
Introduction
The DNA damage response (DDR) pathway has evolved to protect cells from genetic insults in an effort to preserve genomic integrity and cell viability [1, 2]. Anticancer chemotherapy often includes DNA damaging agents which activate the DDR [3]. An important gene in this process is TP53 which can induce G1/S cell cycle arrest and either DNA repair or apoptosis [4]. A hallmark of many tumors is the lack of functional p53 protein with a consequent loss of the G1/S checkpoint leading to a potential increase in reliance on the S and G2/M checkpoints for survival following genotoxic stress [5]. This has therefore stimulated the development of selective G2 checkpoint inhibitors for combination with DNA damaging anticancer drugs [6-9] .
One potential drug target controlling this checkpoint is the serine/threonine kinase CHK1 which has been shown to be involved in the G1 and G2 checkpoints through altering CDC25A stability and CDC25C localization, respectively [3, 10, 11]. CHK1 also maintains replication fork stability (and hence the S-phase checkpoint) and has been implicated in facilitating homologous recombination repair [10, 12, 13]. As a result of promising early studies, several CHK1 inhibitors have been developed and are currently undergoing clinical evaluation in combination with genotoxic drugs [6, 8, 14-16].
Recent studies have also indicated that CHK1 inhibition alone may have therapeutic activity in certain genetic backgrounds [6, 17, 18]. There is increasing evidence that many tumors harbor substantial amounts of DNA damage as a result of replication stress. This process appears to be intimately associated with tumor development and may arise as a result of oncogene-induced increases in the firing of replication origins. As a consequence, depletion of RPA and dNTPs results in the accumulation of stalled replication forks [19, 20]. This in turn leads to an increased requirement for CHK1 to prevent fork collapse and DNA damage. In support of this hypothesis, single-agent CHK1 inhibitor activity has been demonstrated in several tumor types including MYC-driven tumors such as neuroblastoma and lymphoma as well as acute myeloid leukemia and melanoma, all diseases thought to be associated with high levels of replication stress [17, 18, 21, 22] .
We have therefore discovered a novel, potent, orally active CHK1 inhibitor and clinical development candidate: CCT245737. Here we describe the preclinical pharmacology and pharmacodynamics (PD) of this compound together with its therapeutic activity in combination with various genotoxic anticancer drugs in multiple human tumor xenografts. Uniquely, we present clear evidence that the combination of gemcitabine and CCT245737 provides a substantial therapeutic advantage over either agent alone in an antitumor context, thus validating this approach. We describe a novel ELISA for pS296CHK1, which demonstrated target inhibition following CCT245737 treatment at efficacious doses with gemcitabine and carboplatin in a RAS mutant human tumor xenograft model of NSCLC, an area of unmet clinical need in cancer treatment. In addition CCT245737 showed significant antitumor activity as a single-agent in an Eµ-Myc driven mouse model of B-cell lymphoma. Consequently CCT245737 is in late stage preclinical development for scheduled entry into phase I clinical trials.
Results
Structure and kinase selectivity of CCT245737
Figure 1A shows the chemical structure of CCT245737 ((R)-5-((4-((morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyrazine-2-carbonitrile). A model of CCT245737 bound in the ATP pocket of human CHK1 is shown in Supplementary Figure S1 and suggests that key interactions in the ATP binding site have been retained through hydrogen bonding to Glu85 and Cys87 in the hinge region, the hydrogen bonding of the nitrile to Lys38, while the basic nitrogen on the morpholine forms a salt bridge to Glu91. Similar binding poses have been observed for several other 2-amino-pyrazine-5-carbonitrile CHK1 inhibitors [23].
Initial in vitro kinase profiling of CCT245737 (10µM) against 124 kinases showed that only 12 kinases (including CHK1) had > 80% inhibition (Supplementary Table 1). In vitro IC50 values were determined for these 12 kinases and five others including CDK2/CycA and CDK1/CycB (Supplementary Table 2). CCT245737 was a potent inhibitor of recombinant human CHK1 with IC50 of 1.4±0.3nM (mean±SD, n = 3, EZ Reader II assay). There was > 1,000-fold selectivity for CHK1 versus the functionally important kinases CDK1 and CHK2 (IC50 1.26-2.44 and 9.03 µM, respectively), and at least a 90-fold selectivity against cross-reacting kinases such as ERK8, PKD1, RSK1 and 2 (see Supplementary Table 2, 33P radiometric assay); thus demonstrating that CCT245737 is a potent and selective CHK1 inhibitor.
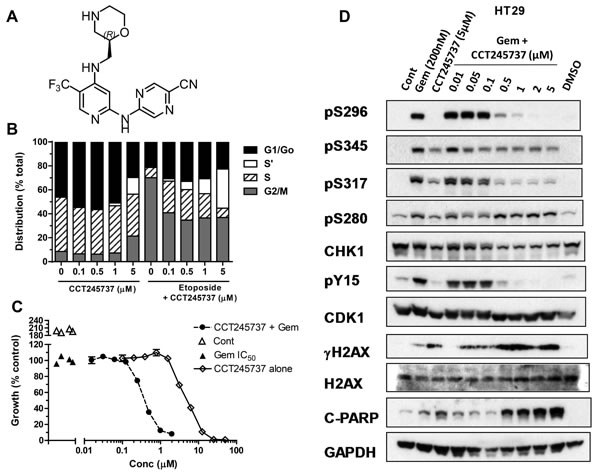
Figure 1: Structure and cellular pharmacology of CCT245737. A. Chemical structure of CCT245737 B. Effects of 24h exposure to CCT245737 either alone or following acute etoposide exposure (50µM x 1h) on the cell cycle distribution in HT29 colon carcinoma cells. BrdU and PI staining were carried out as described in Materials and Methods. Similar results were obtained in a repeat experiment. C. Representative results for a potentiation assay using a fixed concentration of gemcitabine (GI50, ▲ ) and increasing concentrations of CCT245737 either alone ( ◊ ) or in combination with gemcitabine (●) in SW620 colon cancer cells. Vehicle treated cells ( ∆ ). Data points are mean±SE for 4 replicates. D. Characterization of the effects of CCT245737 on gemcitabine-induced biomarker expression in HT29 colon cancer cells. Protein expression was measured using western blotting as described in Materials and Methods with 50μg sample per lane. GAPDH was used as a loading control. Similar results were obtained in a repeat experiment.
