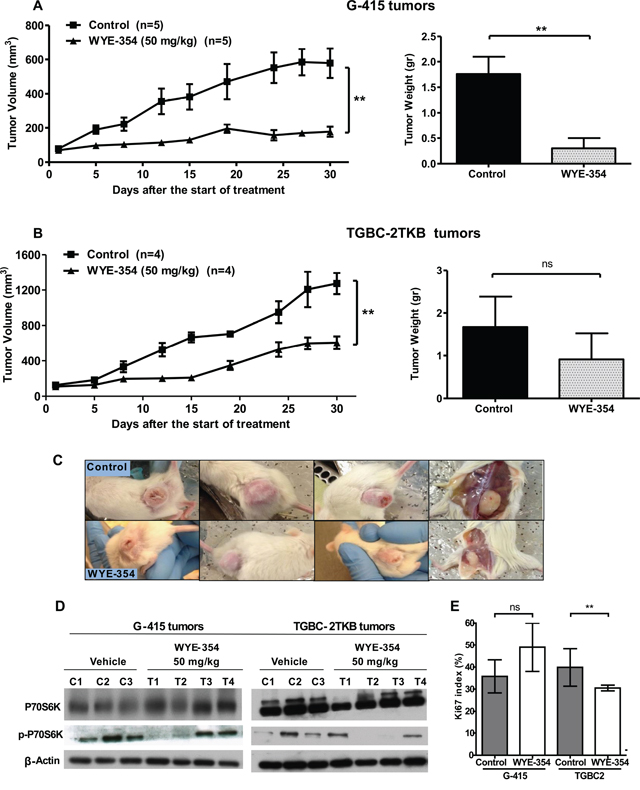
Figure 4: In vivo efficacy of WYE-354 on human gallbladder cancer xenografts. A. and B. Tumor growth of mice harboring G-415 or TGBC2TKB tumors. G-415 or TGBC2TKB cells were injected subcutaneously into NOD-SCID mice. When tumors reached a volume of approximately 100 mm3, they were treated with vehicle or WYE-354, as described in the Materials and Methods section. Animals were sacrificed when tumors reached approximately 1500 mm3 or at day 30. Data are expressed as mean ± SD. WYE-354 exerted a statistically significant antitumor effect (compared with the groups treated with the vehicle (**P < 0.01 at day 30)). C. Representative photographs of mice treated with vehicle or WYE-354. D. Western blot analysis of total P70S6K and phospho-P70S6K of tumor tissue from mice treated with vehicle or WYE-354. E. Ki67 proliferation index was assessed by immunohistochemistry in G-415 and TGBC-2TKB xenografts collected 30 days after the starting the treatment. Ki67 positive cells were counted to calculate the Ki67 proliferation index. Data are expressed as mean ± SD. (**P < 0.01; ns: not significant).
We also investigated the Ki67 proliferative index in tumors from animals treated with both mTOR inhibitors at the end of the in vivo assays. The expression of Ki67 was determined via standard immunohistochemical techniques on serial paraffin sections. The results showed that the number of Ki67 positive cells was decreased in both G-415 and TGBC-2TKB tumors from mice treated with rapamycin, 58.8% and 28.5%, respectively, (P < 0.5; ns) compared with their controls (Figure 3E). The proliferation index in tumors from animals treated with WYE-354 was significantly decreased only in TGBC-2TKB tumors - 23.2%; (P < 0.01) - and interestingly showed a higher Ki67 expression in treated G-415 tumors compared to the control, but this difference did not reach significance (Figure 4E).
DISCUSSION
Deregulation of the mTOR signaling pathway has been associated with the pathogenesis of various human cancers [20, 22, 39–44], and studies have shown its role in angiogenesis, progression, and treatment resistance [45]. In this study, we have evaluated for the first time the therapeutic efficacy of two mTOR inhibitors, rapamycin and WYE-354, in preclinical models of human GBC in NOD-SCID mice.
