
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death worldwide and exerts a considerable emotional and economic burden [1, 2]. Thus, there is an important unmet need for new cardioprotective treatments. In recent years, CVD treatments have made remarkable advances in bridging both medical and surgical approaches. Nevertheless, effective cardiovascular preventive treatments remain limited.
Various studies have demonstrated that lifestyle changes, including cessation of smoking, a healthy diet, and a regular exercise regimen can help prevent or treat CVD [3]. Among these, exercise has been reported to effectively reduce cardiovascular morbidity and mortality [4]. Recently, a study showed that compared to no physical exercise at all, any level of leisure-time physical exercise is associated with decreased rate of sudden death [5]. Those who do less exercise than recommended in the “2008 physical activity guidelines for Americans” still have a 20% lower rate of sudden death. Those people who do exercise and achieve the minimum level recommended have a 31% lower risk than those who do not exercise at all [5]. In addition, exercise has also been reported to improve functional capacity, endothelial function and collateralization in patients with diverse forms of CVD [6]. However, although it appears evident that exercise may confer a cardioprotective phenotype, the strength of the effects and the underlying mechanisms remain elusive.
The main aim of this review is to explore the beneficial effects of exercise in protecting against heart damage, with a special emphasis on the mechanisms underlying these protective effects.
WHAT IS PROPER EXERCISE?
The relationship between physical exercise and CVD has increasingly been in the spotlight in recent years. Generally, participants engaging in exercise enjoy more desirable health outcomes, including better health-related quality of life and functional capacity [7]. However, rigorous exercise is not suited for everyone, and in these individuals, a less stringent form of exercise may help protect against CVD. So, what is proper exercise? In clinical studies, ‘proper exercise’ is defined as several months of mild-to-moderate physical activity or intensity-controlled-exercise training consisting of walking, jogging, swimming, skiing, or cycling 3 to 4 times a week [8]. The control group in these studies is advised to maintain prior exercise habits. The success of the exercise regimen is assessed by the increase in oxygen uptake (VO2) in the training group as compared to the control group [9]. The intensity of exercise is expressed as percent of VO2 max or percent of maximal heart rate (HR). Moderate intensity exercise is that performed at a relative intensity of 40% to 60% of VO2 max, while vigorous-intensity exercise is that performed over 60% of VO2 max [10]. For moderate-intensity exercise, a person's target heart rate should be 50 to 70% of his or her maximum HR. For vigorous intensity exercise, a person's target heart rate may be up to 70 to 85% of his or her maximum HR [11]. This estimate of maximum HR is based on the person's age. It can be obtained by subtracting the person's age from 220 [11]. Thus, the formula of the HR of moderate-intensity exercise is: HR = (50% to 70%)*(220-age).
For basic research using rodent models, there are two types of physical exercise utilized: wheel running and swimming. According to previous studies, both acute (i.e., 1–5 days) and chronic (i.e., weeks to months) exercise conferred benefits for CVD, especially myocardial ischemic injury [8]. However, for different models of myocardial injury, the manner, strength and length of exercise could vary for investigating the beneficial effects of exercise and underlying pathophysiological and molecular mechanisms.
CELLULAR ADAPTATIONS IN THE CONTEXT OF EXERCISE (FIGURE 1)
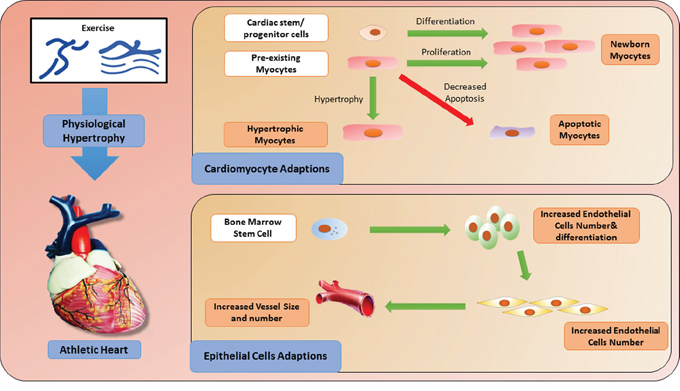
Figure 1: Cellular adaptations induced by exercise.
Exercise-induced cardiac growth: hypertrophy and renewal of cardiomyocytes
Physical exercise-induced cardiac growth comprises a range of changes in heart tissue including cardiac hypertrophy and renewal. Among these, the main morphological and structural adaptation in the myocardium is cardiac hypertrophy, which results in an increase in wall thickness and a growth in the size of cardiomyocytes [12]. Cardiac physiological hypertrophy in response to exercise training differs from pathological hypertrophy in its stimuli, structure and molecular profile [13]. Physiological hypertrophy occurs mainly as a result of regular physical activity or exercise training; in contrast, cardiac pathological hypertrophy occurs in response to a series of stimuli, such as myocardial infarction, valve disease and dilated cardiomyopathy. Physiological hypertrophy is characterized by normal organization of cardiac structure and normal or even elevated cardiac function, and it has a protective effect against CVD. On the other hand, pathological hypertrophy is associated with cardiac dysfuction, myocardial structural disorder, cardiac fibrosis and upregulation of fetal genes, including atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and skeletal α-actin and β-myosin heavy chain (MHC) [14].
Over several decades, the concept that the heart is a post-mitotic organ without any regenerative capacity had taken hold [15]. However, more recent evidence has shown that young hearts have robust growth and regenerative capacity supported by cardiomyocyte division and cardiac stem and progenitor cell activation. In fact, nearly half of the cardiomyocytes are replaced during a normal human lifespan [16]. Human cardiomyocytes retain some proliferative capacity through adulthood, which can be enhanced through physical exercise. For example, it has been reported that endurance swim training in mice induced cardiomyocyte hypertrophy and proliferation through decreased expression of the transcription factor C/EBPβ and increased expression of ED-rich carboxy-terminal domain 4 (CITED4) [17]. This exercise-induced cardiac renewal protects against pathological cardiac remodeling [17, 18].
