
INTRODUCTION
Medullary thyroid carcinoma (MTC) occurs as a sporadic condition in approximately 75% or in an inherited form as a component of the type 2 multiple endocrine neoplasia (MEN 2) syndromes, MEN 2A and MEN 2B, and familial MTC (FMTC) in 25% of all cases [1]. As one subtype of MEN 2, FMTC is operationally diagnosed in families with four or more cases of MTC in the absence of pheochromocytoma or hyperparathyroidism, which occurs in 10–20% of all MEN 2 cases with an older age at onset (often between 20 and 40 years) [1]. FMTC is regularly correlated with germline mutations of the RET proto-oncogene, which is mapped on chromosome 10q11.2 and contains 21 exons [2]. The RET gene encodes a tyrosine kinase transmembrane receptor characterized by three different domains: the extracellular domain, the transmembrane domain, and the intracellular tyrosine kinase domain [3, 4]. Current data indicate that a confirmable mutation has been described in almost all MEN 2 families [1]. Germline mutations in FMTC kindreds are more equally distributed across the RET gene and include mutations at codons 532, 533 (exon 8); 609, 611, 618, 620 (exon 10); 630, 634 (exon 11); 768, 790, 791 (exon 13); V804M, 844 (exon 14); 891 (exon 15); and 912 (exon 16). Mutations at codons 532, 533, 768, 844, and 912 have been identified only in families with FMTC [1]. Recently, some compound mutations of RET (p.V804M/V778I, p.V804M/R844L, and p.C634Y/Y791F) have been reported to have some specific clinical characteristics [1, 5–14]. Additionally, some rare cases have also been reported to be associated with specific RET mutations in many MEN 2 families, such as cutaneous lichen amyloidosis (CLA), Hirschsprung's disease (HSCR), and corneal nerve thickening (CNT) [1, 5, 6, 11, 15–17]. In 2009, the American Thyroid Association (ATA) stratified mutations at codon 891 as ATA-A level, carrying the “least high” risk [1].
The CLA phenotype in MEN 2 usually appears on the upper back as a subtype of cutaneous amyloidosis (CA), and most of the previous cases of CLA in MEN 2A have been anecdotally described [1, 18–29]. The clinical presentation of this MTC-CLA was initially observed only in MEN 2A patients with mutations in the extracellular cysteine 634 codons in exon 11. Another exception is the p.V804M mutation within exon 14, which is in an intracellular tyrosine kinase domain reported in a US female with MTC/CLA [27]. Familial CA mainly includes three clinical types: CLA, macular amyloidosis, and nodular amyloidosis. The pathogenic gene for familial CA is mapped to a locus on 5p13.1-q11.2. Subsequently, OSMR, within this locus, was demonstrated to be the causative gene for familial CA. The OSMR gene encodes oncostatin M receptor OSMRβ, which is an interleukin (IL)-6 family cytokine receptor [30–34].
In this study, we investigated a southeastern Chinese family with FMTC and CA and screened the entire coding sequence of RET in the available family members. We also analyzed the in vitro oncogenic potential of the two RET variants and sequenced the OSMR and IL31RA genes in this family. Finally, we evaluated the correlation between the genotype and phenotype and its potential clinical significance.
RESULTS
Clinical features and phenotypic data
Patients with MTC
The proband (II-2, Figure 1) was a 65-year-old woman with diarrhea for 10 years who was then diagnosed as having MTC in 2012. Biochemical examination revealed an increased level of carcinoembryonic antigen (CEA) (29.7 ng/mL; normal, <5 ng/mL). Doppler ultrasound (USS) and computerized tomography (CT) scanning indicated two hypoechoic nodules in both thyroid lobes with right lymph node enlargement. A bilateral total thyroidectomy (TT) with bilateral level VI and right neck dissection was performed. The histopathologic evaluation suggested bilateral MTC with right neck lymph node metastases (LNMs) (T1N1bM0; Table 1). Two months later, the 45-year-old daughter of the proband (III-2) also underwent TT with bilateral level VI and modified left neck dissection after diagnosis of bilateral thyroid masses with left lymph node enlargement and an elevated CEA level (22.8 ng/ml). Bilateral MTC with LNMs was confirmed by histopathologic examination (T1N1bM0). In 2013, some previously hesitant members of the family (II-4, II-5, III-3, III-5, III-7, III-9, III-12, IV-1~7, and IV-8) agreed to further participate in biochemical testing, imaging studies, and RET screening (Table 1). All of the newly recruited subjects had normal basal serum calcitonin (bCt) levels and USS/CT images, with the exception of II-5 (p.S891A/p.R525W; bCt, 589.6 ng/L [normal for males, < 8.4 ng/L; normal for females, <5.0 ng/L]; CEA, 41.53 ng/ml). Then, the 58-year-old brother of the proband (II-5) accepted and underwent a TT with bilateral level VI and modified bilateral neck dissection. The histopathologic examination revealed bilateral multifocal MTC with LNMs (T1N1bM0; Table 1).