Cellular pharmacology of CCT245737
The ability of CCT245737 to abrogate an etoposide-induced G2 checkpoint (MIA) in four cell lines is shown in Table 1 and IC50 values ranged from 30 to 220nM, confirming potent cellular CHK1 inhibition. There was a greater than 10-fold range in GI50 (0.41 to 5.4µM) for single-agent CCT245737 in these cell lines. Cell cycle studies (Figure 1B) showed that CCT245737 had minimal effects in HT29 cells up to 0.5µM for 24h, but at higher concentrations there was an apparent decrease in the G1/Go population and a concomitant increase in the S’ population, which are unlabelled S-phase cells that may represent a replication crisis [24]. Etoposide alone caused a marked loss of G1/G0 and a corresponding G2/M block. The addition of increasing concentrations of CCT245737 abrogated this G2/M block with a corresponding increase in the S’-phase population, confirming that CCT245737 can abrogate an etoposide-induced G2/M arrest.
Previous studies have established that the antimetabolite gemcitabine and topoisomerase 1 inhibitor SN38 are amongst the best candidate drugs for combination with CHK1 inhibitors [24, 25]. Table 1 confirms that in several human tumor cell lines, CCT245737 significantly enhanced the cytotoxicity of these two anticancer drugs with the greatest potentiation occurring with gemcitabine (Figure 1C). Importantly, similar studies using a more conventional approach to determining the potentiation, with minimally toxic concentrations of CCT245737 and a range of genotoxic drug concentrations, showed a similar relationship (Supplementary Figure 2). However, this conventional assay showed less potentiation and was markedly more sensitive to variations in the CHK1 inhibitor concentration employed, which has implications for early drug development as discussed later. There was no statistically significant difference in cytotoxicity, MIA or PI activity of the S-enantiomer CCT252463 compared with CCT245737 (data not shown).
Cellular PD biomarker studies in HT29 colon tumor cells showed that gemcitabine markedly induced autophosphorylation of CHK1 on S296 and phosphorylation of S280, S317 and S345 (Figure 1D). In addition, pY15 CDK1 was increased, consistent with a drug-induced cell cycle arrest. The addition of ≥0.5µM CCT245737 to gemcitabine caused a marked loss of pS296 signal with complete inhibition at ≥2µM CCT245737. Similar effects were seen on pS317 CHK1 but by contrast there was less marked or minimal inhibition of the other CHK1 phosphorylation sites (pS280 and pS345) or alteration in the expression of total CHK1. Loss of pY15 CDK1 corresponded exactly with loss of pS296 CHK1, consistent with abrogation of cell cycle checkpoint and there was a marked co-ordinate increase in pS139 H2AX (γH2AX) a marker of DNA double-strand breaks and cleaved PARP, a marker of apoptosis. Loss of other inhibitory phosphorylations, such as pT14 CDK1 might further enhance the activity of CCT245737. These observations imply that CCT245737 caused CHK1 inhibition, abrogated gemcitabine induced cell cycle arrest causing DNA damage and cell death. Similar results were obtained in the SW620 colon cancer cell line with either gemcitabine or SN38 (Supplementary Figure 3). These data confirm that CCT245737 can inhibit CHK1 activity and enhance the cytotoxicity of gemcitabine and SN38 in several human cancer cells line in vitro.
Table 1: Summary of in vitro MIA, SRB & PI data for CCT245737
Cells |
MIA (µM) |
GI50 (µM) |
Genotoxic Agent |
Combination GI50 (µM) |
Potentiation Index (PI) |
HT29 |
0.030±0.012 (n = 6) |
0.70±0.29 (n = 6) |
SN38 |
0.39±0.061 (n = 3) |
1.8±0.31* (n = 3) |
Gem |
0.09±0.023 (n = 6) |
7.9±2.1*** (n = 6) |
|||
SW620 |
0.22±0.047 (n = 3) |
5.4±1.4 (n = 5) |
SN38 |
1.8±0.57 (n = 4) |
3.1±1.5* (n = 4) |
Gem |
0.33±0.064 (n = 7) |
17±3.4*** (n = 7) |
|||
MiaPaCa-2 |
0.063±0.011 (n = 3) |
1.3±0.40 (n = 3) |
SN38 |
0.43±0.062 (n = 3) |
3.1±0.43* (n = 3) |
Gem |
0.066±0.025 (n = 3) |
23±9.6 (n = 3) |
|||
Calu6 |
0.084±0.0070 (n = 3) |
0.41±0.059 (n = 3) |
SN38 |
0.32±0.021 (n = 3) |
1.3±0.084* (n = 3) |
Gem |
0.046±0.0069 (n = 3) |
9.1±1.5* (n = 3) |
Mitosis induction assay (MIA IC50) results and cytotoxicity (GI50) values were determined as described in Material and Methods. Potentiation index (PI) is the ratio of GI50 / Combination GI50. Potentiation index (PI) values.1 indicate potentiation of the genotoxic drug. Values are mean±SD of n independent determinations. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Pharmacokinetic and pharmacodynamic relationships of CCT245737 in human tumor xenografts in mice
A critical step in successful drug development is characterization of the pharmacokinetics and metabolism of a molecule, to ensure effective tissue drug exposure [26, 27]. Figure 2A and Supplementary Table 3 summarize the pharmacokinetics of CCT245737 in BALB/c mice following either i.v. or p.o. administration. An i.v. dose of 10mg/kg CCT245737 gave a peak plasma concentration of 4µmol/L, with a half-life of 2.86h, an AUC0-∞ of 9.96µmol.h/L, a plasma clearance of 2.1L/h/kg and a large volume of distribution (0.19L). The equivalent oral dose gave an almost identical profile with an AUC0-∞ of 10.4µmol.h/L showing complete oral bioavailability (F = 105%). Plasma protein binding in nude mice was 75.6% at 10µM CCT245737. In addition, this compound has high cell permeability, as measured by transport across a CaCo2 cell monolayer, with Pe = 25x10-6cm/s. Moreover, there was no evidence of asymmetry in the permeability with apical > basal or basal > apical across the monolayer, indicating that the compound is not a substrate for active efflux by PGP. CCT245737 had linear kinetics in whole blood (Figure 2B). Consistent with the large volume of distribution, concentrations of CCT245737 in the spleen were 10 to 25 times greater than in the plasma for both routes of administration and this was reflected in the correspondingly greater AUC0-∞ (Supplementary Table 3). Similarly high tumor/plasma ratios were obtained in HT29 xenografts treated with a fixed dose of gemcitabine and different doses of CCT245737 (Supplementary Figure 4). Moreover, 24h following a dose of 12.5mg/kg p.o. CCT245737, mean tumor drug concentrations were ≥ 3µmol/L, a value that would be expected to markedly inhibit cellular CHK1 kinase activity (see MIA IC50 Table 1) and gave significant antitumor activity (Supplementary Table 4).