The best characterized downstream substrates of mTORC1 are the ribosomal protein P70S6K and 4E-BP1. Phosphorylation of 4E-BP1 releases eIF4E, allowing the initiation of translation. By acting on P70S6K, mTOR facilitates ribosome biogenesis and translation elongation [46–51]. As a downstream effector of mTOR pathway, the phosphorylation status of P70S6K commonly is used as a marker of mTOR activity and for the pharmacodynamic monitoring of mTOR inhibition [52, 53]. Previously published data from our group shows that phospho-mTOR and phospho-P70S6K expression are significantly higher in human gallbladder carcinoma than in non-neoplastic tissues and that high expression of phospho-mTOR is associated with poor survival in patients with advanced GBC [33, 34]. Similar to our previous in vitro studies with rapamycin in GBC cells [33], WYE-354 inhibited cell viability and decreased cell migration and invasion, accompanied by a markedly decreased phosphorylation status of P70S6K and 4E-BP1 under the treatment conditions assayed. These results indicate that WYE-354 negatively regulated the phosphorylation of effectors downstream from mTOR (mTORC1) and inhibited the mTOR signaling pathway under the study conditions. Although it has been reported that concentrations equal to or greater than 10 nM of WYE-354 have off target-effects on mTOR pathway [38, 54] we did not observe a decrease of in vitro cell survival at a dose of 100 nM. Similar observations have been reported for some tumoral cell lines treated with rapamycin. Concentrations of rapamycin required to reduce in vitro cell viability in breast cancer cells and malignant glioma cells, were significantly higher than the concentrations needed to suppress levels of phospho-P70S6K and phospho-4E-BP1 [55–58]. However, the efficacy of rapamycin is tumour- and cell-type specific, and the precise mechanism behind its actions is not completely understood [59].
Several studies have demonstrated the therapeutic efficacy of mTOR inhibitors on suppressing tumor growth in a number of solid tumors on human subcutaneous growing xenografts [19, 21, 60] and in many phase I–III clinical studies [16, 17, 32]. However, only a few studies have reported the use of mTOR inhibitors on in vivo human GBC. Zong et al. [36] used rapamycin to study the effect of mTOR pathway inhibition on migration and invasion of GBC in tumor metastasis mouse models. Weng et al. used rapamycin as an immunosuppressant that modified host innate or adaptive cellular immunity, facilitating virus infection for oncolytic virotherapy with myxoma virus, but rapamycin was not used as a treatment itself [61]. Our study demonstrated the high therapeutic efficacy of two mTOR inhibitors on suppressing tumor growth on subcutaneous GBC tumor models. At the regimens and doses studied, both rapamycin and WYE-354, were well tolerated with no significant weight loss or adverse effects, suggesting the toxicity of these inhibitors is low at least under the studied conditions. It is likely that the better therapeutic response observed in mice treated with rapamycin compared to WYE-354-treated mice was strongly related to the treatment regimen. Although both treatments significantly inhibited tumor growth compared to their controls, a prolonged-low dose treatment with rapamycin was clearly more effective at decreasing tumor mass than a short-term treatment with a higher dose of WYE-354. Hu et al. [19] observed that the rate of tumor growth in preclinical animal models of head and neck squamous cell carcinoma increased on days without rapamycin treatment. This observation and our results may imply that a continuous a long term treatment may provide better outcomes in inhibiting tumor growth than a short term and cyclical treatment regimen.