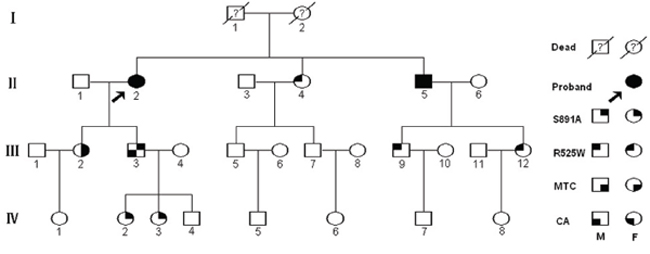
Figure 1: Pedigree of the southern four-generation Chinese family with FMTC and lichen/macular biphasic cutaneous amyloidosis investigated in this study. Circles and squares denote female and male family members, respectively.
Table 1: Clinical characteristics of 9 RET mutation/variant carriers
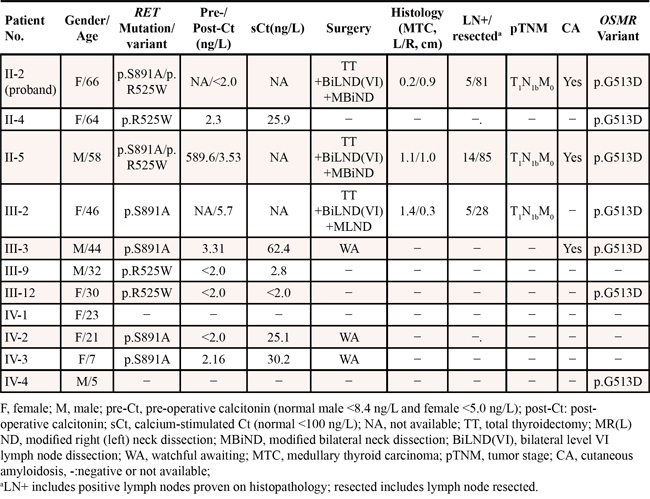
Six other carriers harboring a RET p.S891A mutation or p.R525W variant (III-3, IV-2, and IV-3 or II-4, III-9, and III-12, respectively) were further submitted to stimulated calcitonin (sCt) testing. The peak value (normal, <100 ng/L) was obtained 2 min after calcium stimulation. The mean sCt was 36.40 ng/L (range, 21.7–62.4 ng/L) in the 3 RET p.S891A mutation carriers (mean age, 24 y; range, 7–44 y) and < 2.0, 2.8, or 25.9 ng/L in the 3 RET variant p.R525W carriers (mean age, 42 y; range, 30–64 y; Table 1). None of the 6 carriers had abnormal Ct values.
Patients with CA
Patients II-2, II-5 (p.S891A/p.R525W), and III-3 (p.S891A) were diagnosed as having CA by a dermatologist. None of the other 14 relatives had pruritus or skin lesions. In these three patients, the first symptoms of CA typically began on the lower legs as severe pruritus. The ages at diagnosis were 28, 27, and 31 years, respectively (Figure 1). Once pruritus appeared, the areas with lesions were repetitively scratched and subsequently developed into local skin lichenification and hyperpigmented brown papules. The papules spread to other sites of involvement in the lower legs to thighs, the upper back, shoulders, arms, and forearms (Figure 2A–2F). All three patients also presented with brown macular lesions with a rippled or reticulated appearance on the extremities and the upper back, suggesting the co-existence of both lichen and macular variants (Figure 2A–2D). Additionally, patient II-5 manifested clearer skin lesions with dry, scaly, thickened, and clustered papules, and white patches, which caused epidermal cell damage due to repeated scratching on both lower legs (Figure 2E, 2F). All three patients were treated with glucocorticoid cream, which resulted in a decreased period of itching, but the application was discontinued due to side effects. The histopathologic examination of the skin lesions showed that the overlapping epidermis was hyperkeratotic with mild acanthosis and elongation of rete ridges. Characteristic small globular deposits of amorphous eosinophilic acellular material were present in the papillary dermis (Figure 2G). Little chronic inflammatory infiltrate was noted in the dermis. Globular deposits of amyloid were positive with crystal violet and Congo red staining (Figure 2H, 2I).
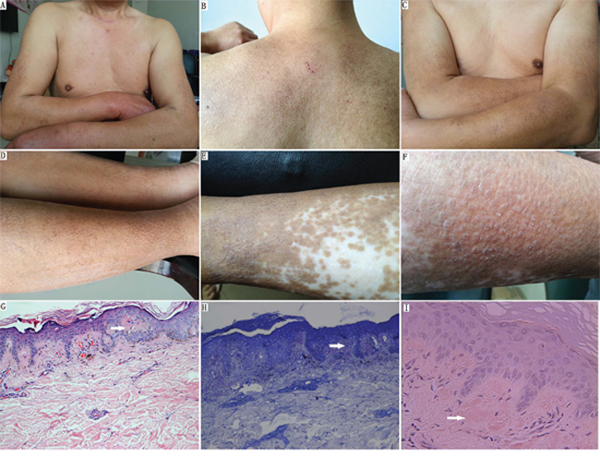
Figure 2: Clinical and histological presentation of FMTC/CA. A–D. Macular amyloidosis: brown hyperpigmented macules on the upper back, shoulders, arms, and legs. (A) Postoperative neck scar and brown hyperpigmented macular skin lesion on the arms (individual II-5). (B–D) Macular amyloidosis: macules showing pigmentation with a rippled or reticulated pattern and with scabby scratches on the upper back and extremities (individual III-3). E. Lichen amyloidosis: dry, scaly, and thickened SKIN with hyperpigmentation and remnant white patches on the lower leg (individual II-5). F. Close-up of lichen amyloidosis: multiple monomorphic skin-colored and clustered lichenified papules with white thin scales on the lower leg (individual II-5). G. Small hyaline deposits of amyloid were situated in the papillary dermis. There is overlying epidermal hyperplasia (hematoxylin & eosin; original magnification, × 100). H. Globular deposits of amyloid were positive for crystal violet staining (original magnification, × 200). I. The papillary dermal deposits were positive for Congo red stain (original magnification, × 400).