An in vivo pharmacokinetic-pharmacodynamic (PK-PD) study in HT29 tumor xenografts taken from mice administered gemcitabine followed by CCT245737 is shown in Figure 2C and 2D. Gemcitabine alone (100mg/kg i.v.) caused marked pS296 CHK1 induction along with pS317 CHK1 and pY15 CDK1. The addition of CCT245737 at 50 or 100mg/kg p.o. significantly reduced the pS296 signal and pY15 CDK1 at the higher dose (P < 0.05, Figure 2D) but not pS317, consistent with CHK1 inhibition in vivo and the in vitro cellular data (see Figure 1D, Supplementary Figure 3). These PK-PD studies show that following oral administration, for a range of drug doses, adequate CCT245737 tumor drug exposure for durable CHK1 inhibition can be achieved.
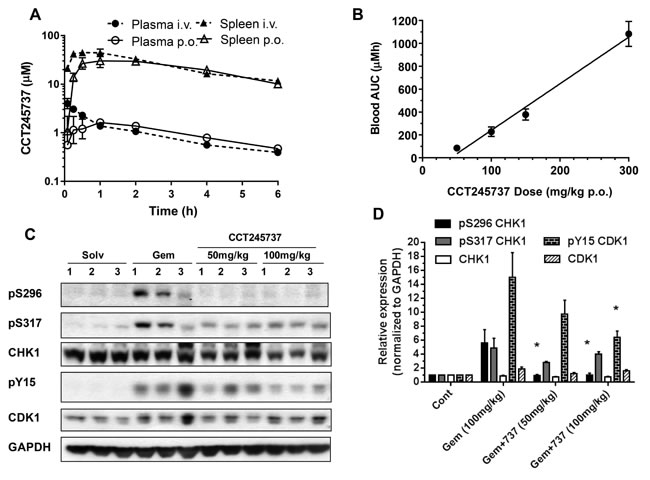
Figure 2: Summary of the pharmacokinetic and pharmacodynamic properties of CCT245737 in vivo. A. Pharmacokinetics of CCT245737 in plasma and spleen following single bolus administration of 10mg/kg either i.v. or p.o. in BALB/c mice. B. Relationship between drug exposure (blood AUC) and drug dose for CCT245737 in BALB/c mice. C. Pharmacodynamic effects of CCT245737 on gemcitabine-induced CHK1 and cell cycle biomarkers at 24h in human HT29 colon carcinoma xenografts. A single dose of gemcitabine was administered on day 0 (100mg/kg i.v.) and a single dose of CCT245737 administered 24h later (50 or 100mg/kg p.o.) and samples were taken 24h following the last drug dose. Protein expression was measured by western blotting as described in Materials and Methods and GAPDH was used as a loading control. Similar results were obtained at the 6h time point. D. Quantification of the pharmacodynamic effects of CCT245737 shown in Figure 2C. Protein expression was quantified using ImageQuant software and normalized to GAPDH expression. Values are mean±SE, n = 3 and statistical analysis was by one-way ANOVA with Dunnett’s correction; *P < 0.05 indicates a significant difference from gemcitabine treatment alone.
Therapeutic activity of CCT245737 combinations in human tumor xenografts
Extensive antitumor studies were carried out using gemcitabine and different doses of CCT245737 in HT29 human colon tumor xenografts (Figure 3A, 3B and Supplementary Table 4). Figure 3A shows that CCT245737 (150mg/kg p.o.) or gemcitabine alone (100mg/kg i.v.) had minimal antitumor activity with growth delays of 0.3±3.4 and -0.6±2.7 days, respectively (mean±SD, Supplementary Table 4). However, the addition of CCT245737 (150mg/kg p.o.) significantly increased the antitumor activity of gemcitabine giving a growth delay of 19.3±1.7 days (mean±SD, P < 0.001, Supplementary Table 4). This enhancement was associated with no body weight loss in drug treated animals (Supplementary Figure 5A). Figure 3B and Supplementary Table 4 show the effect of different doses of CCT245737 up to a maximum of 150mg/kg p.o. on the antitumor effects of a fixed dose of gemcitabine (100mg/kg i.v.) in HT29 xenografts. As expected there appears to be a saturable effect of CCT245737 dose on antitumor activity with the greatest effect occurring at around 100mg/kg p.o. At doses up to 50mg/kg p.o. CCT245737 there was a steep increase in the effects of this CHK1 inhibitor on the tumor growth delay response to gemcitabine indicating that low doses of CCT245737 may still have marked therapeutic activity in this combination. These treatments were minimally toxic with a body weight nadir on day 16 and only 2.5% weight loss in the combination arm (Supplementary Figures 5B & 5C). In addition, there was no antitumor effect of CCT245737 alone at the combination maximum tolerated dose (MTD, 150mg/kg p.o.) and only a small increase in antitumor activity of gemcitabine alone at the MTD of 200mg/kg i.v. from 3 to 6 days (Figure 3B and Supplementary Table 4). Importantly, these results clearly show that the combination of CCT245737 with gemcitabine can readily achieve much greater antitumor activity than either agent alone at their respective MTDs. Moreover there was no increase in toxicity thus validating the therapeutic utility of CHK1 inhibitors in vivo.
These antitumor studies with CCT245737 were extended into combinations employing other anticancer drugs such as irinotecan in the colon cancer HT29 xenograft model (Figure 3C, Supplementary Table 5). Once again there was no antitumor activity of CCT245737 alone at the combination MTD (1.0±1.0 days growth delay, mean±SD), however, irinotecan had substantial antitumor activity alone at the dose employed with a growth delay of 6.2±4.6 days (mean±SD, n = 6). Nevertheless the addition of CCT245737 doubled the growth delay induced by irinotecan to 12.4 days (P < 0.05) with a body weight nadir on day 10 of only 2% loss (Supplementary Figure 5D). Studies in SW620 colon cancer xenografts (Figure 3D) showed that CCT245737 and gemcitabine were minimally active as single agents and the combination showed a significantly enhanced antitumor activity compared with gemcitabine alone (7.3±2.1 versus1.5±2.3 days growth delay, P < 0.001, Supplementary Table 5) with minimal toxicity (4.3% body weight loss on day 9, Supplementary Figure 5E). These results demonstrate that CCT245737 can significantly enhance the antitumor activity of irinotecan and gemcitabine in a number of different human tumor xenograft models. Furthermore, when tested in HT29 tumors, the activity of gemcitabine combined with CCT245737 was greater than could be achieved at the MTD of either agent alone.