In addition to cardiomyocyte division, the newly formed exercise-induced cardiomyocytes might also arise from cardiac stem cells (CSCs) resident in the adult heart [19–22]. CSCs, derived from embryonic stem cells or somatic stem cells [23], are self-renewing, clonogenic and multipotent. They can give rise to cardiomyocytes, smooth muscle cells and endothelial cells [24, 25]. It has been described that physical exercise could activate CSCs, thus giving rise to mature cardiomyocytes and improving the function and regeneration of the cardiovascular system [26]. Interestingly, studies have also demonstrated that the growth of smooth muscle cells and endothelial cells may be regulated by differentiated CSCs in a manner associated with exercise-induced cardiac repair [27].
Apoptosis is a specific form of programmed cell death that plays an important role in development, growth and disease [28]. It has been shown that exercise-induced cardiac growth does not result in cardiac apoptosis [29, 30], and will not induce a reactivation of cardiac fetal genes. Importantly, exercise-induced adaptive changes via cardiac hypertrophy and renewal have been demonstrated to be protective in pathological conditions such as ischemic injury. The protective effect is characterized by reduced cardiac apoptosis and decreased reactivation of cardiac fetal genes [31, 32].
Exercise-induced effects on endothelial progenitor cells
Endothelial progenitor cells (EPCs), a type of circulating progenitor cell derived from bone marrow stem cells (BMSCs) [33, 34], can be activated in response to physical exercise [35]. The importance of EPCs in the context of CVD has been well documented, with the number and function of EPCs being correlated with cardiovascular risk factors [36] and the number of circulating EPCs serving as a predictor of cardiovascular events and death [37]. In addition, during cardiac injury, especially AMI, BMSCs can trans-differentiate into vascular endothelial cells and cardiomyocytes to implant into damaged mycardium and improve cardiac function [38]. Exercise can induce EPCs to proliferate, migrate and differentiate into mature endothelial cells [35] which promote endothelial regeneration and angiogenesis. Thus, the increased concentration of EPCs in response to physical exercise may serve as a physical repair or compensatory mechanism in the setting of cardiac injury.
Overall, cellular adaptations induced by exercise can be attributed to both endogenous and exogenous factors. Physical training switches on the endogenous system and promotes cardiomyocyte self-renewal and cellular turnover. This is of potential importance as it serves as the basis for regeneration of an injured heart [39]. Nevertheless, it is still unknown to what extent the CSCs and/or the proliferation of resident cardiomyocytes contribute to the generation of new cardiomyocytes of the adult mammalian heart [40]. Meanwhile, exercise also promotes an exogenous system, favoring not only the repair of subclinical lesions but also mobilizing exogenous cells against harmful conditions [41–43]. Among exogenous cell types, endothelial cells are the most important ones. For example, exercise reduces the incidence of atherosclerotic heart disease and may promote the formation of vascular branches [44]. Interestingly, since cardiomyocytes are regulated by physical exercise, they can also affect other cell types, such as endothelial cells and smooth muscle cells [45]. Cardiomyocytes and other cell types may be complementary in the context of exercise to improve cardiac function and repair.
EXERCISE PROTECTS AGAINST CARDIOVASCULAR DISEASES (TABLE 1)
Table 1: Exercise protects against cardiovascular diseases
Types of disease |
Effects of exercise |
References |
Myocardial Infarction |
1. Decrease inflammation |
|
Ischemia/Reperfusion Injury |
1. Up-regulate anti-oxidative defense |
|
Diabetic Cardiomyopathy |
1. Enhance mitochondrial biogenesis |
|
Cardiac aging |
1. Decrease cardiac apoptosis |
|
Atherosclerosis |
1. Promote vascular branch formation |
|
Pulmonary hypertension |
1. Increase aerobic capacity |
Exercise protects against myocardial infarction
Acute myocardial infarction (AMI) is a prevailing cause of death worldwide. Cardiac remodeling after AMI refers to changes in the size, shape, structure and composition of the heart [46]. Such injuries result in loss of cardiomyocytes by programmed cell death and are accompanied by compensatory induction of hypertrophic growth and fibrosis. Recent large-scale studies have provided support for the notion that exercise protects against AMI remodeling both in patients and animal models [47–49].
Inflammation plays an important role in AMI-associated cardiac remodeling, and exercise is an effective means to modulate inflammation-associated cytokine activities. Exercise has been shown to reduce pro-inflammatory cytokines levels including C-reactive protein (CRP), IL-1, IL-6 and INF-γ, and increase anti-inflammatory cytokines levels such as IL-10. This blunts inflammation and improves the coronary risk profiles [48, 50]. Besides its effect on inflammation, exercise is also effective in reducing cardiac apoptosis by regulating the mitochondrial-mediated apoptotic pathway, which is assessed via lower terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive staining, caspase-3 cleavage and a decreased ratio of Bax (pro-apoptotic)/Bcl-2(anti-apoptotic) protein levels [51]. Additionally, exercise can also reduce myocardial infarct size and fibrosis by down-regulating the fibrosis-related factor TGF-β [49]. Other potential mechanisms underlying the protective effect of exercise against AMI include increased regeneration of cardiomyocytes, metabolic regulation and decreased risk of re-infarction.
Exercise protects against cardiac ischemia/reperfusion injury
Cardiac ischemia/reperfusion (I/R) Injury is defined as myocardial damage when oxygenated blood returns to the heart after a period of ischemia or lack of oxygen [52]. The absence of oxygen or nutrients from blood creates a condition in which the restoration of circulation results in apoptosis and oxidative damage through the induction of oxidative stress [52]. Previous studies have demonstrated that exercise training can protect against I/R injury in clinical patients and SD rats [53].