Other MEN2-associated disease
The 17 individuals had no evidence of pheochromocytoma, hyperparathyroidism, CNT, HSCR, or other endocrine tumors.
Identification of the RET germline mutations/variants
A heterozygous missense mutation within exon 15 of RET, c.2671T>G (p.S891A), was confirmed in the proband (II-2) and five other members (II-5, III-2, III-3, IV-2, and IV-3; Figure 3A). Another RET novel variant, p.R525W (c.1573C>T), within exon 8 was identified in five individuals (II-2, II-4, II-5, III-9, and III-12). Two patients with MTC (II-2 and II-5; Figure 3B and Table 1) underwent RET compound mutations (p.S891A/p.R525W). Individuals III-9 and III-12 only carry p.R525W, and this compound mutation did not co-segregate with MTC, so we can conclude that p.S891A/p.R525W passed in a trans inherited pattern. Meanwhile, we found five recurrent exonic single nucleotide polymorphisms (SNPs): c.135A > G (p.A45A), c.1296A > G (p.A432A), c.2071G > A (p.G691S), c.2307T > G (p.L769L), and c.2712C > G (p.S904S) within exons 2, 7, 11, 13, and 15, respectively. A SNP in intron 2 (IVS2+9A > G) was also identified (Figure 1; data not shown).
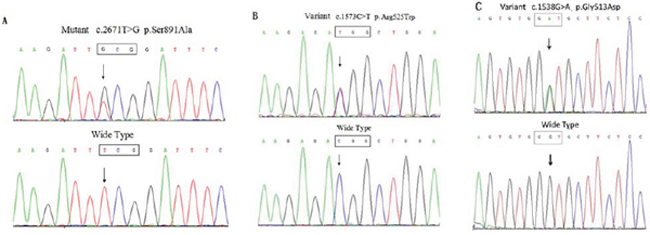
Figure 3: RET and OSMR variants in this pedigree were confirmed by direct sequencing. A. Direct sequencing of PCR products from the proband demonstrated a heterozygous T-to-G substitution at nucleotide position 2671 within exon 15, resulting in a missense mutation designated p.S891A. B. RET p.R525W (c.1573C > T) within exon 8. C. OSMR p.G513D (c.1538G > A) within exon 11 was confirmed.
Additional screening indicated that two RET p.S891A mutation carriers with CA (II-5 and III-3; Figure 3C) also carry heterozygous OSMR c.1538G > A (p.G513D/exon10). Several OSMR SNPs, i.e., p.D535N (c.1657G > A/exon11), p.N703N (c.2019C > T/exon13), and p.T732T (c.2196G > A/exon14), and IL31RA SNPs p.P24P (c.72C > T/exon 2) and p.S529N (c.1586G > A/exon13) were also identified and are listed in Supplementary Table 1. Further sequencing of exon 10 of OSMR identified p.G513D in four of the other eight affected individuals with RET mutations/variants and one unaffected member (IV-4; Table 1).
Functional significance of RET mutants (p.S891A/p.R525W and p.R525W)
The transforming capacity was assessed in HEK293 and HEK293T cells transfected with wild type-RET, p.R525W, and p.S891A/p.R525W. We found that RET p.S891A/p.R525W and p.S891A had a significant effect on the promotion of cell proliferation rates, and p.S891A/p.R525W showed a stronger effect than p.S891A (Figure 4A). RET p.R525W, p.S891A, p.S891A/p.R525W, and p.C634Y all increased the level of Akt phosphorylation. The phosphorylation level of p.R525W was 20%, 23%, and 30% weaker than that of p.S891A, p.S891A/p.R525W, and p.C634Y, respectively (Figure 4B). To confirm whether the proliferative effect resulted from apoptosis, we detected any alteration in an important apoptosis marker, caspase 3. Western blotting indicated that caspase 3 was also downregulated by RET and its mutants (Figure 4C). RET glycosylation was completely inhibited by p.R525W, p.S891A, p.S891A/p.R525W, and p.C634Y (Figure 4C). Disulfide-bridge-mediated RET dimerization was observed only in p.C634Y mutants (Figure 4D, 4E), which is consistent with the previous conclusion that RET mutations affecting extracellular cysteines lead to constitutive dimerization [35], and the p.S891A and p.S891A/p.R525W mutants together functioned as a monomeric receptor. Immunostaining indicated that RET p.R525W, p.S891A, and p.S891A/p.R525W were located mainly in the cytoplasm but rarely in the cellular membranes, suggesting an effect on the location and thus the function of RET by these mutations. Collectively, these data indicated that RET p.S891A can facilitate cell proliferation through promotion of the anti-apoptotic effect of Akt and incoming mitogenic stimulators. Meanwhile, the oncogenic activities of RET p.S891A are lower than those of p.C634Y.