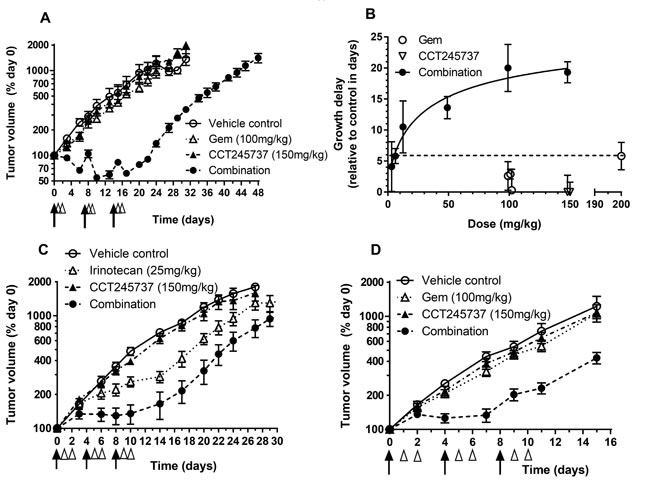
Figure 3: Therapeutic activity of CCT245737 in combination with gemcitabine or irinotecan in human tumor xenografts. A. CCT245737 enhances the antitumor effects of gemcitabine in HT29 human colon cancer xenografts. Symbols and treatments: o vehicle; ∆ gemcitabine alone (100mg/kg i.v. days 0,7 and 14); ▲ CCT245737 alone (150mg/kg p.o. days 1,2; 8,9 and 15,16) and ● combination of gemcitabine + CCT245737 at doses and schedules shown for single agents. Values are mean±SE with 5-6 mice per point. B. Summary of the antitumor effects of different oral doses of CCT245737 combined with a fixed dose of gemcitabine (100mg/kg i.v.) in HT29 human colon carcinoma xenografts. The activity of the MTD for single dose gemcitabine (200mg/kg i.v.) and CCT245737 (150mg/kg p.o.) are also shown (dashed line). Growth delays (days) were determined in several independent experiments and are shown as mean±SE for n = 6-10 mice per point. C. Antitumor efficacy of irinotecan or CCT245737 alone or in combination in HT29 human colon carcinoma xenografts. Symbols and treatments: o vehicle; ∆ irinotecan alone (25mg/kg i.p. days 0,4 and 8); ▲ CCT245737 alone (150mg/kg p.o. days 1,2; 5,6; 9 and 10) and ● gemcitabine + CCT245737 combination at times and doses shown for single agents. D. Antitumor activity of gemcitabine or CCT245737 alone or combined in SW620 human colon cancer xenografts. Symbols and treatments: o vehicle; ∆ gemcitabine alone (100mg/kg i.v. days 0,4 and 8); ▲ CCT245737 alone (150mg/kg p.o. days 1,2; 5,6; 9 and 10) and ● gemcitabine + CCT245737 combination at times and doses shown for single agents. Values are mean±SE, n = 5-10 mice per point for C. and D.. Tumor size and growth delay were determined as described in Materials and Methods. The black arrows in A., C. and D. indicate when the genotoxic agent was administered and the open arrow heads represent CCT245737 administration with day 0 being the start of therapy of established tumors.
A novel, quantitative and sensitive biomarker assay for CHK1 activity
CCT245737 may be evaluated in several clinical settings, including solid tumors treated with genotoxic drugs, such as lung cancers. This will require a validated assay for a suitable PD biomarker to confirm that CHK1 inhibition has occurred. We therefore developed an ELISA for measuring S296 CHK1 autophosphorylation (pS296) in human tumor material as this is the most sensitive and specific proximal biomarker of CHK1 kinase activity (see Materials and Methods for details). Figure 4A shows that CCT245737 alone or gemcitabine plus carboplatin combined (a standard treatment for lung cancer) had minimal antitumor activity in the Calu6 RAS mutant NSCLC human tumor xenograft model (Supplementary Table 5). The addition of CCT245737 to the genotoxic agents resulted in a statistically significant 9 day increase in tumor growth delay (Supplementary Table 5, P < 0.001) with minimal weight loss (nadir on day 3 = 2%, Supplementary Figure 5F). Western blotting for PD biomarker changes in Calu6 xenograft tumors taken from individual mice (Figure 4B) showed that the combination of gemcitabine and carboplatin markedly induced pS296 CHK1 but had minimal effects on pS317 and pS345 CHK1 signals, consistent with our previous studies (Figure 1D [24, 28]). The addition of CCT245737 completely abolished the pS296 signal but actually enhanced both pS317 and pS345 CHK1 levels - confirming once again that pS296CHK1 is a sensitive, robust and reproducible biomarker of CHK1 inhibition. There were minimal changes in total CHK1 expression. Using the same Calu6 tumor lysates from these studies we show that the ELISA assay for pS296 CHK1 accurately reproduced this signal as detected by western blotting (Figure 4C). In addition the ELISA for total CHK1 showed minimal changes consistent with the immunoblotting results (Figure 4D). Both assays were capable of detecting recombinant pS296 and total CHK1 down to a concentration of 0.4nM (assuming a signal:noise ratio of 2:1). These results show that we have developed an accurate and sensitive ELISA for pS296 CHK1, a biomarker of CHK1 activity, which can be quantified in human tumor tissue.
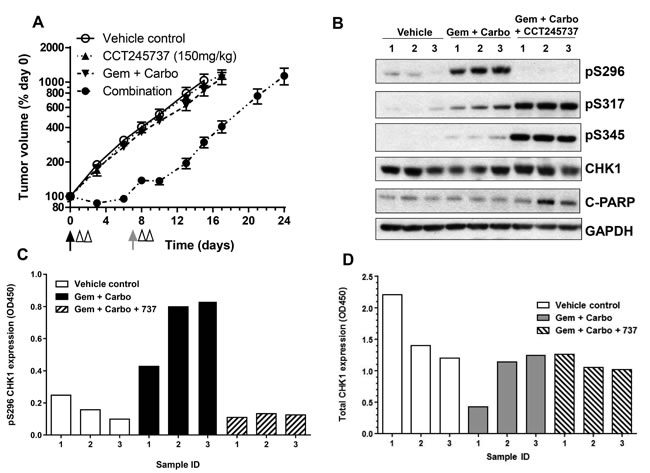
Figure 4: Development and validation of a pharmacodynamic biomarker assay for CCT245737 activity in human lung tumor xenografts. A. Enhanced activity of gemcitabine and carboplatin combined with CCT245737 compared with either CCT245737 alone (150mg/kg p.o.) or gemcitabine and carboplatin without CCT245737 (100mg/kg i.v. and 5mg/kg i.p., respectively) in Calu6 human lung cancer xenografts. Values are mean±SE, n = 6-8 mice per point. The black arrows indicate the time of gemcitabine and carboplatin drug administration, the gray arrow when gemcitabine was administered and the open arrow heads the time of CCT245737 administration. B. Western blot of biomarker expression in Calu6 tumor xenografts treated as in A.. Samples were taken 6h after the last dose of CCT245737 from individual mice and analysed by western blotting as described in Materials and Methods with GAPDH as a loading control. C. Expression of pS296 CHK1 and D. total CHK1 expression measured in the same human tumor xenograft samples as shown in B. using novel ELISA methods the development of which are described in Methods and Materials and Results.