Oxidative stress is an important component participating in I/R injury, which is characterized by accumulation of reactive species and damage in the heart and other organs such as the lung and brain. Studies have demonstrated that regular exercise leads to up-regulation of anti-oxidative defense mechanisms, which help minimize oxidative stress following I/R [54]. For example, during five weeks of five weekly exercise sessions, an increased antioxidant capacity has been observed in the heart. The prevention of excessive nitric oxide synthesis limited its binding to O2 and consequent formation of peroxynitrite [55]. Exercise also induces EPCs to promote angiogenesis. The growth of new blood vessels is an important natural process required for healing wounds and for restoring blood flow to tissues after I/R injury [56]. Finally, during the process of I/R, there is significant cardiac dysfunction and myocardial apoptosis [57]. Exercise results in decreased apoptosis and improved cardiac function by increasing the expression of AKT, AKT phosphorylation and glycogen synthase kinase (GSK)-3β phosphorylation. Treatment with a PI3K kinase inhibitor abolished the beneficial effects of exercise, providing mechanistic insights into the effects of exercise on cardiac apoptosis and function [53].
Exercise protects against diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM) is a disorder of the heart muscle that occurs in the setting of type I or type II diabetes. DCM can compromise the ability of the heart to effectively pump and circulate blood throughout the body and can lead to fluid accumulation in the lungs or legs [58]. DCM is a major cause of morbidity and mortality among diabetics, encompassing structural, morphological, functional, and metabolic abnormalities in the heart. Recently, it has been shown that exercise can protect against DCM, especially in type II diabetes [59].
Mitochondrial dysfunction is one of the major mechanisms underlying DCM. DCM associated-downregulation of PGC-1α and its downstream components leads to decreased mitochondrial biogenesis [59]. Increasing evidence has shown that exercise can increase the number and size of mitochondria by upregulating PGC-1α and nuclear respiratory factor 1 (NRF-1) signaling pathways. Improved mitochondrial biogenesis can also increase the efficient oxidation of carbohydrates, thus improving glucose tolerance and insulin sensitivity [60]. Notably, exercise has been shown to improve cardiac mitochondrial biogenesis in DCM due to activation of PGC-1α, providing compelling evidence of exercise-associated protection for cardiac metabolism [59].
Exercise protects against cardiac aging
Progressive aging induces several structural and functional disorders in the cardiovascular system characterized by increased myocyte size, reduced number of cardiomyocytes and increased interstitial collagen fibers [61]. A growing literature suggests that exercise has a positive effect on cardiovascular performance in both elderly patients and animal models of aging [62, 63].
Aging-related cardiomyocytes loss is mediated by apoptosis and necrosis. The mitochondrial-mediated apoptotic pathway in particular is the best characterized and believed to play a critical role in regulating apoptosis due to aging [62]. As discussed earlier, exercise can decrease the level of apoptosis in cardiac and skeletal muscles of young adult rats by changing the expression of apoptosis regulatory factors such as Bcl-2 and apoptotic protease activating factor-1 (apaf-1) [64]. Physical exercise training may also increase the antioxidant defense and decrease oxidative stress in the elderly. Furthermore, exercise protects against aging-associated cardiac fibrosis by reducing fiber production via downregulated TGF-β1, which is a potential contributor to fibrosis, in addition to increasing collagen degradation via increased MMP (MMP-1, MMP-2, MMP-3, and MMP-4) and decreased TIMP expression [65].
Exercise protects against pulmonary hypertension
Pulmonary hypertrophy (PH) is a disease with increased blood pressure in the pulmonary arteries, pulmonary veins or capillaries. PH can cause a number of symptoms including shortness of breath, dizziness, fainting and leg swelling [66]. More recently, a body of literature has shown the safety and efficacy of exercise training in PH.
In clinical studies, exercise training has been identified to confer numerous clinical benefits in patients with PH, including increasing aerobic capacity, rasing muscle strength, enhancing exercise tolerance and ultimately improving quality of life [67]. Exercise also plays a key role in a subtype of PH, pulmonary artery hypertension (PAH). In rats, PAH or right ventricular failure can be induced by a single injection of monocrotaline (MCT). Voluntary exercise by running has been demonstrated to retain exercise capacity and delay the onset of heart failure, thereby improving the survival rate in rats with PAH [68, 69]. However, even though the positive phenotype of exercise on PH or PAH is obvious, the underlying mechanism needs to be studied in further detail.
Molecular mechanisms underlying exercise-induced beneficial effects in the heart
Exercise-induced cardiac protection has been appreciated for many decades, and several molecular mechanisms have been proposed to mediate exercise-associated cardioprotective phenotypes (Figure 2).
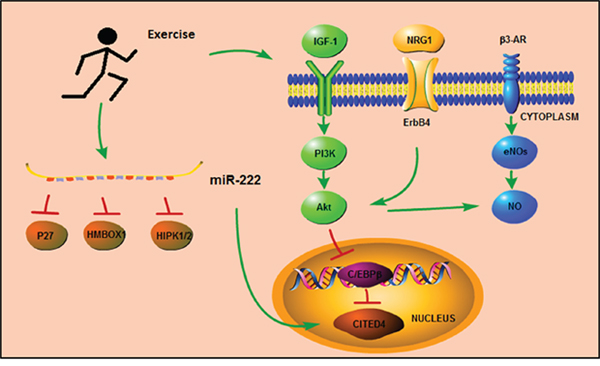
Figure 2: Signaling pathways mediates in exercise training.