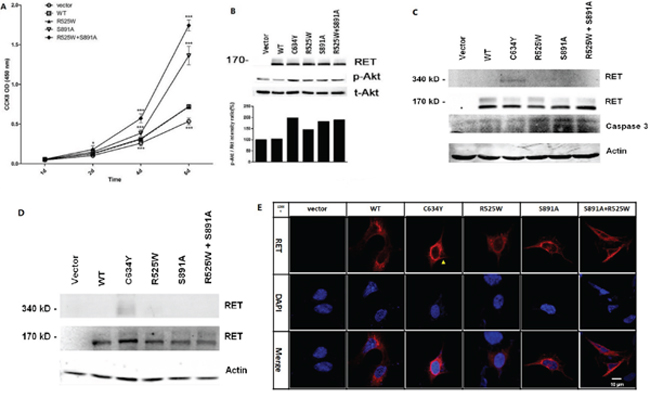
Figure 4: In vitro assays performed to characterize RET mutants p.R525W, p.C634Y, and p.S891A/p.R525W. A. The promotion effect of RET and its mutants on HEK293 proliferation. *P < 0.05; ***P < 0.001. B. Upper panel: Western blotting was performed in HEK293T cells expressing RET and mutants using antibodies against RET, phospho-Akt (Ser473), and Akt. Lower panel: Relative phosphorylation levels were calculated as ratios of phosphorylated to total protein levels using densitometry. C. Western blot shows complete inhibition of glycosylation in wild-type (WT), p.R525W, p.S891A, p.S891A/p.R525W, and p.C634Y. β-actin was used as a loading control. D. Presence of active disulfide-bound RET homodimers in p.R525W, p.S891A, p.S891A/p.R525W, and p.C634Y cells in comparison to RET-WT cells. E. Immunostaining showed that RET p.C634Y (red) were mainly localized in the cell membrane and cytoplasm (yellow arrow), whereas p.R525W, p.S891A, and p.S891A/p.R525W were mainly located in the cytoplasm and rarely in the membranes. Original magnification, × 1200; scale bar, 10 um.
DISCUSSION
In this study, we explored a recurrent intracellular p.S891A mutation and a novel extracellular variant p.R525W of RET in a southern Chinese family with FMTC/CA. All three patients with CA had the RET p.S891A mutation and a novel OSMR variant p.G513D, which provide possible new insight into the mechanism underlying FMTC/CA.
Previous studies have shown that the p.S891A mutation accounts for < 5% of all patients with RET mutations. Most patients with the p.S891A mutation have manifested FMTC, whereas just a few have manifested as MEN 2A [11]. Recently, 93 carriers of the p.S891A mutation were summarized [11, 12, 36]: MTC was present in 74.2% (69/93), and pheochromocytoma, hyperparathyroidism, and CNT were present in 3.2% (3/93). The mean age at MTC diagnosis in those carriers was 42.1 years. Age-related penetrance of MTC in 36 patients with the p.S891A mutation was 20.0%, 71.4%, 92.3%, and 100.0% in those aged 0–20, 21–40, 41–60, and > 60 years, respectively [11]. In our study, three of the six p.S891A mutation carriers presented with MTC as the sole clinical endocrine tumor, whereas the other 3 carriers (mean age, 24 y; range, 7–44 y) still had consistently undetectable elevations in bCt or sCt and chose a watchful waiting approach to treatment [12, 36, 37]. The clinical data in this family is consistent with that in previous patients with the RET p.S891A mutation and other FMTC patients with ATA-A level mutations carrying “moderate risk” (ATA-MOD) as reported worldwide recently [1, 37].
Two patients (II-2 and II-5) with RET p.S891A/p.R525W presented with MTC, whereas three carriers with p.R525W (II-4, III-9, and III-12) had no evidence of thyroid/skin or bCt/sCt abnormalities (Figure 1 and Table 1). There are mutations in the extracellular cysteine domain of RET that are reported to cause comparatively mild FMTC or FMTC/MEN 2A, such as p.C515S, p.C531R, and p.G533C [6, 9, 10, 14]. In this study, it is still inclusive that RET p.R525W is causative for MTC. RET double mutations associated with MEN 2 were also previously reported to have specific clinical characteristics [14]. For example, it appears that p.V778I, p.Y806C, and the p.Y791F polymorphisms have additive effects to p.V804M and p.C634Y, whereas p.R844L has an inhibitory modifying effect on p.V804M [14]. Although the oncogenic activity of p.S891A was slightly enhanced by p.R525W, two patients with trans p.S891A/p.R525W in our study only presented with MTC, which shows similar clinical features to p.S891A described previously [11, 12, 36]. Nonetheless, the follow-up study needs to be validated to avoid misinterpretation and irreversible clinical outcomes [38].