Single-agent antitumor activity of CCT245737 in an Eµ-Myc driven mouse model of B-cell lymphoma
The deregulation of certain oncogenes such as MYCN in neuroblastoma and C-MYC in lymphoma appears to be associated with an increase in single-agent CHK1 sensitivity, possibly through enhanced replication stress. We therefore evaluated the antitumor activity of CCT245737 in an Eµ-Myc driven transgenic mouse transplant model of B-cell lymphoma that infiltrates the lymph glands. Figure 5A clearly shows that 9 days oral administration of 150mg/kg CCT245737 significantly reduced the weight of the inguinal, brachial/axillary and mesenteric lymph nodes compared with vehicle treated control animals (P < 0.05). Moreover there were minimal effects on normal tissues such as the lung (Figure 5B, p = 0.904) and bone marrow (Figure 5C, p = 0.0838) or kidney (p = 0.959, not shown). There was a significant decrease in thymus weight following CCT245737 treatment (p < 0.01) suggesting some tumor involvement and spleen weight was increased significantly (p < 0.01) suggesting some splenomegaly (not shown). The treatment was well tolerated as evidenced by stable body weights (Figure 5D) and negligible effects on other parameters such as water consumption and body temperature (data not shown). Consequently these results show that CCT245737 has significant antitumor activity as an oral single-agent in a Myc-driven mouse model of B-cell lymphoma.
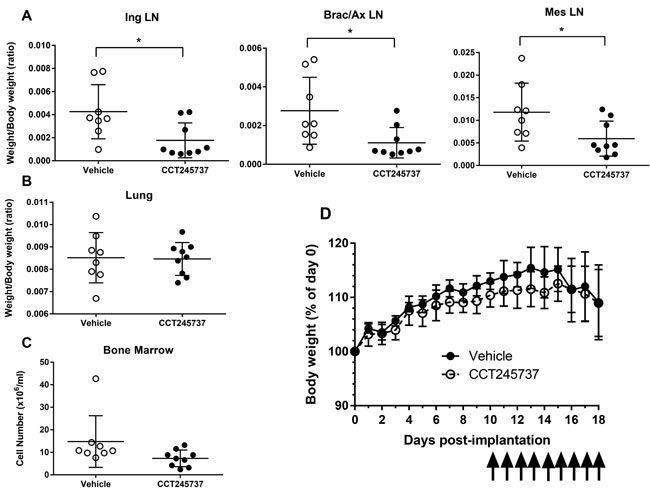
Figure 5: Antitumor activity of single-agent CCT245737 in an Eµ-Myc mouse model of human B-cell lymphocytic leukemia. A. Effect of CCT245737 treatment on: inguinal (Ing), brachial/axillary (Brac/Ax) and mesenteric (Mes) lymph node (LN) mass compared with controls in Eµ-Myc tumor transplanted mice. These sites are commonly associated with infiltrating B-cell lymphoma in this model system. B. Effect of CCT245737 treatment on normal lung tissue mass and C. bone marrow cell number in Eµ-Myc tumor transplanted mice. D. Effect of CCT245737 treatment on mouse body weight. Animals were administered CCT245737 (150mg/kg p.o.) or vehicle, once daily for 9 days (as indicated by arrows) starting 10 days post-implantation. Mice were culled 24h after the last treatment. Values are mean±SD, n = 7-9 and statistical analysis was by Students t-test: *P < 0.05 indicates a significant difference from vehicle control treated animals.
Discussion
CCT245737 is a novel, potent, selective and well-tolerated orally active CHK1 inhibitor, and our clinical development candidate. Enzyme kinetic and modeling data clearly show that CCT245737 is a potent, ATP-competitive inhibitor of human CHK1 kinase with an IC50 of 1.4nM. Initial screening against other kinases demonstrated an appropriate selectivity profile with subsequent studies showing ≥ 500-fold selectivity for all but five kinases. Importantly there was minimal inhibition of CHK2 which has little therapeutic utility in genotoxic combinations [29, 30]. There was also no cross-reactivity with CDK1 and 2, inhibition of which might antagonize the cytotoxic effects of CHK1 inhibition, as CDK1 activation has been implicated in CHK1 inhibitor-based cell killing [30]. This selectivity profile compares favorably with other CHK1 inhibitors, such as AZD7762 [31], GDC-0575 [32], PF00477736 [33] and LY2603618 [34]. There was evidence of potent intracellular CHK1 inhibition in several human tumor cell lines following genotoxic stress (Table 1) and this was associated with abrogation of a late S, G2/M checkpoint (Figure 1C) consistent with CHK1 inhibition and similar to other CHK1 inhibitors [28, 33] . Several previous studies have established that certain classes of genotoxic anticancer drugs are potentiated more effectively than others by CHK1 inhibitors e.g. antimetabolites and topoisomerase 1 inhibitors [25, 35].
Using a rapid and sensitive assay for determining the combination GI50 (where a fixed concentration of the genotoxic agent is combined with a range of CCT245737 concentrations) we derived a potentiation index (ratio of IC50 for CCT245737 alone: combination GI50) and showed that the cytotoxicity of both gemcitabine and SN38 were significantly enhanced (Table 1). This technique was validated by comparison with the more conventional approach where a non-toxic concentration of a CHK1 inhibitor is combined with increasing concentrations of the genotoxic drug (Supplementary Figure 3) with qualitatively similar results. The rapid technique presented here has the advantage of quickly establishing a genotoxic GI50 in contrast to the most active non-toxic CHK1 inhibitor concentration. Moreover, this rapid technique was more sensitive than the conventional approach with 2 to 3-fold greater PI values, allowing evaluation and ranking of relatively non-potent and selective compounds in the early stages of the CHK1 inhibitor drug discovery project.
It is clear from these and other available data that antimetabolites such as gemcitabine are potentiated by several CHK1 inhibitors to a greater extent than topoisomerase 1 inhibitors [24, 25, 35]. This may reflect a greater ability of the former to deplete nucleotide pools and enhance replication stress with an increased reliance on CHK1 to avoid replication fork collapse [36, 37]. Our cellular PD biomarker studies reproducibly showed that there was a concentration-dependent inhibition of genotoxic induced pS296 CHK1 autophosphorylation by CCT245737. Moreover this pS296 CHK1 signal was a more sensitive and robust readout of CHK1 inhibition in our systems than other phospho-CHK1 sites. This is in contrast to other reports [38], possibly due to the fact that pS296 is a specific and direct readout of CHK1 activity [39], an essential requirement for PD biomarkers of kinase inhibitors. This CCT245737 concentration-dependent loss of pS296 CHK1 signal was coincident with loss of cell cycle arrest, increased DNA damage and apoptosis implying that CHK1 inhibition induced cell death [3, 40].
