
Introduction
Prostate cancer (PCa) is the most common cancer type among men in Western countries and the second leading cause of cancer-related death in males [1]. In more than 80% of cases, PCa is diagnosed at the local stage and is often a low-risk disease with indolent clinical course and favorable survival [2]. The management of localized PCa is controversial as the identification of the subset of patients with aggressive, high-risk disease remains challenging. Considering histopathological features (i.e. Gleason grade, extent of the tumor), serum prostate specific antigen (PSA) level, clinical stage, age and co-morbidity of the patient, treatment of localized PCa includes active surveillance, radical prostatectomy or radiotherapy [2, 3].
Local recurrent PCa after failure of primary surgery/radiotherapy and hormone-naive metastatic disease are treated with androgen deprivation therapy (ADT) [4]. Luteinizing hormone-releasing hormone (LHRH) agonists or LHRH antagonists, eventually combined with anti-androgens as bicalutamide, are used to achieve hormonal depletion [4]. Initially, the reduction of circulating androgens decreases androgen receptor (AR)-mediated proliferation and survival of tumor cells. Therefore, ADT leads to remission of the tumor lasting for up to a few years and results in a decline of serum PSA [5]. However, after initial response to ADT, tumor cells ultimately become castration-resistant resulting in progression of the disease despite anorchid serum androgen levels [6]. Of all patients diagnosed of PCa, 10-20% develop castration-resistant disease, mostly within a few months to a few years after initiation of ADT [7].
The underlying molecular basis how PCa cells escape from the growth control by exogenous androgens is still poorly understood. Adaptive mechanisms of PCa cells include molecular alterations in response to androgen ablation leading to re-activation of the AR despite low circulating androgens after initial response to ADT [8]. Over the past years, several studies give evidence for selection advantages resulting in clonal outgrowth of initial androgen-independent cells [9]. This theory of castration-resistance is based on the observation that aggressive castration-resistant cells show distinct gene expression patterns and molecular properties, which are not present in parental PCa cells [10].
Mechanisms mediating castration-resistance comprise the re-activation of AR signaling despite low levels of circulating androgens on the one hand, as well as the activation of alternative AR-independent pathways on the other hand.
Maintained AR activity under androgen deprivation is based on genetic and functional aberrations affecting components of the AR signaling axis which have been observed in CRPC cells [11]. Genetic alterations such as AR gene amplification resulting in increased AR expression occur in about one third of CRPC tumors [11, 12], while AR mutation or alternative splice variants allowing the tumor to respond to very low androgen levels can be observed in a smaller subset of CRPC cases [11, 13]. Persistent transcriptional AR activity can also be mediated through alternative ligands as progesterone or by ligand-independent transcriptional activity [14]. Furthermore, the intratumoral conversion of androgen precursors as well as the de novo steroidogenesis provides persistent intraprostatic androgen concentrations sufficient to activate the AR despite low serum testosterone [15]. Additionally, alternative AR activation eventuates from alterations of coactivators and corepressors of the AR signaling as well as cross-activation through bypass pathways [16, 17]. Genetic alterations frequently found in CRPC contributing to dysregulated survival signaling involve c-myc amplification, PTEN loss, as well as alterations of genes implicated in the growth factor receptor signaling such as PI3K, Src kinase, Ras/MAPK [6, 8].
Admittedly, no treatment options with curative intent are available for castration-resistant prostate cancer (CRPC) to date [4]. Current palliative therapeutic strategies for CRPC comprise docetaxel as conventional chemotherapy, the immunostimulant sipuleucel-T and the inhibitor of androgen synthesis abiraterone acetate [4]. Cabazitaxel as chemotherapeutic agent, the AR antagonist enzalutamide and the radiopharmaceutical radium-223 are available as second line therapy after docetaxel treatment [4]. Additional options for patients with metastatic CRPC include the bone-targeting agents zoledronic acid and the receptor activator of nuclear factor kB ligand inhibitor denosumab [4]. Various pre-clinical approaches identified promising strategies to prevent rapid progression to castration-resistance [18]. Different classes of agents targeting components involved in survival pathways [19], DNA damage repair [20], angiogenesis [21], tumor microenvironment [22] or the immune system [4, 18] have reached phase III in clinical trials. Future studies will reveal whether these agents have the potential to significantly increase survival of patients with CRPC.