CLA is verified to be rarely associated with the MEN 2-related specific RET genotype. Eng et al. reported that the frequency of CLA in MEN 2A families was approximately 9% (18/199), and all 18 families were frequently associated with RET codon 634 mutation, but there was no specific analysis of the correlation between the RET genotype and CLA phenotype or of case distribution of CLA in each family [15]. In 2009, a p.V804M RET germline mutation was identified in a MTC/CLA subject [27]. Accumulating evidence indicates that CLA occurred in 42.3% (33/78) of 78 RET mutation carriers in 14 MEN 2/CLA families, similar to the approximately 50% morbidity for pheochromocytoma in MEN 2A [1, 15, 39]. Gender-related predominance in the prevalence of CLA was observed as indicated by the male-to-female ratio of approximately 2:9 (6:27) (Table 2). The mean age at the time of diagnosis of CLA with a RET mutation was 31.4 y (range, 5–60 y). Pruritus seems to be the first clinical manifestation of CLA because of its early onset in infancy or adolescence, and most patients present with pruritic symptoms before the diagnosis of MTC [23, 26, 40].
Table 2: Clinical data of patients with an association between the specific RET mutations and CLA in MEN 2
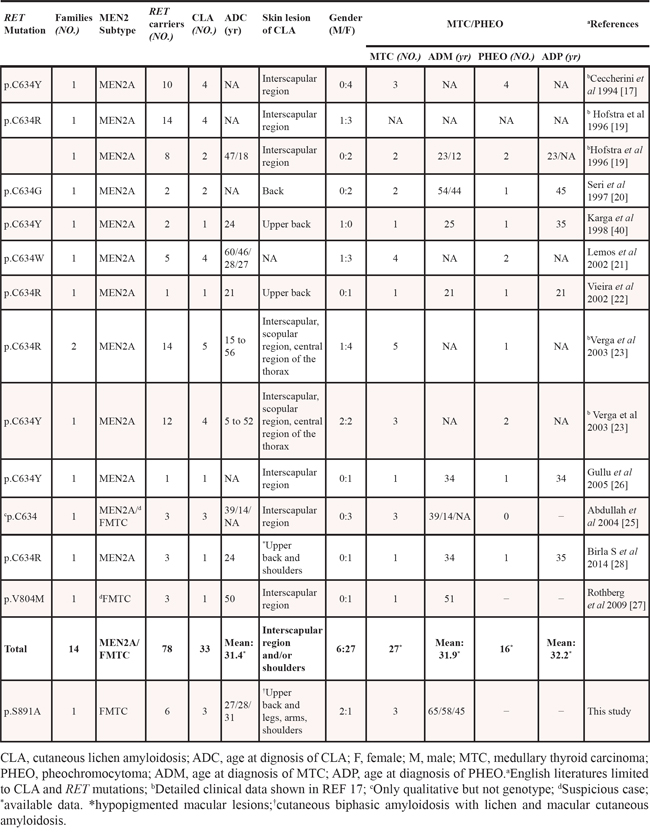
In the present family, of the three patients with CA, the two with the p.S891A/p.R525W mutation (II-2, and II-5) had MTC, whereas the p.S891A carrier (III-3; 44 years old) did not but did have a high Ct level (Table 1). Moreover, another patient with the p.S891A mutation with MTC (III-2; 46 years old) and five carriers (three individuals with p.R525W and two individuals with p.S891A) had no CA lesions and skin pruritus. Family members with the same RET mutation may have different clinical phenotypes, and younger individuals might show the CA phenotype earlier [23, 40]. Our observations also indicated that the driving course of CA was independent from the clinical evolution of the disease and was not associated with MTC [23, 26, 27]. The phenotype of CA co-segregated with RET p.S891A implied that the germline RET p.S891A mutation possibly caused the FMTC/CA. SNPs within non-hot spot regions show no association with MTC or CA, which indicated that genetic screening of RET hot spot regions is adequate for the diagnosis [1, 6, 15]. Similar to CNT, CA appears to be another rare clinical characteristic of the RET p.S891A mutation [11, 14, 36]. It should be noted that families with CLA only did not have the RET mutation [15, 19], whereas all of the patients with FMTC/CA and the RET p.S891A mutation have OSMR p.G513D. The other four OSMR p.G513D carriers including a 64-year-old female (II-4), had no evidence of CA (Table 1). Therefore, OSMR p.G513D may play a role in modifying the evolutionary process of CA with RET mutation. Interestingly, however, all three of these CA patients presented a more expanded pathologic region than previously described, particularly the scapular region of the upper back corresponding to dermatomes T2-T6 in MEN 2-CLA, manifesting as co-existence of papular and macular forms known as cutaneous biphasic amyloidosis (Figure 2 and Table 2) [17, 23, 30]. This could be indirect evidence that the cases presented here are more like a “neurodermatitis” or a common clinical variant of familial CA disease and that the treatment of CA is most disappointing [32, 33]. Although the underlying molecular mechanism of CA/MEN 2 pathogenesis remains largely unknown, it may occur due to the accumulation of genetic disruptions, either through errors in chromosomal replication, or the interaction of other modifying factors and different expression patterns of the same RET mutations, and/or through PI3K/Akt pathways to modify disease susceptibility and the clinical phenotype [26, 32, 40, 41].
MATERIALS AND METHODS
Subjects
We investigated a four-generation southeastern Chinese pedigree including 17 individuals with FMTC/CA from Zhejiang Province, China (Figure 1). The present study was conducted in accordance with the Helsinki Declaration and approved by the Ethics Committee of the 117th PLA Hospital (Hangzhou, China). Written informed consent was provided by all of the subjects in this study.