It has been widely studied that the positive regulation of AKT by mTORC2 leads to mTOR acting both upstream and downstream from AKT [16, 62]. Unlike WYE-354, rapamycin does not directly inhibit mTORC2, due to the FRB domain on mTORC not being accessible on mTORC2 [63, 64]. However, the remarkable therapeutic response observed in our study with rapamycin treatment may be in part due to the fact that a prolonged rapamycin treatment inhibits mTORC2 in some types of human cancer cells by preventing mTORC2 complex assembly, blocking the phosphorylation of its substrate AKT at Ser473 [51, 60]. The fact that the better anti-tumor effect observed in rapamycin-treated mice was accompanied with a greater decrease in the phosphorylation status of P70S6K suggests a more effective mTOR pathway inhibition [19, 52, 53]. However, both inhibitors only partially inhibited phospho-P70S6K in most tumors treated. This could suggest that the antitumor effect of these drugs is likely to be indirect. Some studies have shown that the effect might be via targeting normal cells [65]. Lkb1+/− mice treated with rapamycin showed a reduction in gastric tumor burden and in the number of polyps per mouse. Yet, the phosphorylation of ribosomal P70S6 kinase in the polyps from the treated mice was maintained. However, a significant reduction in microvessel density was seen in these polyps [59]. Systemic rapamycin treatment of mammary tumors grown in Cav-1 KO mice not only inhibited tumor growth but also stromal content and angiogenesis [66]. Checkley et al., reported that rapamycin inhibited skin tumor promotion accompanied with a significantly decrease in the number of infiltrating macrophages, T cells, neutrophils, and mast cells in the dermis [67].
Tumor mass reduction observed in both rapamycin and WYE-354 treated groups was associated with fewer vascularized tumors than in the controls. Several studies have established that the antitumor efficacy of rapamycin and its rapalogs as well as pyrazolopyrimidines such as WYE-354 can be attributed partly to a reduction in HIF1-alpha expression, which is an activator of vascular endothelial growth factor (VEGF) gene transcription [38, 46, 68–71]. In conclusion, our findings provide a proof of concept for the use of mTOR pathway-targeted therapies as potentially good candidates in clinical trials in advanced GBC patients.
MATERIALS AND METHODS
Cell culture
The human GBC cells G-415 and TGBC2KB were obtained from Riken BioResource Center (Ibaraki, Japan). G-415 were grown in Roswell Park Memorial Institute (RPMI) 1640 medium and TGBC-2TKB were grown in Dulbecco's Modified Eagle's Medium (high glucose), supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin and maintained in a 37°C atmosphere containing 5% CO2.
mTOR inhibitors
Rapamycin inhibitor (R-5000) was purchased from LC Laboratories (Woburn, MA, USA) and WYE-354 (A10114) was purchased from Adooq Bioscience LLC (Irvine, CA, USA). For in vitro assays the inhibitors were dissolved at 10 mM in dimethylsulfoxide as stock solutions and stored at −20°C. Inhibitors were diluted in culture medium before each in vitro experiment and 0.01% dimethylsulfoxide in culture medium was used as a vehicle control. For in vivo studies rapamycin was formulated in 4% ethanol, 5.2% Tween 80 and 5.2% PEG 400, while WYE-354 was formulated in 5% ethanol, 5% Tween 80 and 5% PEG 400 and stored at −20°C.
Antibodies
Rabbit monoclonal antibodies, anti-P70S6K (#2708) and anti-phospho-P70S6K (Thr389, #9234) anti-eIF4E (#2067), anti-phospho-eIF4E (Ser209, #9741), anti-4E-BP1 (#9644), anti-phospho-4E-BP1 (Thr37/46, #2855) and anti-β-actin (#4970) were purchased from Cell Signaling Technology and used for Western blot analysis. The secondary antibody was goat anti-rabbit IgG horseradish peroxidase (Santa Cruz Biotechnology Inc.). Mouse monoclonal antibody anti-Ki67 (Clone MIB1 DAKO, Glostrup, Denmark) was obtained from Dako, Agilent Technologies and rabbit monoclonal antibody anti-phospho-4E-BP1 (Thr37/46, #2855) was purchased from Cell Signaling Technology and used for immunohistochemical staining.