This review will highlight the current knowledge about adaptive mechanisms of the AR signaling as well as the significance of its interaction partners contributing to the development of castration-resistance.
Adaptive responses in AR signaling through AR mutants and AR variants
An adaptive response in AR signaling may also occur at the level of mutations and splice variants of the AR that occur under the selective pressure of ADT. Structurally, the AR is organized in 4 different domains: the N-terminal transactivation domain (TAD), a central DNA-binding domain (DBD), a hinge region (HR) which connects the DBD to the carboxy-terminal ligand binding domain (LBD) (Figure 1A). Upon androgen binding, the AR dissociates from heat shock proteins (HSP) and translocates to the nucleus where it dimerizes with another AR molecule. Subsequently, this AR dimer binds to chromatin and androgen response elements in the promoter regions of androgen-dependent genes, thereby activating/inhibiting their transcription. The implication of HSP in adaptation of AR signaling in PCa has recently been reviewed and suggests an increased expression of HSP70 and HSP27 that correlates with PCa aggressiveness and CRPC [23].
AR mutations are very rare in early stages of PCa. However, approximately 10-30% of CRPC patients carry AR mutations, especially when treated with ADT, indicating an adaptation to ADT by changing AR function [24]. In CRPC almost 50% of AR mutations cluster to 4 discrete regions of the AR LBD (Figure 1A). Somatic mutations in the AR LBD usually result in decreased receptor specificity, thereby broadening the number of steroids that can bind and activate the receptor. In addition, many of these mutated AR can be activated by anti-androgens. A prototype for this promiscuous gain of function mutants is AR-T877A. Initially identified in LNCaP cells, T877A (now T878A, according to the AR Mutations Database at http://androgendb.mcgill.ca, [25] was repeatedly found in flutamide-treated CRPC patients [26-28]. Functional studies demonstrated that T877A is strongly activated by the anti-androgens flutamide/hydroxyflutamide and by progesterone. Interestingly, the CYP17A1 inhibitor abiraterone was shown to increase intracellular progesterone levels, thereby allowing progesterone-inducible T877A to circumvent abiraterone-mediated inhibition of AR signaling in CRPC cells [29]. While T877A diminishes the efficacy of abiraterone, another interesting mutation, F876L, is able to convert AR antagonists like enzalutamide and ARN-509 to AR agonists [30, 31]. Most importantly, F876L still remained sensitive to the effects of bicalutamide [30].
Early functional in vitro studies showed a high constitutive transcriptional activity of AR constructs in which the LBD has been artificially deleted [32]. Due to the deletion of the functional LBD situated in the AR C-terminus, these AR variants are generally referred to as AR∆LBD. Blocking of the androgen/AR signaling axis was shown to induce a rapid increase of AR∆LBD in PCa cells [33]. So far, 17 AR∆LBD variants have been isolated from castration-resistant tumor cell lines/xenografts or clinical tumor specimens. Although AR∆LBD are predominantly products of alternative splicing (AR-V), they can also be products of nonsense mutations (AR-Q640X) or proteolytic cleavage (tr-AR) [34] (Figure 1B). Although all AR∆LBD lack a functional LBD, they can be subdivided into 2 structurally different subgroups, depending on the presence or absence of a HR (Figure 1B). Besides its function as a flexible linker between the DBD and the LBD, the HR carries a nuclear localization signal and a microtubule binding domain.