All individuals underwent clinical and biochemical examinations according to the published criteria in three scenarios. (i) The biochemical evaluation consisted of CEA, parathyroid hormone, and serum and/or 24-h urinary determination of catecholamines, along with USS, CT, and/or emission CT scans. Surgical thyroidectomy was performed after confirmation of a RET mutation and elevated bCt for the diagnosis. (ii) Levels of bCt and sCt were measured using a fully-automated chemiluminescence immunoassay (Immulite 2000 Immunoassay System; Siemens Ltd., USA). Eight carriers of the RET mutation/variation participated in calcium-sCt testing as follows: Ca gluconate was administered intravenously at a dose of 25 mg/kg at 10 ml/min (2.3 mg of elemental Ca), and the blood samples were obtained before and at 2, 5, and 15 min from the end of the Ca infusion via an indwelling intravenous cannula. (iii) CA was diagnosed clinically based on persistent pruritic, cutaneous papules with some scales or macular pigmentation showing a rippled or reticulated pattern and was characterized histopathologically by amyloid deposits in the papillary dermis [17, 23]. Follow-up was then carried out.
Histopathologic analysis
The diagnosis of MTC was further confirmed by histopathology. Tumor staging was performed according to the American Joint Committee on Cancer (AJCC, 7th edition) tumor-node-metastasis (TNM) classification system [42].
The skin biopsy specimens were obtained from the cutaneous lesions of patients II-5 and III-3, embedded in paraffin wax, and fixed in formalin. Hematoxylin-eosin, crystal violet, and Congo red stains were applied to 4-μm-thick sections.
RET mutation analysis
Genomic DNA was isolated from peripheral blood leukocytes of available family members as previously described (8), followed by polymerase chain reaction (PCR) amplification and sequencing of the entire exons and the flanking splice junctions of RET, OSMR, and IL31RA. One hundred unrelated healthy matched controls and 6 affected individuals with the RET p.S891A mutation along with matched controls were included as previously reported [12].
Construction of expression vector
The full-length open reading frame of RET9 was cloned to the pCIG vector. For the RET p.C634Y, p.R252W, p.S891A, and p.S891A/R525W mutations, we use the KOD-Plus-Mutagenesis Kit (Toyobo, Japan) following the manufacturer's protocol. The sequence and orientation of the RET open reading frame and its mutants were confirmed by direct sequencing.
Cell cultures and transfections
The human embryonic kidney cell lines HEK293T and HEK293 were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco's Modified Eagle's Medium (Invitrogen, Grand Island, NY, USA) supplemented with 10% fetal bovine serum. Various constructs were introduced into HEK293 cells through retroviral or lentiviral infection using standard protocols. To obtain stable transfectants, the transfected cells were grown in medium with G418 (400 μg/ml), and resistant clones were confirmed using Western blotting.
Cell proliferation assay
Cell viability was determined with the Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Japan) following the manufacturer's protocol. Briefly, HEK293 cell were seeded into 96-well plates (2000 cells/well). Then, 10 μl of the CCK-8 solution was added to each well of each of the plates, and the plates were incubated for 2 h in an incubator at every time point (24 h, 48 h, 72 h and 96 h, respectively) The absorbance at 450 nm was measured using a microplate reader. Each experiment was independently repeated in triplicate wells three times. Statistical analysis was carried out with one-way ANOVA (GraphPad Prism 5; GraphPad Software, Inc., La Jolla, CA, USA). Results are presented as mean ± SD. P < 0.05 was considered as statistically significant.
Western blotting
Cell lysates were prepared by incubating cell pellets in lysis buffer (50 mmol/l Tris-HCl, pH 8.0; 150 mmol/l NaCl, 0.5% NP-40) for 30 min on ice, followed by centrifugation at 14,000 × g for 15 min at 4°C. For Western blot analysis, membranes were incubated with primary antibodies for 1 h at room temperature or overnight at 4°C, followed by incubation with secondary antibodies at room temperature for 1 h. Immunoreactive bands were detected using Western blot luminol reagent (GE Healthcare, Waukesha, WI, USA). Antibodies used were anti-Caspase3 (#9664), anti-RET (#3223), anti-p-Akt (#4060), anti-Akt (#9272) (all, Cell Signaling, Boston, MA, USA), and anti-Actin (#sc-69879; Santa Cruz, Dallas, TX, USA).
Indirect immunofluorescence
Cells grown on coverslips were stained by indirect immunofluorescence as published elsewhere [43]. Briefly, cells were incubated with primary antibody against RET and RET mutants (p.C634Y, p.R525W, p.S891A, and p.S891A/R525W) and then incubated with Alexa Fluor 594- (Invitrogen Molecular Probes, Carlsbad, CA, USA) secondary antibody against mouse or rabbit IgG. Cells were then counterstained with DAPI and imaged with a laser scanning confocal microscope (Fluoview FV1000, Olympus Co., Japan).
ACKNOWLEDGMENTS
The authors thank all the patients and their families who agreed to participate in this study. This work was supported by the National Natural Science Foundation of China (81472861); the Key Project of Zhejiang Province Science and Technology Plan, China (2014C03048-1); the Key Scientific Research Project of Nanjing Military Command, China (10Z036); the Medical Science and Technology Project of Zhejiang Province, China (2014KYB219).