Western blot analysis
For in vitro assay, cells were seeded on 75 cm2 culture plates. At 60–70% confluency the cells were treated with WYE-354 (1 μM), rapamycin (50 nM), and 0.01% dimethylsulfoxide (as control) for 18 hours. Cell lysate was prepared using RIPA buffer (Sigma-Aldrich Co, St Louis, MO, USA). Briefly, cells were washed three times with cold phosphate-buffered saline (PBS) and lysed on ice with RIPA buffer containing a phenylmethylsulfonyl fluoride (PMSF), protease and phosphatase inhibitor cocktail (Sigma-Aldrich Co, St Louis, MO, USA). For in vivo experiments, tumor lysates were prepared on ice using cell lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl pH 7.4, 2 mM EDTA) containing a protease and phosphatase inhibitor cocktail (Sigma-Aldrich Co, St Louis, MO, USA). Whole lysates were collected after centrifugation at 8,000 g for 10 minutes at 4°C. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific Inc, Rockford, IL, USA) according to the manufacturer's instructions. Equal amounts of total cellular protein (70 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in 4%–12% NuPAGE® Bis-Tris Precast Gels (Novex, Life Technologies Corporation) and electrotransferred to polyvinylidene difluoride membranes (Immobilon®-P membrane Millipore, Bedford, MA, USA). The membranes were blocked with 1 × Tris-buffered saline containing 0.05% Tween (TBST) and 5% fat-free milk for 1 hour at room temperature and incubated overnight at 4°C with primary antibodies. After washing with TBST, the membranes were further incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Antibody-bound protein bands were detected with enhanced chemiluminescence reagent SuperSignal West Pico Substrate (Pierce). Images were acquired on a Thermo Scientific myECL Imager. β-actin expression was used as a loading control.
Cell viability assays
G-415 and TGBC-2TKB cell lines were plated onto 96 well plates at a density of 2 × 103 cells per well. After an overnight attachment period cells were treated with WYE-354. The number of viable cells was determined at certain intervals using CellTiter 96 Aqueous One Solution Cell Proliferation assay (Promega Corp., Madison, WI). Briefly, 20 μl CellTiter 96 solution was added to each well and the plates were incubated for 2 hour after which the absorbance of each well was read at a wavelength of 490 nm using a multiwell plate reader (Autobio Labtec Instruments Co, Ltd, Zhengzhou City, People's Republic of China). All assays were performed in quintuplicate, and each assay was repeated three times.
Transwell cell migration and invasion assays
Migration assays were performed using 24-well Transwell™ plates containing polycarbonate filters with an 8 μm pore size (BD Biosciences, Bedford, MA, USA). Cells were exposed to WYE-354 (1 μM), rapamycin (50 nM) and 0.01% dimethylsulfoxide (as control) for 12 hours at 37°C. Complete medium was placed in the lower chamber to act as a chemoattractant and G-415 or TGBC-2TKB cells were seeded at a density of 2.5 × 104 or 5 × 104 cells, respectively, with serum-free medium into the upper chamber. After 24 hours, the cells were fixed in methanol for 15 minutes and then stained with 0.05% crystal violet in 25% methanol/PBS for 15 minutes. Cells on top of the membrane were removed using a cotton swab, and the filters were washed with PBS. Cells on the underside of the filters were viewed and counted under a microscope in 6 randomly selected fields. Invasion assays were performed as for the migration assays described above, except inserts were pre-coated with Matrigel (Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, #356230) at concentrations of 20 μg for G-415 and 10 μg for TGBC2-TKB cell lines.