Signaling pathways underlying protective effects of exercise
Growth factor neuregulin1 (NRG1)-ErbB4-C/EBPβ is one of the main pathways implicated in mediating the changes in cardiomyocytes induced by exercise [70, 71]. The effect of exercise on cardiomyocyte hypertrophy and proliferation involves reduction in the expression of the transcription factor C/EBPβ, and a linked increase in the expression of CITED4 [17]. The reduction of C/EBPβ expression level results in upregulation of hypertrophy-related genes, such as Gata4, Tbx5, Nkx2.5, α-MHC, TnI, and TnT [17]. Of note, Gata4 was recently reported to regulate cell proliferation during cardiac regeneration [72]. Moreover, C/EBPβ is also an important functional target of NRG1-related signaling pathways. Injection of NRG1 in adult mice induces cardiomyocyte proliferation and promotes myocardial regeneration, leading to improved function following myocardial infarction [73].
The insulin-like growth factor (IGF)-1-PI3k-Akt signaling pathway also plays an important role in the protective effects conferred by regular physical exercise [74, 75]. Exercise increases cardiac expression of IGF-1, and treatment with IGF-1 up-regulates myocardial telomerase activity and increases expression of phosphorylated Akt protein kinase, thereby regulating cardiac hypertrophy, viability and homeostasis.
AMP-activated protein kinase (AMPK) is an enzyme participating in cellular energy homeostasis. Recently, exercise was identified to be one of the factors upregulating AMPK, and the activation of AMPK is also responsible for most of the benefical effects resulting from physical exercise [76]. One study demonstrated that swimming training decreased isoproterenol (ISO)-induced cardiac fibrosis by inhibiting the ROS-NADPH oxidase pathway mediated by AMPK activation [77]. Another study also reported that activated AMPK induced by endurance exercise conferred positive effects against type 2 diabetes by increasing fatty acid and glucose metabolism, mitochondrial content and insulin sensitivity. Interestingly, the benefical effects of exercise may also be related to PGC-1α, another key factor which is also increased in exercise training [78].
Endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) seem to be closely related to the positive effects of exercise on endothelial cells [79, 80]. Interestingly, exercise alters the expression and phosphorylation status of eNOS in a tissue-specific manner. In the heart, exercise increases the expression of eNOS-PSer1177 and decreases the expression of eNOS-PThr495 without changing the expression of total eNOS, whereas in the skeletal muscle, exercise increases the expression of total eNOS and decreases the expression of eNOS-PThr495 [81]. Recent studies also reported that the activation of vascular eNOS during exercise could be caused by several signaling pathways involving Akt (protein kinase B), protein kinase A (PKA) and/or AMPK, and inhibition of Akt signaling decreases the expression of eNOS-PSer1177 in mice subjected to endurance exercise in comparison to vehicle-treated exercised mice, but without changing the expression of phosphorylated CREB (PKA signaling) or AMPK [82]. Previous studies demonstrated that β3-adrenergic receptors (β3-AR) played a major role in regulating the phosphorylation of eNOS at serine 1177 as well as in maintaining the basal expression of myocardial eNOS during exercise [81, 83]. The relationship between β3-AR, eNOS, and Akt during exercise may yield promising insights into therapeutic targets against myocardial injury.
MicroRNAs responsible for cardiac protection effects of exercise (Table 2)
Table 2: microRNAs in response to exercise training
MicroRNAs |
Targets |
Function |
References |
miR-1, miR-133a, miR-133b (down) |
RhoA, Cdc42, NELFA |
1. Increase cell differentiation & growth |
|
miR-214 (down) |
SERCA2a |
1. Increase left ventricular myocyte mass |
|
miR-29a, miR-29c(up) |
COL1A1, COL3A |
1. Increase ventricular compliance |
|
miR-222 (up) |
P27, HMBOX1, HIPK1/2 |
Increase cardiac growth |
|
miR-126 (up) |
MAPK PI3K/Akt/eNOS |
Increase cardiac angiogenesis |
MicroRNAs (miRNAs, miRs) are highly-conserved, non-protein coding RNAs which can inhibit target gene expressions and functions by binding to the 3’ untranslated region (3’ UTR) of messenger RNA (mRNA) and thereby repressing their translation and/or promoting their degradation [84]. miRNAs regulate cardiac development, hypertrophy, and angiogenesis [85–87]. miR-1, -133a, and -133b are the well-known miRNAs explored in heart [88, 89]; these three families are down-regulated in three cardiac models of hypertrophy including transverse aortic constriction (TAC), Akt transgenic mice and endurance exercise. In addition, during 12 weeks of physical exercise, miR-1, -133a, and -133b were shown to decrease cell differentiation and growth by inhibiting Ras homolog gene family-A (RhoA), cell division control protein 42 (Cdc42) and wolf-Hirschhorn syndrome candidate (NELFA) [90]. These data indicate that miR-1, -133a, and -133b have a close relationship with cardiac hypertrophy both in physiological and pathological contexts, with further investigation required to document the function of these three miRNA families in cardiac hypertrophy. Another miRNA, miR-214, is also decreased upon resistance training, leading to increased left ventricular myocyte width and volume and faster cell contraction in rats during eight weeks of training. SERCA2a, a target gene of miR-214, was increased in the exercise group [91].
Recent reports document that physical training induces up-regulation of the expression of miR-29a and -29c in the heart, which are associated with a significant decrease in left ventricu1ar collagen (COLIAI and COLIIIAI) gene and protein levels [92]. The up-regulation of the miR-29 family is also accompanied by improvement of ventricular compliance and cardiac function [93].
More recently, miR-222 has been reported to be an important mediator of exercise-induced cardiac growth by inhibiting Cyclin-dependent kinase inhibitor 1B (p27), Homeobox containing 1 (HMBOX1) and Homeodomain-interacting protein kinase 1/2 (HIPK1/2). In ischemic injury models, miR-222 protects against adverse cardiac remodeling and cardiac dysfunction [94]. Interestingly, miR-222 has also been linked to CITED4; the up-regulation of miR-222 in swimming mice is associated with increased expression of CITED4, indicating cross-interaction between miR-222 and the C/EBPβ/CITED4 signaling pathway [95].