Pharmacokinetic studies with CCT245737 showed complete oral bioavailability with linear pharmacokinetics and high tumor/plasma ratios consistent with extensive tumor exposure. Pharmacodynamic studies showed that CCT245737 concentrations required for inhibition of gemcitabine-induced pS296 CHK1 and pY15 CDK1 were readily achieved in tumors at 24h following a single oral dose of 50mg/kg (Figure 2C). We and others have shown that an effective antitumor combination strategy involves delaying CHK1 inhibitor administration by 24h following a genotoxic drug dose and then extending effective CHK1 exposure for a further 48h [7, 24, 35]. Moreover there were no detectable pharmacokinetic interactions with this CHK1 inhibitor chemical class and the genotoxic agents studied. The PK-PD relationship described here for CCT245737 and its high oral bioavailability, make this an ideal compound for clinical evaluation. `
Efficacy studies in a number of different xenograft tumor models showed that CCT245737 enhanced the activity of both gemcitabine and irinotecan. Importantly and uniquely, we show for the first time that a CHK1 inhibitor can markedly enhance the antitumor activity of gemcitabine to a greater extent than can be achieved by the MTD of either agent alone. Moreover the dose-response curve had a relatively steep initial phase, suggesting that a therapeutically useful increase in gemcitabine activity is feasible at modest doses of CCT245737, as might be achieved in early stage clinical trials. This response appeared to plateau at doses above 50mg/kg, implying that very high concentrations of CCT245737 may not be required for optimal therapeutic activity and consistent with the idea that CHK1 inhibition in tumors may enhance DNA damage or stalled replication fork collapse [8, 13]. These observations further support the clinical development and evaluation of CCT245737 in combination with genotoxic agents such as gemcitabine.
To this end we have shown that CCT245737 can significantly enhance the antitumor activity of both gemcitabine and carboplatin in a RAS mutant NSCLC human tumor xenograft model. This is an area of unmet clinical need and a disease setting in which clinical testing of CCT245737 is proposed. In order to ensure that these antitumor effects are a result of CHK1 inhibition we also carried out PD studies and showed once again that pS296 CHK1 inhibition is a more sensitive, robust and reproducible marker of CHK1 inhibition than either pS317 or pS345 CHK1. Indeed, induction/inhibition of pS317 and pS345 CHK1 by CCT245737 appeared to be either concentration and/or time dependent (Figure 1D and Supplementary Figure 3) as well as context dependent (Figure 2C and 4B) possibly through differences in feedback repression of ATR/ATM, DNA damage and repair capacity or PP2A and WIP1 phosphatase activity. Consequently we developed a novel, sensitive and quantitative ELISA for pS296 CHK1 which will facilitate the clinical evaluation of this combination and allow target inhibition monitoring in the patient. Nevertheless other indirect readouts such as CDC25A or pY15 CDK1 loss, apoptosis markers and γH2AX and RAD51 foci formation may prove useful in confirming that functionally significant CHK1 inhibition has occurred [8, 24, 41, 42].
An intriguing aspect of CHK1 inhibitor development is the realization that these compounds may exhibit single-agent activity in particular malignancies. It appears that tumors with deregulated MYC expression or high levels of replication stress are very sensitive to single-agent CHK1 inhibition [17, 18, 21, 22]. To expand the potential clinical utility of CCT245737 monotherapy we have shown that it has significant antitumor activity in an Eµ-Myc driven transgenic murine model of infiltrating B-cell lymphoma (Burkitt’s-type lymphoma) [43]. MYC gene alterations are common in other B-cell neoplasms and are often associated with poor outcomes [44]. CCT245737 as a single-agent was well tolerated in the Eµ-Myc transgenic model and had minimal effects on normal tissues such as lung and bone marrow and kidney, although there was some evidence of splenomegaly. However the use of an isogenic MYC inducible model would be even more definitive. Nevertheless, these results are consistent with studies using the CHK1 inhibitor PF-0477736 in a variety of Eµ-Myc driven transgenic murine lymphoma cell lines [18] and are directly comparable with antitumor studies using UCN01 (5mg/kg x 9 days), a non-selective CHK1 inhibitor in the same model [45]. In the latter case clear evidence of tumor growth inhibition was associated with a decreased blood cell count, possibly due to off-target effects of UCN01 and in marked contrast to this study. Should CHK1 inhibitors prove successful in the clinic it will be important to identify biomarkers of sensitivity to facilitate patient selection and to establish other molecular targets, which upon inhibition give a synthetically lethal phenotype [46].
In conclusion, we have investigated the preclinical pharmacology, PK-PD relationships and antitumor activity of the novel, potent, selective and orally active CHK1 clinical development candidate CCT245737. We clearly show that CCT245737 enhanced the antitumor activity of several anticancer agents in vivo and uniquely demonstrate that for gemcitabine this was greater than the activity achievable by dose escalation of either agent alone. The lack of additional toxicity in the combinations thus provides a valuable therapeutic gain. We also show that pS296 CHK1 is a robust PD biomarker of CHK1 activity and provide a novel, quantitative ELISA-based assay suitable for the measurement of this biomarker in clinical material. We demonstrate that CCT245737 has single-agent antitumor activity in a Myc driven model of B-cell lymphoma, providing preclinical evidence of a potential adult clinical monotherapy setting. Consequently CCT245737 is in late stage preclinical development in preparation for a first-in-man phase I clinical trial.
Materials and Methods
Drugs and compounds
CCT245737 (Figure 1A) and its S-enantiomer CCT252463 were synthesized as described [47]. Irinotecan was purchased from Pfizer and its active metabolite (SN38) from LKT laboratories. Gemcitabine was obtained from Eli Lilly. All other compounds were purchased from Sigma.
Human tumor cell lines
The colon tumor cell lines HT29 (p53-/-, APC+/-, PIK3CA+/-, BRAF+/- and SMAD4-/-) and SW620 (p53-/-, KRAS-/-, APC-/-, SMAD4-/- and MAP2K4-/-), the pancreatic cancer cell line MiaPaCa-2 (p53-/-, KRAS+/-, KDM6A-/-, CDKN2a; p14-/- and CDKN2A) and the non-small cell lung cancer Calu6 cell line (p53-/- and KRAS+/-) were purchased from ATCC (Lot numbers 4487729, 3924081, 57866607 and 58683029, respectively and Sanger Centre data http://www.sanger.ac.uk/genetics/CGP/cosmic). Cells were grown in Dulbecco’s modified Eagles medium containing 10% fetal calf serum and 2mM glutamine and were mycoplasma free [24].