In vivo studies on subcutaneous tumors
8 to 12-week-old NOD-SCID mice (obtained from the animal facility of the Georg Speyer Haus, Frankfurt, Germany) were subcutaneously injected in one flank with either 2 × 106 or 5 × 106 cells of G-415 or TGBC2TKB, respectively, and re-suspended in 200 μl of PBS with 30% of Matrigel (Matrigel™ Basement Membrane Matrix, BD Biosciences). When the average tumor reached 100 mm3, mice were randomly separated into four groups and treated with rapamycin or WYE-354 and its respective vehicles. Rapamycin was administered at a daily intraperitoneal (i.p) dose of 10 mg/kg for 5 days per week for 3 weeks, while WYE-354 was administrated at a daily i.p dose of 50 mg/kg for 5 days. Tumor volumes were estimated twice a week from caliper measurements (volume = 0.52 × (width)2 × length).
Ethics statement
Mouse husbandry and animal experiments has been conducted in accordance with the ethical standards and according to the local animal protection law.
Immunohistochemical staining
Xenograft tumors were freshly removed, fixed in 10% buffer formalin and subsequently embedded in paraffin. The sections were manually stained with hematoxylin and eosin (HE) for normal histological evaluation. 3 μm-thick sections were deparaffinized with Histo-Clear (National Diagnostics; Atlanta, GA, USA) and rehydrated through alcohol gradient. Heat-induced antigen retrieval was performed by immersing slides in Tris/EDTA pH 9.0 buffer (10 mMTris, 1 mM EDTA) for 20 min at 99°C in a thermo-regulated bath.
Automated staining was performed using an antibody against Ki67 antigen (Clone MIB1 DAKO, Glostrup, Denmark) at a dilution of 1:100. Ki67 index was measured using the open access web application ImmunoRatio for automated image analysis [40] using 3–10 images per sample, depending on tumor size and excluding necrotic areas. Statistical analyses were performed using an unpaired Mann Whitney test using GraphPad Prism v. 5 (GraphPad Software, Inc. San Diego CA).
An immunohistochemical analysis of phospho-4EBP1 was performed on the xenograft tumors tissues using the antibody Phospho-4E-BP1 (Thr37/46) (236B4) Rabbit mAb #2855 (Cell Signaling Technology) at a dilution of 1:400. All samples were processed simultaneously under the same conditions using an autostainer.
Statistical analysis
Statistical analyses of in vitro and in vivo experiments were performed by analysis of variance (one way ANOVA) followed by Tukey's multiple comparisons test. P values less than 0.05 were considered significant. Data analysis was performed with the GraphPad Prism 5 (GraphPad Software, Inc. San Diego CA).
ACKNOWLEDGMENTS
We would like to thank Dr. Sebastian Newrzela (Senckenberg Institute of Pathology; Goethe-University of Frankfurt Medical School; Frankfurt am Main, Germany) and Tanya Syzonenko (Institute for Tumor Biology and Experimental Therapy, Georg-Speyer-Haus. Frankfurt. Germany) for technical support.
GRANT SUPPORT
FONDECYT (National Fund for Scientific and Technological Development) grants 1130204, 11130515 and 1151008; CEGIN (Center of Genetic and Immunological Studies) grant 09CN14-5960.
CONFLICTS OF INTEREST
The authors report no conflicts of interest in this work.
REFERENCES
1. Wistuba II, Gazdar AF. Gallbladder cancer: Lessons from a rare tumour. Nat Rev Cancer. 2004; 4:695–706.
2. Wiedmann M, Feisthammel J, Bluthner T, Tannapfel A, Kamenz T, Kluge A, Mossner J, Caca K. Novel targeted approaches to treating biliary tract cancer: the dual epidermal growth factor receptor and ErbB-2 tyrosine kinase inhibitor NVP-AEE788 is more efficient than the epidermal growth factor receptor inhibitors gefitinib and erlotinib. Anticancer Drugs. 2006; 17:783–795.
3. Fiteni F, Nguyen T, Vernerey D, Paillard MJ, Kim S, Demarchi M, Fein F, Borg C, Bonnetain F, Pivot X. Cisplatin/gemcitabine or oxaliplatin/gemcitabine in the treatment of advanced biliary tract cancer: a systematic review. Cancer Med. 2014; 3:1502–1511.