As AR∆LBD do not express a functional LBD, they are insensitive to all currently available hormonal therapies targeting directly (anti-androgens) or indirectly (inhibitors of androgen synthesis) the LBD [35, 36]. These in vitro observations could explain at least in part the well described cross-resistance between abiraterone and enzalutamide in the clinical setting [37, 38]. In addition, there is experimental evidence that different AR∆LBD variants are able to determine the sensitivity in PCa cells towards first generation taxanes like docetaxel or paclitaxel [39]. Both compounds were shown to impair nuclear localization of the AR via modulation of the microtubule AR network [39, 40]. As suggested by Thadani-Mulero, only AR forms expressing a microtubule binding domain situated in the HR of the receptor (e.g. wild type AR, ARv567es) are susceptible to taxane-mediated microtubule stabilization that is abrogating nuclear translocation and transcriptional activity of these receptors [40]. In contrast, HR-negative AR∆LBD like AR-V7 that do not associate with the microtubule machinery accumulate in the nucleus via a yet unknown mechanism thereby activating the transcriptional machinery [40]. However, the ability of taxanes to modulate AR/AR∆LBD-signaling in the clinical setting has been discussed with some controversy [41, 42]. Enzalutamide-resistant tumors often exhibit a cross-resistance with docetaxel. By contrast, cabazitaxel, a second generation taxane, remains highly effective in enzalutamide-resistant tumors, indicating that the inhibitory effects of docetaxel on AR/AR∆LBD-signaling represent only a minor part of its antitumor activity [41]. Moreover, the concentrations reported to affect AR and AR∆LBD translocation in vitro [40] are far beyond the effective taxane concentrations achieved in the clinical setting [41, 42]. As a result, a more thorough analysis of the mechanisms involved in regulation of AR/AR∆LBD-signaling by taxanes is needed.
The occurrence and detection of AR point mutations and AR∆LBD are of prognostic and therapeutic significance. In order to guide initial treatment selection or sequential therapies, there is an urgent need for new markers. Serum biomarker studies may be difficult to compare because of different procedures in various laboratories. Current research is focusing on the non-invasive retrieval of tumor DNA from blood/serum samples, i.e. circulating tumor DNA (ctDNA) or DNA from circulating tumor cells (CTC) to analyze aberrant AR variants. AR mutants like F876L or T877A have already been successfully isolated from blood and CTC samples isolated from patients suffering from advanced CRPC [31, 43, 44]. Recently, Antonarakis et al. were able to demonstrate that patients with AR-V7 expression in CTCs had statistically shorter time to PSA and radiographic progression and shorter overall survival [45]. These findings are supported by a recent study from our group, which analyzed both AR-V7 and AR point mutations in CTCs of patients suffering from advanced PCa [44].
The discovery of constitutively active AR∆LBD that do not express a LBD has led to the development of promising novel experimental approaches targeting AR-signaling in a LBD-independent manner [46-51]. Recent efforts to develop drugs targeting the TAD situated in the N-terminus of the AR have led to the discovery of the small molecule inhibitor EPI-001, a bisphenol A-derivative that binds covalently and inhibits the AR amino-terminal TAD [46]. Low toxicity combined with the ability to block transactivation of both AR and AR∆LBDs makes EPI-001 the most promising third generation compound for the treatment of CRPC.
The analysis of AR-point mutations and/or AR-splice variants has led to the discovery of new prognostic and therapeutic targets in CRPC. In summary, the combination of new prognostic parameters able to guide treatment selection along with novel therapeutic approaches will establish a new era of personalized, targeted therapies.
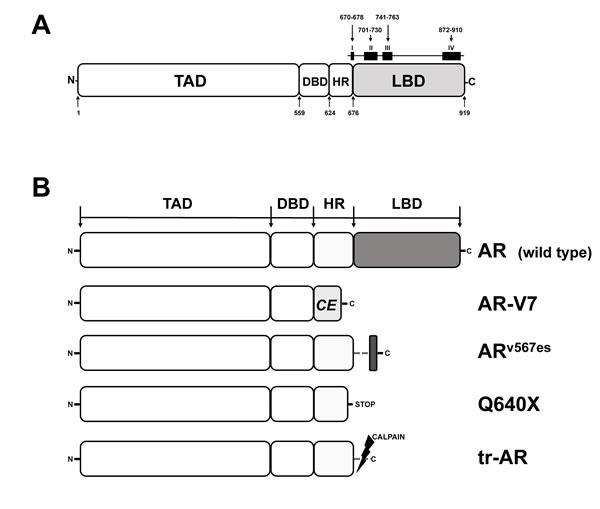
Figure 1: Functional domains of the human AR and AR variants expressed in PCa. A. Functional regions of the AR. TAD, transactivation domain; DBD, DNA-binding domain; HR, hinge region; LBD, ligand binding domain. Squares (I-IV) on top of the LBD delineate clusters of AR mutations, numbers indicate amino acid (aa) positions. B. AR and AR∆LBD variants identified in PCa. AR, full length AR wild type; AR-V7, product of alternative splicing, CE, new cryptic exon; ARv567es, product of altered splicing, exon 5, 6, 7 skipped during splicing; Q640X, AR with a nonsense mutation leading to a truncated AR of 640 aa; tr-AR, truncated AR, enzymatically cleaved by calpain.