The expression of miR-126 has also been shown to be increased in aerobic training in rats during ten weeks of swimming, leading to cardiac angiogenesis via targeting MAPK and PI3K/Akt/eNOS [96]. Similarly, miR-150 has been reported to be upregulated while miR-26b and -143 downregulated in the heart upon exercise [97]. However, the specific relationship and mechanisms underlying changes in miRNA levels upon physical exercise still need to be studied in further detail.
In addition to local miRNAs, plasma-based “circulating” miRNAs (c-miRNAs) are significantly changed in the setting of regular exercise in humans, including miR-106a, -30b, -146, -338 and -21 [96, 98, 99]. c-miRNAs may become potent biomarkers of physiological exercise and provide novel insight into the physical exercise process.
CONCLUSION
In this review, we summarize the protective effects of exercise against cardiac injury and address the underlying cellular and molecular mechanisms. Exercise increases myocardial oxygen supply and extraction, reduces myocardial fibrosis and cardiomyocyte apoptosis, promotes angiogenesis, and also improves cardiac function and cardiomyocyte survival in various CVD. For the cellular mechanisms, we provide an overview of both exercise-induced cardiac hypertrophy and renewal, and exercise-induced mobilization of EPCs to proliferate, migrate and differentiate into mature endothelial cells capable of regeneration and angiogenesis as primary contributors. For the molecular mechanisms, we summarize C/EBPβ-Cited4 and IGF-1-PI3k-Akt as the main signaling pathways mediating the changes in cardiomyocytes induced by exercise. Exercise also has established relationships with miRNAs, with recent reports documenting that exercise induced upregulation of miRNAs-29a and -29c correlates with a significant decrease in left ventricular collagen gene and protein concentration [92]. In addition, the expression of miR-222 was increased upon exercise to regulate myocyte growth via targeting of p27, HMBOX1, and HIPK1/2. Also, downregulation of miRNA-1, -133a, -133b and -214 were observed in the exercised heart [92]. Thus, the protective effects of exercise against myocardial damage are clearly pleiotropic in their mechanisms.
Although evidence of the protective effects of physical exercise against cardiac injury is strong, certain caveats need to be noted. First, the optimal level of exercise for preventing CVD is unclear. In some studies, the reduction in risk from increased levels of exercise activity appeared to be linear up to a certain level, above which there was no further benefit; in others, the effect was restricted to the highest categories of total energy expenditure [100]. The response of the heart is not dependent on the type of exercise applied, but rather on the duration and intensity at which the exercise is performed [101]. Moderate versus vigorous exercise may have completely different effects following myocardial damage. There is an urgent need to determine to what extent exercise can ameliorate disease processes and reduce morbidity and mortality. Likewise, the origin of cardiomyocyte regeneration induced by exercise is still elusive.
In summary, exercise is a potent functional intervention to protect against CVD. Future studies are needed to explore the specific strength and intensity of exercise required to protect against cardiac injuries, to investigate which cell types specifically contribute to exercise-induced cardiac adaptations, and to define the complex interplay between these cell types.
FINANCIAL SUPPORT
This work was supported by the grants from National Natural Science Foundation of China (81370332 and 81170201 to XL Li, 81200169 to JJ Xiao), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD to XL Li), Innovation Program of Shanghai Municipal Education Commission (13YZ014 to JJ Xiao), Innovation fund from Shanghai University (sdcx2012038 to JJ Xiao), and Program for the integration of production, teaching and research for University Teachers supported by Shanghai Municipal Education Commission (to JJ Xiao). Dr XL Li is an Associate Fellow at the Collaborative Innovation Center For Cardiovascular Disease Translational Medicine.
CONFLICTS OF INTEREST
The authors declare there are no conflicts of interest.
REFERENCES
1. Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: A review of global methodologies of mortality measurement. Circulation. 2013; 127:749–756.
2. Chen M, Davignon J, Baass A. Diagnosis and cardiovascular risk factors in dysbetalipoproteinemia: A large cohort study. J Clin Lipidol. 2014; 8:302–303.
3. Eckel RH, Jakicic JM, Ard JD, Miller NH, Hubbard VS, Nonas CA, de Jesus JM, Sacks FM, Lee I-M, Smith SC. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular riska report of the american college of cardiology/american heart association task force on practice guidelines. J Am Coll Cardiol. 2013.
4. Figueiredo PA, Appell Coriolano HJ, Duarte JA. Cardiac regeneration and cellular therapy: Is there a benefit of exercise? Int J Sports Med. 2013; 35:181–90.
5. Arem H, Moore SC, Patel A, Hartge P, Berrington de Gonzalez A, Visvanathan K, Campbell PT, Freedman M, Weiderpass E, Adami HO, Linet MS, Lee IM, Matthews CE. Leisure time physical activity and mortality: A detailed pooled analysis of the dose-response relationship. JAMA Intern Med. 2015; 175:959–67.
6. Bleumink GS, Knetsch AM, Sturkenboom MC, Straus SM, Hofman A, Deckers JW, Witteman JC, Stricker BH. Quantifying the heart failure epidemic: Prevalence, incidence rate, lifetime risk and prognosis of heart failure the rotterdam study. Eur Heart J. 2004; 25:1614–1619.
7. Gielen S, Laughlin MH, O’Conner C, Duncker DJ. Exercise training in patients with heart disease: Review of beneficial effects and clinical recommendations. Prog Cardiovasc Dis. 2015; 57:347–355.
8. Lennon SL, Quindry J, Hamilton KL, French J, Staib J, Mehta JL, Powers SK. Loss of exercise-induced cardioprotection after cessation of exercise. J Appl Physiol (1985). 2004; 96:1299–1305.
9. Hambrecht R, Fiehn E, Weigl C, Gielen S, Hamann C, Kaiser R, Yu J, Adams V, Niebauer J, Schuler G. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation. 1998; 98:2709–2715.