4. Lee J, Park SH, Chang HM, Kim JS, Choi HJ, Lee MA, Jang JS, Jeung HC, Kang JH, Lee HW, Shin DB, Kang HJ, Sun JM, Park JO, Park YS, Kang WK, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012; 13:181–188.
5. Williams KJ, Picus J, Trinkhaus K, Fournier CC, Suresh R, James JS, Tan BR. Gemcitabine with carboplatin for advanced biliary tract cancers: a phase II single institution study. HPB (Oxford). 2010; 12:418–426.
6. Woo SM, Lee WJ, Kim JH, Kim DH, Han SS, Park SJ, Kim TH, Lee JH, Koh YH, Hong EK. Gemcitabine plus Cisplatin versus Capecitabine plus Cisplatin as First-Line Chemotherapy for Advanced Biliary Tract Cancer: A Retrospective Cohort Study. Chemotherapy. 2013; 59:232–238.
7. Lamarca A, Benafif S, Bridgewater JA, Ross PJ, Valle JW. Efficacy and safety of cisplatin and gemcitabine (CG) chemotherapy for advanced biliary tract cancer (ABC) in jaundiced patients (pts). J Clin Oncol. 2014; 32.
8. Valle JW, Furuse J, Jitlal M, Beare S, Mizuno N, Wasan H, Bridgewater J, Okusaka T. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann Oncol. 2014; 25:391–398.
9. Tajima H, Ohta T, Shinbashi H, Hirose A, Tsukada T, Okamoto K, Nakanuma S, Sakai S, Furukawa H, Makino I, Nakamura K, Hayashi H, Oyama K, Inokuchi M, Nakagawara H, Miyashita T, et al. Successful treatment of unresectable gallbladder cancer with low-dose paclitaxel as palliative chemotherapy after failure of gemcitabine and oral S-1: A case report. Oncol Lett. 2012; 4:1281–1284.
10. Suyama K, Ikeda M, Suzuki E, Kojima M, Mitsunaga S, Shimizu S, Ohno I, Takahashi H, Okuyama H, Kuwahara A, Okusaka T, Furuse J. Early relapse of unresectable gallbladder cancer after discontinuation of gemcitabine monotherapy administered for 5 years in a patient who had complete response to the treatment. Case Rep Oncol. 2013; 6:531–537.
11. Bizama C, Garcia P, Espinoza JA, Weber H, Leal P, Nervi B, Roa JC. Targeting specific molecular pathways holds promise for advanced gallbladder cancer therapy. Cancer Treat Rev. 2015; 41:222–234.
12. Matsushita S, Onishi H, Nakano K, Nagamatsu I, Imaizumi A, Hattori M, Oda Y, Tanaka M, Katano M. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014; 105:272–280.
13. Zhu AX, Hezel AF. Development of Molecularly Targeted Therapies in Biliary Tract Cancers: Reassessing the Challenges and Opportunities. Hepatology. 2011; 53:695–704.
14. Espinoza JA, García P, Bizama C, Roa JC. Molecular Genetics of Gallbladder Cancer. eLS John Wiley & Sons, LtdChichester: 2015.
15. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012; 7:183–188.
16. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR Signaling Network for Cancer Therapy. J Clin Oncol. 2009; 27:2278–2287.
17. Grozinsky-Glasberg S, Pavel M. Inhibition of mTOR in carcinoid tumors. Target Oncol. 2012; 7:189–195.
18. Pavlidou A, Vlahos NF. Molecular Alterations of PI3K/Akt/mTOR Pathway: A Therapeutic Target in Endometrial Cancer. Sci World J. 2014.
19. Hu M, Ekshyyan O, Ferdinandez LH, Rong XH, Caldito G, Nathan CAO. Efficacy and Comparative Effectiveness of Sirolimus as an Anticancer Drug. Laryngoscope. 2011; 121:978–982.