Adaptive responses in AR signaling by translational regulation of the AR
The upregulation of AR protein is a hallmark of CRPC and seems to represent an adaptive response to ADT [52]. Presumably, the increased AR protein level expedites the reactivation of the AR signaling. On the one hand, the AR gene amplification is observed in about one third of these cases [53] as one mechanism while on the other hand post-translational regulation represents another underlying molecular mechanism with increasing importance for enhancing AR signaling. Here the factors and pathways at the level of translational regulation of the AR are described in the context of PCa. Interestingly, the AR transcript has a long 3’untranslated region (3’UTR), which is predestinated for post-translational regulation via RNA binding proteins that modulate mRNA stability or translation efficiency. RNA binding proteins Hu antigen R and polyC-binding proteins (PCBPs) 1 and 2 were found to bind to a UC rich motif in the 3’UTR of AR mRNA and regulate its translation [54, 55]. PCBP1 was identified as a blocker of AR translation in dedifferentiating endometrial cells and this role was confirmed in LNCaP PCa cells [56].
An UC-rich 3’UTR motif is also the target site of another AR post-transcriptional inhibitor, ErbB3 binding protein (EBP1) that in addition also binds to a RNA stem-loop formed by the CAG repeats encoding the poly-glutamine stretch in the AR N-terminus [57]. Whereas EBP1 interaction with the UC region in the 3’UTR promotes mRNA decay, its binding to the CAG stem-loop seems to attenuate translation of AR mRNA. This post-transcriptional inhibition of AR protein synthesis of EBP1 is an additional function to its described activity as a repressor protein of AR transcriptional activity [58, 59]. In line with its interference with the AR axis, EBP1 was found downregulated in advanced stages of PCa [60] and inhibiting PCa cell growth when overexpressed [58].
The nucleic acid binding protein called heterogeneous nuclear ribonucleoprotein K (hnRNP-K) is an additional inhibitor of AR mRNA translation and an inhibitor of PCa cell proliferation via binding to several sites in the AR mRNA including sites in both untranslated terminal regions and in the coding region [61]. Further support for its inhibitory role in PCa comes from its inverse correlation with AR protein in primary prostate tumors and its altered pattern of expression in tumor metastases.
A mechanism for enhancement of AR translation was identified recently and is based on a reciprocal link between the AR and the ribonucleoprotein transcriptional enhancer complex assembled by midline 1 (MID1), a protein that is mutated in the developmental syndrome Opitz G/BBB [62, 63]. Overexpression of MID1 in PCa cells results in an upregulation of AR protein and in line with this an increase of AR transcriptional activity whereas MID1 knockdown decreases AR protein levels [64]. Within the AR N-terminus there is a poly-glutamine and a poly-glycine repeat both encoded by purine-rich trinucleotid repeats, a CAG and a GGY repeat, respectively. Notably, both trinucleotide repeats interact with the MID1 protein complex. The MID1 protein complex binds the AR mRNA via both trinucleotide repeats and enhances AR translation [64]. In addition to MID1, this transcriptional regulator complex contains the regulatory and the catalytic subunits of protein phosphatase 2A (alpha-4 and PP2Ac) [65] and MID1 also has an ubiquitin ligase activity that targets PP2Ac in the complex to proteasomal degradation thus regulating PP2A activity [66]. As PP2A is a major antagonist of protein kinases involved in growth factor signaling cascades, its degradation further stimulates tumor cells [67].