10. Thompson PD, Buchner D, Pina IL, Balady GJ, Williams MA, Marcus BH, Berra K, Blair SN, Costa F, Franklin B, Fletcher GF, Gordon NF, Pate RR, Rodriguez BL, Yancey AK, Wenger NK. American Heart Association Council on Clinical Cardiology Subcommittee on Exercise R, Prevention, American Heart Association Council on Nutrition PA, Metabolism Subcommittee on Physical A.: Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease: A statement from the council on clinical cardiology (subcommittee on exercise, rehabilitation, and prevention) and the council on nutrition, physical activity, and metabolism (subcommittee on physical activity). Circulation. 2003; 107:3109–3116.
11. Tanaka H, Monahan KD, Seals DR. Age-predicted maximal heart rate revisited. J Am Coll Cardiol. 2001; 37:153–156.
12. Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, Abel ED. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. 2014; 34:3450–3460.
13. McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007; 34:255–262.
14. Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. Pathological cardiac hypertrophy. Cardiovasc Res. 2011; 90:234–242.
15. Mohl W, Gangl C, Jusic A, Aschacher T, De Jonge M, Rattay F. Picso: From myocardial salvage to tissue regeneration. Cardiovasc Revasc Med. 2015; 16:36–46.
16. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009; 324:98–102.
17. Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. C/EBP beta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010; 143:1072–1083.
18. Kubin T, Pöling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lörchner H, Schimanski S. Oncostatin m is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell stem cell. 2011; 9:420–432.
19. Chamuleau SA, Vrijsen KR, Rokosh DG, Tang XL, Piek JJ, Bolli R. Cell therapy for ischaemic heart disease: Focus on the role of resident cardiac stem cells. Neth Heart J. 2009; 17:199–207.
20. Windmolders S, De Boeck A, Koninckx R, Daniels A, De Wever O, Bracke M, Hendrikx M, Hensen K, Rummens JL. Mesenchymal stem cell secreted platelet derived growth factor exerts a pro-migratory effect on resident cardiac atrial appendage stem cells. J Mol Cell Cardiol. 2014; 66:177–188.
21. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114:763–776.
22. Van Vliet P, Roccio M, Smits A, Van Oorschot A, Metz C, van Veen T, Sluijter J, Doevendans P, Goumans M. Progenitor cells isolated from the human heart: A potential cell source for regenerative therapy. Neth Heart J. 2008; 16:163–169.
23. Kocher AA, Schlechta B, Gasparovicova A, Wolner E, Bonaros N, Laufer G. Stem cells and cardiac regeneration. Transpl Int. 2007; 20:731–746.
24. Rani KGA, Jayakumar K, Srinivas G, Nair RR, Kartha CC. Isolation of ckit-positive cardiosphere-forming cells from human atrial biopsy. Asian Cardiovasc Thorac Ann. 2008; 16:50–56.
25. Van Vliet P, Smits AM, De Boer TP, Korfage TH, Metz CH, Roccio M, Van Der Heyden MA, Van Veen TA, Sluijter JP, Doevendans PA. Foetal and adult cardiomyocyte progenitor cells have different developmental potential. J Cell Mol Med. 2010; 14:861–870.
26. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM. Bone marrow cells regenerate infarcted myocardium. Nature. 2001; 410:701–705.
27. Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells a paradigm shift in cardiac biology. Circulation. 2006; 113:1451–1463.
28. Goussetis E, Spiropoulos A, Tsironi M, Skenderi K, Margeli A, Graphakos S, Baltopoulos P, Papassotiriou I. Spartathlon, a 246 kilometer foot race: Effects of acute inflammation induced by prolonged exercise on circulating progenitor reparative cells. Blood Cells Mol Dis. 2009; 42:294–299.
29. Jin H, Yang R, Li W, Lu H, Ryan AM, Ogasawara AK, Van Peborgh J, Paoni NF. Effects of exercise training on cardiac function, gene expression, and apoptosis in rats. Am J Physiol Heart Circ Physiol. 2000; 279:H2994–H3002.
30. Kang PM, Yue P, Liu Z, Tarnavski O, Bodyak N, Izumo S. Alterations in apoptosis regulatory factors during hypertrophy and heart failure. Am J Physiol Heart Circ Physiol. 2004; 287:H72–H80.
31. Xu T, Tang H, Zhang B, Cai C, Liu X, Han Q, Zou L. Exercise preconditioning attenuates pressure overload-induced pathological cardiac hypertrophy. Int J Clin Exp Pathol. 2015; 8:530–540.
32. Tanno AP, das Neves VJ, Rosa KT, Cunha TS, Giordano FC, Calil CM, Guzzoni V, Fernandes T, de Oliveira EM, Novaes PD, Irigoyen MC, Moura MJ, Marcondes FK. Nandrolone and resistance training induce heart remodeling: Role of fetal genes and implications for cardiac pathophysiology. Life Sci. 2011; 89:631–637.
33. Leone AM, Valgimigli M, Giannico MB, Zaccone V, Perfetti M, D’Amario D, Rebuzzi AG, Crea F. From bone marrow to the arterial wall: The ongoing tale of endothelial progenitor cells. Eur Heart J. 2009; 30:890–899.
34. Möbius-Winkler S, Höllriegel R, Schuler G, Adams V. Endothelial progenitor cells: Implications for cardiovascular disease. Cytometry A. 2009; 75:25–37.
35. Black MA, Cable NT, Thijssen DH, Green DJ. Impact of age, sex, and exercise on brachial artery flow-mediated dilatation. Am J Physiol Heart Circ Physiol. 2009; 297:H1109–H1116.
36. Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001; 89:e1–e7.
37. Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005; 353:999–1007.
38. Henning RJ. Stem cells in cardiac repair. Future Cardiol. 2011; 7:99–117.
39. Golbidi S, Laher I. Molecular mechanisms in exercise-induced cardioprotection. Cardiol Res Pract. 2011; 2011.
40. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011; 331:1078–1080.