20. Jerusalem G, Rorive A, Collignon J. Use of mTOR inhibitors in the treatment of breast cancer: an evaluation of factors that influence patient outcomes. Breast Cancer (Dove Med Press). 2014; 6:43–57.
21. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010; 70:288–298.
22. Battelli C, Cho DC. mTOR inhibitors in renal cell carcinoma. Therapy. 2011; 8:359–367.
23. Sivendran S, Agarwal N, Gartrell B, Ying J, Boucher KM, Choueiri TK, Sonpavde G, Oh WK, Galsky MD. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treat Rev. 2014; 40:190–196.
24. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005; 65:7052–7058.
25. Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015; 4:165–176.
26. Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015; 137:173–179.
27. Manfredi GI, Dicitore A, Gaudenzi G, Caraglia M, Persani L, Vitale G. PI3K/Akt/mTOR signaling in medullary thyroid cancer: a promising molecular target for cancer therapy. Endocrine. 2015; 48:363–370.
28. Sun Z, Wang Z, Liu X, Wang D. New development of inhibitors targeting the PI3K/AKT/mTOR pathway in personalized treatment of non-small-cell lung cancer. Anticancer Drugs. 2015; 26:1–14.
29. Zhang X, Shi H, Tang H, Fang Z, Wang J, Cui S. miR-218 inhibits the invasion and migration of colon cancer cells by targeting the PI3K/Akt/mTOR signaling pathway. Int J Mol Med. 2015; 35:1301–1308.
30. Xing X, Zhang L, Wen X, Wang X, Cheng X, Du H, Hu Y, Li L, Dong B, Li Z, Ji J. PP242 suppresses cell proliferation, metastasis, and angiogenesis of gastric cancer through inhibition of the PI3K/AKT/mTOR pathway. Anticancer Drugs. 2014; 25:1129–1140.
31. Arcaro A. Targeting PI3K/mTOR Signaling in Cancer. Front Oncol. 2014; 4:84.
32. Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN. The role of mTOR in the management of solid tumors: An overview. Cancer Treat Rev. 2009; 35:148–159.
33. Leal P, Garcia P, Sandoval A, Buchegger K, Weber H, Tapia O, Roa JC. AKT/mTOR substrate P70S6K is frequently phosphorylated in gallbladder cancer tissue and cell lines. Oncotargets Ther. 2013; 6:1373–1384.
34. Leal P, Garcia P, Sandoval A, Letelier P, Brebi P, Ili C, Alvarez H, Tapia O, Roa JC. Immunohistochemical Expression of Phospho-mTOR Is Associated With Poor Prognosis in Patients With Gallbladder Adenocarcinoma. Arch Pathol Lab Med. 2013; 137:552–557.
35. Wu Q, Kiguchi K, Kawamoto T, Ajiki T, Traag J, Carbajal S, Ruffino L, Thames H, Wistuba I, Thomas M, Vasquez KM, DiGiovanni J. Therapeutic effect of rapamycin on gallbladder cancer in a transgenic mouse model. Cancer Res. 2007; 67:3794–3800.
36. Zong H, Yin B, Zhou H, Cai D, Ma B, Xiang Y. Inhibition of mTOR pathway attenuates migration and invasion of gallbladder cancer via EMT inhibition. Mol Biol Rep. 2014; 41:4507–4512.
37. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014; 4:64.
38. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, et al. Biochemical, Cellular, and In vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res. 2009; 69:6232–6240.
39. Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009; 34:620–627.
40. Dobbin ZC, Landen CN. The Importance of the PI3K/AKT/MTOR Pathway in the Progression of Ovarian Cancer. Int J Mol Sci. 2013; 14:8213–8227.
41. Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, Oien KA, McKay CJ, Carter CR, Gillen G, Champion S, Pimlott SL, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014; 63:1481–1489.
42. Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009; 9:237–249.