Author contributions
Conceived and designed the experiments: X.P.Q. Performed the experiments: X.P.Q., J.Q.Z., Z.G.C, J.L.C., F.L., M.S., E.F. Analyzed the data: X.P.Q., Z.G.C., J.L.C, J.D., N.F.L., J.G., H.J. Contributed reagents/materials/analysis tools: J.G., H.J., Y.M.Z., J.M.M. Wrote the paper: X.P.Q.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells SA Jr. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid: official journal of the American Thyroid Association. 2009; 19:565–612.
2. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA Jr. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Human molecular genetics. 1993; 2:851–856.
3. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985; 42:581–588.
4. Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988; 3:571–578.
5. Wells SA Jr., Pacini F, Robinson BG, Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. The Journal of clinical endocrinology and metabolism. 2013; 98:3149–3164.
6. Elisei R, Alevizaki M, Conte-Devolx B, Frank-Raue K, Leite V, Williams GR. 2012 European thyroid association guidelines for genetic testing and its clinical consequences in medullary thyroid cancer. European thyroid journal. 2013; 1:216–231.
7. Ahmed SR, Ball DW. Clinical review: Incidentally discovered medullary thyroid cancer: diagnostic strategies and treatment. The Journal of clinical endocrinology and metabolism. 2011; 96:1237–1245.
8. Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, Mao R. Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Human mutation. 2009; 30:548–556.
9. Fazioli F, Piccinini G, Appolloni G, Bacchiocchi R, Palmonella G, Recchioni R, Pierpaoli E, Silvetti F, Scarpelli M, Bruglia M, Melillo RM, Santoro M, Boscaro M, Taccaliti A. A new germline point mutation in Ret exon 8 (cys515ser) in a family with medullary thyroid carcinoma. Thyroid: official journal of the American Thyroid Association. 2008; 18:775–782.
10. Da Silva AM, Maciel RM, Da Silva MR, Toledo SR, De Carvalho MB, Cerutti JM. A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. The Journal of clinical endocrinology and metabolism. 2003; 88:5438–5443.
11. Schulte KM, Machens A, Fugazzola L, McGregor A, Diaz-Cano S, Izatt L, Aylwin S, Talat N, Beck-Peccoz P, Dralle H. The clinical spectrum of multiple endocrine neoplasia type 2a caused by the rare intracellular RET mutation S891A. The Journal of clinical endocrinology and metabolism. 2010; 95:E92–97.
12. Qi XP, Zhang RX, Cao JL, Chen ZG, Jin HY, Yang RR. The rare intracellular RET mutation p.S891A in a Chinese Han family with familial medullary thyroid carcinoma. Journal of biosciences. 2014; 39:505–512.
13. Kasprzak L, Nolet S, Gaboury L, Pavia C, Villabona C, Rivera-Fillat F, Oriola J, Foulkes WD. Familial medullary thyroid carcinoma and prominent corneal nerves associated with the germline V804M and V778I mutations on the same allele of RET. Journal of medical genetics. 2001; 38:784–787.
14. Qi XP, Ma JM, Du ZF, Ying RB, Fei J, Jin HY, Han JS, Wang JQ, Chen XL, Chen CY, Liu WT, Lu JJ, Zhang JG, Zhang XN. RET germline mutations identified by exome sequencing in a Chinese multiple endocrine neoplasia type 2A/familial medullary thyroid carcinoma family. PloS one. 2011; 6:e20353.
15. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. Jama. 1996; 276:1575–1579.
16. Eng C, Mulligan LM. Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumours, and hirschsprung disease. Human mutation. 1997; 9:97–109.
17. Ceccherini I, Romei C, Barone V, Pacini F, Martino E, Loviselli A, Pinchera A, Romeo G. Identification of the Cys634→Tyr mutation of the RET proto-oncogene in a pedigree with multiple endocrine neoplasia type 2A and localized cutaneous lichen amyloidosis. Journal of endocrinological investigation. 1994; 17:201–204.
18. Anitha B, Mysore V. Lichen Amyloidosis: Novel Treatment with Fractional Ablative 2,940 nm Erbium: YAG Laser Treatment. Journal of cutaneous and aesthetic surgery. 2012; 5:141–143.
19. Hofstra RM, Sijmons RH, Stelwagen T, Stulp RP, Kousseff BG, Lips CJ, Steijlen PM, Van Voorst Vader PC, Buys CH. RET mutation screening in familial cutaneous lichen amyloidosis and in skin amyloidosis associated with multiple endocrine neoplasia. The Journal of investigative dermatology. 1996; 107:215–218.
20. Seri M, Celli I, Betsos N, Claudiani F, Camera G, Romeo G. A Cys634Gly substitution of the RET proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis. Clinical genetics. 1997; 51:86–90.
21. Lemos MC, Carrilho F, Rodrigues FJ, Santos P, Carvalheiro M, Ruas MA, Regateiro FJ. Early onset of medullary thyroid carcinoma in a kindred with multiple endocrine neoplasia type iia associated with cutaneous lichen amyloidosis. Endocrine practice: official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2002; 8:19–22.
22. Vieira AE, Mello MP, Elias LL, Lau IF, Maciel LM, Moreira AC, Castro M. Molecular and biochemical screening for the diagnosis and management of medullary thyroid carcinoma in multiple endocrine neoplasia type 2A. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2002; 34:202–206.