In vitro kinase assays
Commercial in vitro 33P radiometric kinase assays were carried out against 124 human kinases using 10µM CCT245737 at ATP concentrations corresponding to the kinase Km,ATP (MRC Phosphorylation Unit, Dundee). Other kinase IC50 determinations for CHK2 and FLT3 were performed using a commercial assay (Z’-Lyte, Invitrogen) or in-house with recombinant human CHK1 on a LabChip® EZ Reader II (PerkinElmer) or CDK1 in a DELFIA assay (PerkinElmer).
Cellular cytotoxicity, G2 checkpoint abrogation and potentiation assays
Assays were performed as described [24]. Cytotoxicity was determined as the drug concentration that gave 50% inhibition of tumor cell proliferation (GI50) using a 96h (i.e. 4-doublings) Sulforhodamine B (SRB) assay. Inhibition of intracellular CHK1 activity was measured using a cell based ELISA for the abrogation of an etoposide induced G2 checkpoint (mitosis induction assay, MIA [24]). The IC50 for G2 checkpoint abrogation (MIA) was determined in the presence of nocodazole using UCN01 as a positive control. The activity index (AI) was used as a measure of the compounds ability to induce mitosis relative to its toxicity (i.e., ratio of MIA IC50: 96h SRB GI50). Routine potentiation studies were carried out using a fixed concentration (GI50) of either gemcitabine or SN38 in combination with a range of CCT245737 concentrations to determine the combination GI50 of CCT245737. The ability of CCT245737 to enhance gemcitabine or SN38 cell killing was expressed as a potentiation index (PI) equal to the ratio of the GI50 for CCT245737 alone versus the combination GI50 for CCT245737 (see above). PI values > 1 indicate potentiation of the genotoxic activity. In addition, a series of experiments was carried out using fixed, non- or minimally toxic concentrations of CCT245737 (≤GI20) with a range of different concentrations of gemcitabine or SN38 to determine the extent to which CCT245737 enhanced drug cytotoxicity compared with the genotoxic agent alone, i.e. conventional PI (ratio GI50 genotoxic alone: GI50 genotoxic combined with non-toxic CCT245737 concentration, Con PI).
Immunoassays
Western blotting was carried out as described [24]. ELISA assays for pS296 CHK1 and total CHK1 were developed and validated for use in vitro and in tumor homogenates. Tumor samples for ELISA and western blotting were lysed in CHAPS buffer (1% CHAPS in 150mM NaCl, 50mM Tris-HCl pH 8.0, 1mM EDTA, 0.2mM PMSF and 1:50 Sigma Phosphatase inhibitors 2 and 3 and 1:100 Sigma protease cocktail) and immunoblotted. ELISA assays were based on commercially available kits with modifications (Cell Signalling Technologies). For pS296 CHK1 detection the commercial pS317 CHK1 antibody was replaced with a pS296 CHK1 antibody (Cell Signalling Technology). Xenograft tumor homogenate was added in duplicate to 96 well plates (100µl containing 40ug protein) and quantification of the colorimetric readout was determined at 450nm using a PerkinElmer 2103 EnVision Multilabel plate reader.
Cell cycle analysis
Cell cycle analysis using propidium iodide and bromodeoxyuridine staining was as described [28].
Animal studies
Compound tolerability and pharmacokinetic investigations were carried out in female BALB/c mice (Charles River). Human tumor xenografts were established s.c. in female CRTac:Ncr-Fox1(nu) athymic mice and treated as previously described [24, 28]. The vehicle for oral administration of CCT245737 was DPTW (10% DMSO, 20% PEG400, 5% Tween 80 and 65% water) and gemcitabine and irinotecan were administered in their respective clinical vehicles. Treatments were generally initiated when tumors reached a mean diameter of 5-6mm (day 0). For combination studies, CCT245737 was given orally 24 and 48h after genotoxic drug administration, previously determined as an optimal schedule for CHK1 inhibitor and genotoxic drug combinations [24]. In HT29 xenograft studies, gemcitabine was administered at 100mg/kg i.v. on days 0,7 and 14 and CCT245737 at the indicated doses on days 1,2,8,9,15 and16. Irinotecan was administered at 25mg/kg i.p. on days 0,4 and 8 with CCT245737 administered at 150mg/kg p.o. on days 1,2,5,6,9 and 10. In SW620 and Calu6 xenograft studies, gemcitabine was administered at 100mg/kg i.v. on days 0,4 and 8 and CCT245737 subsequently at 150mg/kg on days 1,2,5,6,9 and 10. For the Calu6 xenograft studies involving gemcitabine and carboplatin, drugs were administered at 100mg/kg i.v. and 5mg/kg i.p., respectively on day 0 with gemcitabine alone at 100mg/kg i.v. on day 7 with CCT245737 at 150mg/kg p.o. on days 1,2,8 and 9. The genotoxic drug doses employed were sub-maximally active to facilitate detection of subsequent potentiation. Initial treatment groups contained from 6 to 10 mice and animals were inspected daily and tumor size and volume measured every 2 or 3 days. Tumor volume and growth delay were determined as previously described [28].
Transgenic Eµ-Myc mice which develop aggressive infiltrating lymphoma were established and monitored as previously described [48, 49]. To generate transgenic Eµ-Myc driven lymphoma allografts, tumor cells from 3 separate tumors were harvested, cells counted and injected via a tail vein. Six mice were set up per tumor to provide 3 control and 3 treated animals, giving a maximum of 9 mice per treatment group. Animals were monitored daily and continuously using RFID transponders to measure temperature, activity and water consumption as previously described [49]. For studies of single-agent CCT245737 activity in mice injected with transgenic Eµ-Myc lymphoid tumor cells, CCT245737 was administered at 150mg/kg p.o. for 9 successive days with culling 24h after the last dose. Lymph nodes and other tissues were removed from vehicle and CCT245737 treated mice and their weights and tissue/body weight ratios compared to assess antitumor activity. Bone marrow cellularity was also determined to check for tumor cell involvement. All mice were handled in compliance with local and national animal welfare guidelines [50].
Pharmacokinetics
Compounds were extracted from whole blood, plasma and tissue homogenates with methanol-containing internal standards using established protocols. Drug concentrations were determined using liquid chromatography/tandem mass spectrometry (LC/MS-MS) and pharmacokinetics were calculated using Pharsight WinNonLin software (version 5.2.1) as previously described [28].