43. Tapia O, Riquelme I, Leal P, Sandoval A, Aedo S, Weber H, Letelier P, Bellolio E, Villaseca M, Garcia P, Roa JC. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014; 465:25–33.
44. Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014; 5:49–66.
45. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23:53–62.
46. Al-Batran SE, Ducreux M, Ohtsu A. mTOR as a therapeutic target in patients with gastric cancer. Int J Cancer. 2012; 130:491–496.
47. Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010; 22:169–176.
48. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21–35.
49. Hall MN. mTOR-what does it do?. Transplant Proc. 2008; 40:S5–8.
50. Caron E, Ghosh S, Matsuoka Y, Ashton-Beaucage D, Therrien M, Lemieux S, Perreault C, Roux PP, Kitano H. A comprehensive map of the mTOR signaling network. Mol Syst Biol. 2010; 6:453.
51. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006; 22:159–168.
52. Hartmann B, He X, Keller F, Fischereder M, Guba M, Schmid H. Development of a sensitive phospho-p70 S6 kinase ELISA to quantify mTOR proliferation signal inhibition. Ther Drug Monit. 2013; 35:233–239.
53. Hartmann B. p70S6 kinase phosphorylation for pharmacodynamic monitoring. Clin Chim Acta. 2012; 413:1387–1390.
54. Dormond-Meuwly A, Roulin D, Dufour M, Benoit M, Demartines N, Dormond O. The inhibition of MAPK potentiates the anti-angiogenic efficacy of mTOR inhibitors. Biochem Biophys Res Commun. 2011; 407:714–719.
55. Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, Rodrik-Outmezguine V, Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle. 2011; 10:3948–3956.
56. Saqcena M, Patel D, Menon D, Mukhopadhyay S, Foster DA. Apoptotic effects of high-dose rapamycin occur in S-phase of the cell cycle. Cell Cycle. 2015; 0.
57. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005; 65:3336–3346.
58. Foster DA, Toschi A. Targeting mTOR with rapamycin: one dose does not fit all. Cell Cycle. 2009; 8:1026–1029.
59. Robinson J, Lai C, Martin A, Nye E, Tomlinson I, Silver A. Oral rapamycin reduces tumour burden and vascularization in Lkb1(+/-) mice. J Pathol. 2009; 219:35–40.
60. Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013; 3:3230.
61. Weng M, Gong W, Ma M, Chu B, Qin Y, Zhang M, Lun X, McFadden G, Forsyth P, Yang Y, Quan Z. Targeting gallbladder cancer: oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol Cancer. 2014; 13:82.
62. Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012; 149:274–293.
63. Boulbes D, Chen CH, Shaikenov T, Agarwal NK, Peterson TR, Addona TA, Keshishian H, Carr SA, Magnuson MA, Sabatini DM, Sarbassov dos D. Rictor phosphorylation on the Thr-1135 site does not require mammalian target of rapamycin complex 2. Mol Cancer Res. 2010; 8:896–906.
64. Yu K, Toral-Barza L. Biochemical and pharmacological inhibition of mTOR by rapamycin and an ATP-competitive mTOR inhibitor. Methods Mol Biol. 2012; 821:15–28.
65. Blagosklonny MV. Rapalogs in cancer prevention: anti-aging or anticancer? Cancer Biol Ther. 2012; 13:1349–1354.
66. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, Blagosklonny MV, Lisanti MP. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181:278–293.
67. Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Prev Res (Phila). 2011; 4:1011–1020.
68. Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011; 71:1573–1583.
69. Del Bufalo D, Ciuffreda L, Trisciuoglio D, Desideri M, Cognetti F, Zupi G, Milella M. Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006; 66:5549–5554.
70. Lionello M, Lovato A, Staffieri A, Blandamura S, Turato C, Giacomelli L, Staffieri C, Marioni G. The EGFR-mTOR pathway and laryngeal cancer angiogenesis. Eur Arch Otorhinolaryngol. 2014; 271:757–764.
71. Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006; 12:122–127.