23. Verga U, Fugazzola L, Cambiaghi S, Pritelli C, Alessi E, Cortelazzi D, Gangi E, Beck-Peccoz P. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clinical endocrinology. 2003; 59:156–161.
24. Ferrer JP, Halperin I, Conget JI, Alsina M, Martinez-Osaba MJ, Palou J, Bombi JA, Vilardell E. Primary localized cutaneous amyloidosis and familial medullary thyroid carcinoma. Clinical endocrinology. 1991; 34:435–439.
25. Abdullah F, Udelsman R. Cutaneous lichen amyloidosis in a family with familial medullary thyroid cancer. Surgery. 2004; 135:563–564.
26. Gullu S, Gursoy A, Erdogan MF, Dizbaysak S, Erdogan G, Kamel N. Multiple endocrine neoplasia type 2A/localized cutaneous lichen amyloidosis associated with malignant pheochromocytoma and ganglioneuroma. Journal of endocrinological investigation. 2005; 28:734–737.
27. Rothberg AE, Raymond VM, Gruber SB, Sisson J. Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid: official journal of the American Thyroid Association. 2009; 19:651–655.
28. Birla S, Singla R, Sharma A, Tandon N. Rare manifestation of multiple endocrine neoplasia type 2A & cutaneous lichen amyloidosis in a family with RET gene mutation. The Indian journal of medical research. 2014; 139:779–781.
29. Lee DD, Huang JY, Wong CK, Gagel RF, Tsai SF. Genetic heterogeneity of familial primary cutaneous amyloidosis: lack of evidence for linkage with the chromosome 10 pericentromeric region in Chinese families. The Journal of investigative dermatology. 1996; 107:30–33.
30. Vijaya B, Dalal BS, Manjunath GV. Primary cutaneous amyloidosis: a clinico-pathological study with emphasis on polarized microscopy. Indian journal of pathology & microbiology. 2012; 55:170–174.
31. Lee DD, Lin MW, Chen IC, Huang CY, Liu MT, Wang CR, Chang YT, Liu HN, Liu TT, Wong CK, Tsai SF. Genome-wide scan identifies a susceptibility locus for familial primary cutaneous amyloidosis on chromosome 5p13.1-q11.2. The British journal of dermatology. 2006; 155:1201–1208.
32. Arita K, South AP, Hans-Filho G, Sakuma TH, Lai-Cheong J, Clements S, Odashiro M, Odashiro DN, Hans-Neto G, Hans NR, Holder MV, Bhogal BS, Hartshorne ST, Akiyama M, Shimizu H, McGrath JA. Oncostatin M receptor-beta mutations underlie familial primary localized cutaneous amyloidosis. American journal of human genetics. 2008; 82:73–80.
33. Sakuma TH, Hans-Filho G, Arita K, Odashiro M, Odashiro DN, Hans NR, Hans-Neto G, McGrath JA. Familial primary localized cutaneous amyloidosis in Brazil. Archives of dermatology. 2009; 145:695–699.
34. Wang WH, Li LF, Huang ES, Zhang Q, Sun TT, Song QH, Xu ML, Chen XR. A new c.1845A→T of oncostatin M receptor-beta mutation and slightly enhanced oncostatin M receptor-beta expression in a Chinese family with primary localized cutaneous amyloidosis. European journal of dermatology: EJD. 2012; 22:29–33.
35. Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science (New York, NY). 1995; 267:381–383.
36. Pelizzo MR, Torresan F, Boschin IM, Nacamulli D, Pennelli G, Barollo S, Rubello D, Mian C. Early, Prophylactic Thyroidectomy in Hereditary Medullary Thyroid Carcinoma: A 26-year Monoinstitutional Experience. American journal of clinical oncology. 2015; 38:508–513.
37. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid: official journal of the American Thyroid Association. 2015; 25:567–610.
38. Erlic Z, Hoffmann MM, Sullivan M, Franke G, Peczkowska M, Harsch I, Schott M, Gabbert HE, Valimaki M, Preuss SF, Hasse-Lazar K, Waligorski D, Robledo M, Januszewicz A, Eng C, Neumann HP. Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. The Journal of clinical endocrinology and metabolism. 2010; 95:308–313.
39. Castinetti F, Qi XP, Walz MK, Maia AL, Sanso G, Peczkowska M, Hasse-Lazar K, Links TP, Dvorakova S, Toledo RA, Mian C, Bugalho MJ, Wohllk N, Kollyukh O, Canu L, Loli P, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. The Lancet Oncology. 2014; 15:648–655.
40. Karga HJ, Karayianni MK, Linos DA, Tseleni SC, Karaiskos KD, Papapetrou PD. Germ line mutation analysis in families with multiple endocrine neoplasia type 2A or familial medullary thyroid carcinoma. European journal of endocrinology / European Federation of Endocrine Societies. 1998; 139:410–415.
41. Plaza Menacho I, Koster R, van der Sloot AM, Quax WJ, Osinga J, van der Sluis T, Hollema H, Burzynski GM, Gimm O, Buys CH, Eggen BJ, Hofstra RM. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a Src/JAK/STAT3 pathway, independent of glial cell line-derived neurotrophic factor. Cancer research. 2005; 65:1729–1737.
42. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Annals of surgical oncology. 2010; 17:1471–1474.
43. Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell reports. 2012; 2:1233–1243.