Statistics
Statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001) was determined using an unpaired, one-tailed or two-tailed t-test or one-way ANOVA with either Tukey or Dunnett test, as appropriate using GraphPad Prism 5 software.
Acknowledgments
We gratefully acknowledge help from Gowri Vijayaraghavan with flow cytometry, Nathan Brown with modelling studies and useful discussions with other members of the Cell Cycle Control Team at The ICR.
Grant support
This work was supported by Cancer Research UK [CUK] grant numbers C309/A8274, C309/A11566. We acknowledge the support received from The Wellcome Trust, grant number 091763Z/10/Z and also NHS funding to the NIHR Biomedical Research Centre.
Conflicts of interest
M I Walton, P D Eve, A Hayes, A T Henley, M R Valenti, A K De Haven Brandon, G Box, K J Boxall, M Tall, K Swales, T P Matthews, T McHardy, M Lainchbury, J Osborne, G W Aherne, S A Eccles, F I Raynaud, I Collins and M D Garrett are current or former employees of The Institute of Cancer Research, which has a commercial interest in CHK1 inhibitors.
JC Reader is an employee of Sareum Ltd which has a commercial interest in CHK1 inhibitors. Sareum Ltd is a wholly owned subsidiary of Sareum Holdings PLC, of which JC Reader is a shareholder.
Both Sareum and The Institute of Cancer Research have been involved in a commercial collaboration with Cancer Research Technology Ltd (CRT) to discover and develop inhibitors of CHK1.
JE Hunter and ND Perkins declare no conflict of interest.
References
1. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179-204.
2. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071-8.
3. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245-55.
4. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413-31.
5. Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88-96.
6. McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142:1-10.
7. Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308-16.
8. Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: how mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76:358-69.
9. Lainchbury M, Collins I. Checkpoint kinase inhibitors. A patent review (2009-2010). Exp Opin Ther Patents. 2011;21:1191-210.
10. Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16:376-83.
11. Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134:1013-23.
12. Cho SH, Toouli CD, Fujii GH, Crain C, Parry D. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle. 2005;4:131-9.
13. Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195-201.
14. Calvo E, Chen VJ, Marshall M, Ohnmacht U, Hynes SM, Kumm E, et al. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with Pemetrexed and cisplatin in patients with advanced cancer. Invest New Drugs. 2014;32:955-68.
15. Seto T, Esaki T, Hirai F, Arita S, Nosaki K, Makiyama A, et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol. 2013;72:619-27.
16. Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin Cancer Res. 2012;18:6723-31.
17. Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proceedings of the National Academy of Sciences. 2011;108:3336-41.
18. Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 2012;31:1661-72.
19. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220-8.
20. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2-9.
21. Brooks K, Oakes V, Edwards B, Ranall M, Leo P, Pavey S, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2013;32:788-96.
22. Cavelier C, Didier C, Prade N, Mansat-De Mas V, Manenti S, Recher C, et al. Constitutive Activation of the DNA Damage Signaling Pathway in Acute Myeloid Leukemia with Complex Karyotype: Potential Importance for Checkpoint Targeting Therapy. Cancer Research. 2009;69:8652-61.
23. Matthews TP, Jones AM, Collins I. Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies. Expert Opin Drug Discov. 2013;8:621-40.
24. Walton MI, Eve PD, Hayes A, Valenti MR, De Haven Brandon AK, Box G, et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin Cancer Res. 2012;18:5650-61.
25. Xiao Y, Ramiscal J, Kowanetz K, Delnagro C, Malek S, Evangelista M, et al. Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Mol Cancer Ther. 2013;12:2285-95.
26. Sarker D, Workman P. Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res. 2007;96:213-68.
27. Workman P. How much gets there and what does it do?: The need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr Pharm Des. 2003;9:891-902.
28. Walton MI, Eve PD, Hayes A, Valenti M, De Haven Brandon A, Box G, et al. The Preclinical Pharmacology and Therapeutic Activity of the Novel CHK1 Inhibitor SAR-020106. Molecular Cancer Therapeutics. 2010;9:89-100.
29. Anderson VE, Walton MI, Eve PD, Boxall KJ, Antoni L, Caldwell JJ, et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011;71:463-72.
30. Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10:591-602.
31. Oza V, Ashwell S, Almeida L, Brassil P, Breed J, Deng C, et al. Discovery of checkpoint kinase inhibitor (S)-5-(3-fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by structure-based design and optimization of thiophenecarboxamide ureas. J Med Chem. 2012;55:5130-42.
32. Davies KD, Humphries MJ, Sullivan FX, von Carlowitz I, Le Huerou Y, Mohr PJ, et al. Single-agent inhibition of Chk1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo. Oncol Res. 2011;19:349-63.
33. Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394-404.
34. King C, Diaz H, Barnard D, Barda D, Clawson D, Blosser W, et al. Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Invest New Drugs. 2014;32:213-26.
35. Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427-38.
36. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435-46.
37. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17 Suppl 5:7-12.
38. Parsels LA, Qian Y, Tanska DM, Gross M, Zhao L, Hassan MC, et al. Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res. 2011;17:3706-15.
39. Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem J. 2005;388:705-12.
40. Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477-86.
41. Rawlinson R, Massey AJ. gammaH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments. BMC Cancer. 2014;14:483-95.
42. Del Nagro CJ, Choi J, Xiao Y, Rangell L, Mohan S, Pandita A, et al. Chk1 inhibition in p53-deficient cell lines drives rapid chromosome fragmentation followed by caspase-independent cell death. Cell Cycle. 2014;13:303-14.
43. Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt’s lymphoma. Lancet. 2012;379:1234-44.
44. Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Hematology Am Soc Hematol Educ Program. 2013;2013:575-83.
45. Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331-5.
46. Carden CP, Sarker D, Postel-Vinay S, Yap TA, Attard G, Banerji U, et al. Can molecular biomarker-based patient selection in Phase I trials accelerate anticancer drug development? Drug Discov Today. 2010;15:88-97.
47. Collins I, Matthews TP, Faria da Fonseca McHardy T, Osborne J, Lainchbury M, Walton MI, et al. 5-[[4-[[Morpholin-2-yl]methylamino]-5-(trifluoromethyl-2-pyridyl]amino]pyrazine-2-carbonitrile and therapeutic uses thereof. Intl Patent Appl. 2013;WO2013/171470A1.
48. Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eµ-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353-71.
49. Hunter JE, Butterworth J, Perkins ND, Bateson M, Richardson CA. Using body temperature, food and water consumption as biomarkers of disease progression in mice with Eµ-myc lymphoma. Br J Cancer. 2014;110:928-34.
50. Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555-77.