41. Wonders KY, Hydock DS, Greufe S, Schneider CM, Hayward R. Endurance exercise training preserves cardiac function in rats receiving doxorubicin and the her-2 inhibitor gw2974. Cancer Chemother Pharmacol. 2009; 64:1105–1113.
42. Powers SK, Locke M, Demirel HA. Exercise, heat shock proteins, and myocardial protection from ischemia reperfusion injury. Med Sci Sports Exerc. 2001; 33:386–392.
43. Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med. 2008; 44:193–201.
44. Sniehotta FF, Scholz U, Schwarzer R. Bridging the intention-behaviour gap: Planning, self-efficacy, and action control in the adoption and maintenance of physical exercise. Pharmacol Ther. 2005; 20:143–160.
45. Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res. 1994; 74:349–353.
46. Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following st-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014; 63:1593–1603.
47. Kim C, Choi HE, Lim MH. Effect of high interval training in acute myocardial infarction patients with drug-eluting stent. Am J Phys Med Rehabil. 2015.
48. Goldhammer E, Tanchilevitch A, Maor I, Beniamini Y, Rosenschein U, Sagiv M. Exercise training modulates cytokines activity in coronary heart disease patients. Int J Cardiol. 2005; 100:93–99.
49. McElroy CL, Gissen SA, Fishbein MC. Exercise-induced reduction in myocardial infarct size after coronary artery occlusion in the rat. Circulation. 1978; 57:958–962.
50. Kjolhede T, Dalgas U, Gade AB, Bjerre M, Stenager E, Petersen T, Vissing K. Acute and chronic cytokine responses to resistance exercise and training in people with multiple sclerosis. Scand J Med Sci Sports. 2015.
51. Rodrigues B, Lira FS, Consolim-Colombo FM, Rocha JA, Caperuto EC, De Angelis K, Irigoyen MC. Role of exercise training on autonomic changes and inflammatory profile induced by myocardial infarction. Mediators Inflamm. 2014; 2014:702473.
52. Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000; 190:255–266.
53. Zhang KR, Liu HT, Zhang HF, Zhang QJ, Li QX, Yu QJ, Guo WY, Wang HC, Gao F. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and akt-mediated mechanism. Apoptosis. 2007; 12:1579–1588.
54. Gomes EC, Silva AN, de Oliveira MR. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid Med Cell Longev. 2012; 2012:756132.
55. Otani H. The role of nitric oxide in myocardial repair and remodeling. Antioxid Redox Signal. 2009; 11:1913–1928.
56. Li J, Zhang H, Zhang C. Role of inflammation in the regulation of coronary blood flow in ischemia and reperfusion: Mechanisms and therapeutic implications. J Mol Cell Cardiol. 2012; 52:865–872.
57. Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg. 2005; 18:335–350.
58. Chen L, Pei JH, Kuang J, Chen HM, Chen Z, Li ZW, Yang HZ. Effect of lifestyle intervention in patients with type 2 diabetes: A meta-analysis. Metabolism. 2015; 64:338–347.
59. Wang H, Bei Y, Lu Y, Sun W, Liu Q, Wang Y, Cao Y, Chen P, Xiao J, Kong X. Exercise prevents cardiac injury and improves mitochondrial biogenesis in advanced diabetic cardiomyopathy with pgc-1alpha and akt activation. Cell Physiol Biochem. 2015; 35:2159–2168.
60. Le Douairon Lahaye S, Bekono FR, Broderick T. Physical activity and diabetic cardiomyopathy: Myocardial adaptation depending on exercise load. Curr Diabetes Rev. 2014; 10:371–390.
61. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015; 6:109–120.
62. Kwak HB. Effects of aging and exercise training on apoptosis in the heart. J Exerc Rehabil. 2013; 9:212–219.
63. Bouzid MA, Filaire E, McCall A, Fabre C. Radical oxygen species, exercise and aging: An update. Sports Med. 2015.
64. Siu PM, Bryner RW, Martyn JK, Alway SE. Apoptotic adaptations from exercise training in skeletal and cardiac muscles. FASEB J. 2004; 18:1150–1152.
65. Kwak HB. Aging, exercise, and extracellular matrix in the heart. J Exerc Rehabil. 2013; 9:338–347.
66. Vaidya B, Gupta V. Novel therapeutic approaches for pulmonary arterial hypertension: Unique molecular targets to site-specific drug delivery. J Control Release. 2015; 211:118–133.
67. Arena R, Cahalin LP, Borghi-Silva A, Myers J. The effect of exercise training on the pulmonary arterial system in patients with pulmonary hypertension. Prog Cardiovasc Dis. 2015; 57:480–488.
68. Natali AJ, Fowler ED, Calaghan S, White E. Voluntary exercise delays heart failure onset in rats with pulmonary artery hypertension. Am J Physiol Heart Circ Physiol. 2015; 309:H421–4.
69. Hargett LA, Hartman LJ, Scruggs AK, McLendon JM, Haven AK, Bauer NN. Severe pulmonary arterial hypertensive rats are tolerant to mild exercise. Pulm Circ. 2015; 5:349–355.
70. Marini M, Lapalombella R, Margonato V, Ronchi R, Samaja M, Scapin C, Gorza L, Maraldi T, Carinci P, Ventura C, Veicsteinas A. Mild exercise training, cardioprotection and stress genes profile. Eur J Appl Physiol. 2007; 99:503–510.
71. Nizielski SE, Arizmendi C, Shteyngarts AR, Farrell CJ, Friedman JE. Involvement of transcription factor c/ebp-beta in stimulation of pepck gene expression during exercise. Am J Physiol. 1996; 270:R1005–1012.
72. Kikuchi K, Niikura Y, Kitagawa K, Kikuchi A. Dishevelled, a wnt signalling component, is involved in mitotic progression in cooperation with plk1. EMBO J. 2010; 29:3470–3483.
73. Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/erbb4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009; 138:257–270.
74. Weeks KL, McMullen JR. The athlete’s heart vs. The failing heart: Can signaling explain the two distinct outcomes? Physiology. 2011; 26:97–105.
75. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol Ther. 2010; 128:191–227.
76. Grochowska E, Jarzyna R. Physical activity in the prevention and treatment of diseases of affluence—the key role of amp-activated protein kinase (ampk). Postepy Hig Med Dosw. 2014; 68:1114–1128.
77. Ma X, Fu Y, Xiao H, Song Y, Chen R, Shen J, An X, Shen Q, Li Z, Zhang Y. Cardiac fibrosis alleviated by exercise training is ampk-dependent. PLoS One. 2015; 10:e0129971.
78. Marcinko K, Steinberg GR. The role of ampk in controlling metabolism and mitochondrial biogenesis during exercise. Exp Physiol. 2014; 99:1581–1585.
79. Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, Maloney RE, Bharti A, Rodriguez J, Feelisch M. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005; 1:290–297.
80. Brown MB, Chingombe TJ, Zinn AB, Reddy JG, Novack RA, Cooney SA, Fisher AJ, Presson RG, Lahm T, Petrache I. Novel assessment of hemodynamic kinetics with acute exercise in a rat model of pulmonary arterial hypertension. Exp Physiol. 2015; 100:742–54.
81. Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL, Sindler AL, Gundewar S, Seals DR, Barouch LA, Lefer DJ. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: Role of nitrite and nitrosothiols. Circ Res. 2011; 108:1448–1458.
82. Zhang Q-J, McMillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD. Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: Role of vascular signalling kinases. J Physiol. 2009; 587:3911–3920.
83. Kou R, Michel T. Epinephrine regulation of the endothelial nitric-oxide synthase roles of rac1 and β3-adrenergic receptors in endothelial no signaling. J Biol Chem. 2007; 282:32719–32729.
84. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microrna. Science. 2007; 316:575–579.
85. Latronico MV, Catalucci D, Condorelli G. Emerging role of micrornas in cardiovascular biology. Circ Res. 2007; 101:1225–1236.
86. Liu N, Olson EN. Microrna regulatory networks in cardiovascular development. Dev Cell. 2010; 18:510–525.
87. Wang S, Olson EN. Angiomirs—key regulators of angiogenesis. Curr Opin Genet Dev. 2009; 19:205–211.
88. Chen J-F, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang D-Z. The role of microrna-1 and microrna-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2005; 38:228–233.
89. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang M-L, Segnalini P, Gu Y, Dalton ND. Microrna-133 controls cardiac hypertrophy. Nat Med. 2007; 13:613–618.
90. Fernandes-Silva MM, Carvalho VO, Guimaraes GV, Bacal F, Bocchi EA. Physical exercise and micrornas: New frontiers in heart failure. Arq Bras Cardiol. 2012; 98:459–466.
91. Melo SF, Barauna VG, Junior MA, Bozi LH, Drummond LR, Natali AJ, de Oliveira EM. Resistance training regulates cardiac function through modulation of mirna-214. Int J Mol Sci. 2015; 16:6855–6867.
92. Soci UPR, Fernandes T, Hashimoto NY, Mota GF, Amadeu MA, Rosa KT, Irigoyen MC, Phillips MI, Oliveira EM. Micrornas 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiol Genomics. 2011; 43:665–673.
93. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. Mir-29b as a therapeutic agent for angiotensin ii-induced cardiac fibrosis by targeting TGF-beta/smad3 signaling. Mol Ther. 2014; 22:974–985.
94. Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F, Xiao C, Bezzerides V, Bostrom P, Che L, Zhang C, Spiegelman BM, Rosenzweig A. Mir-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015; 21:584–595.
95. Uchida S, Dimmeler S. Exercise controls non-coding rnas. Cell Metab. 2015; 21:511–512.
96. Baggish AL, Park J, Min PK, Isaacs S, Parker BA, Thompson PD, Troyanos C, D’Hemecourt P, Dyer S, Thiel M, Hale A, Chan SY. Rapid upregulation and clearance of distinct circulating micrornas after prolonged aerobic exercise. J Appl Physiol (1985). 2014; 116:522–531.
97. Martinelli NC, Cohen CR, Santos KG, Castro MA, Biolo A, Frick L, Silvello D, Lopes A, Schneider S, Andrades ME, Clausell N, Matte U, Rohde LE. An analysis of the global expression of micrornas in an experimental model of physiological left ventricular hypertrophy. PLoS One. 2014; 9:e93271.
98. Baggish AL, Hale A, Weiner RB, Lewis GD, Systrom D, Wang F, Wang TJ, Chan SY. Dynamic regulation of circulating microrna during acute exhaustive exercise and sustained aerobic exercise training. J Physiol. 2011; 589:3983–3994.
99. Sawada S, Kon M, Wada S, Ushida T, Suzuki K, Akimoto T. Profiling of circulating micrornas after a bout of acute resistance exercise in humans. PLoS One. 2013; 8:e70823.
100. Fletcher GF, Balady GJ, Amsterdam EA, Chaitman B, Eckel R, Fleg J, Froelicher VF, Leon AS, Piña IL, Rodney R. Exercise standards for testing and training a statement for healthcare professionals from the american heart association. Circulation. 2001; 104:1694–1740.
101. Leon AS, Franklin BA, Costa F, Balady GJ, Berra KA, Stewart KJ, Thompson PD, Williams MA, Lauer MS. Cardiac rehabilitation and secondary prevention of coronary heart disease an american heart association scientific statement from the council on clinical cardiology (subcommittee on exercise, cardiac rehabilitation, and prevention) and the council on nutrition, physical activity, and metabolism (subcommittee on physical activity), in collaboration with the american association of cardiovascular and pulmonary rehabilitation. Circulation. 2005; 111:369–376.