AR regulation by MID1 occurs via enhancement of AR translation. However, the interaction of the MID1 complex and the AR axis is not uni- but bi-directional. Whereas MID1 regulates AR protein through translation control, AR is a negative regulator of MID1 via several AR binding sites in the MID1 gene. This mutual interaction forms a classical regulatory feedback loop that was suggested as a fine-tuning mechanism for homeostasis of AR protein level. ADT of PCa would disrupt this mechanism and result in MID1 and subsequently AR protein upregulation. In line with this, MID1 is significantly overexpressed in PCa in a stage-dependent manner [64].
Noteworthy, the assembly of the MID1-alpha4/PP2Ac protein complex is disrupted by the antidiabetic drug metformin as first reported by Kickstein et al. [68]. Besides decreasing blood glucose levels metformin inhibits many types of cancer and non-malignant cells, among them also PCa cells [69, 70]. In addition it was suggested as a tumor preventive drug, although these data are still inconclusive [71-74]. Testing its effect on the MID1-AR feedback loop revealed disruption of AR mRNA association with the protein complex and subsequent downregulation of AR protein in PCa cells treated with metformin [75]. The inhibitory effect of metformin was mimicked by disruption of the MID1-alpha4/PP2As protein complex by siRNA knockdown of MID1 or alpha4 whereas activation of another target of metformin, AMP kinase was not required.
Thus, the inhibition of AR protein levels by metformin suggests its use in the treatment of hormone-naïve PCa and CRPC. In support of this, a metformin treatment study in 44 men with progressive metastatic CRPC who received metformin until disease progression resulted in disease stabilization and prolongation of PSA doubling time [76]. Retrospective analysis revealed that the risk of progression to CRPC in patients treated for localized disease with external-beam radiation therapy was reduced in metformin users compared to patients treated with other anti-diabetic drugs [77]. In contrast another recent study did not confirm a reduced risk for adverse outcome in PCa patients by metformin alone [78], which calls for further thorough evaluation of a potential benefit of metformin in the treatment of PCa. Perhaps, the combination of metformin and AR antagonists, such as enzalutamide, that is currently under clinical investigation (NCT02339168), may provide a benefit for patients.
Last but not least, a new class of post-transcriptional regulators also acting on AR, are micro-RNAs (miRNAs) that control gene expression by inhibition of protein translation or induction of mRNA cleavage. These small RNAs of about 22 bases in length are generated by processing of mostly untranslated RNA and each miRNA can regulate a variety of target mRNAs [79, 80]. Several miRNAs have been reported to target various sites in AR mRNA and inhibit androgen receptor-positive PCa cell lines, e.g. miR488*, miR125, miR205, mir185, miR1, miR31 [81-86]. A systematic combined experimental and in silico screen for miRNAs targeting the long 3’UTR of the AR performed in PCa cell lines identified 75 miRNAs that regulate AR protein level [87]. Fifteen miRNAs downregulating AR were confirmed to decrease androgen-induced proliferation of PCa cells. This number underscores the complexity of post-transcriptional regulation by miRNAs and offer new strategies for therapeutic intervention.
In conclusion, the diverse mechanisms of AR modulation at the level of mRNA and translation into protein can result in adaptation of AR signaling. The variety of proteins and miRNAs involved define a complex regulatory network for fine-tuning of AR protein level in PCa that might be used as drug targets and calls for efforts to develop methods to interfere with post-transcriptional AR regulation for improving inhibition of the AR axis in PCa therapy.
Adaptive responses in AR signaling through growth factors and tyrosine kinases
There are several levels of interaction between growth factor receptors and AR in PCa. Transcription activation function of the AR could be enhanced by growth factors and growth factor-related receptors in a synergistic and ligand-independent manner [88, 89].
Mechanistically, growth factors and related receptors lead to activation of MAPK that phosphorylate specific amino acids in the N-terminal region of the AR [90]. In consequence, HER-2, which activates kinases of the MAPK group, leads to AR-dependent progression of PCa, as evidenced in the LAPC-4 model [91]. The effect of HER-2 on AR could be explained by modulation of receptor DNA binding and AR stability [92]. Activation of the AR by HER-2 and HER-3 was reported also in a PCa recurrent cell line CWR-R1 [93]. Induction of HER-2 occurs in vitro in conditions of androgen depletion and pointing to possible adaptive and compensatory effects of endocrine therapy [94]. Consistently with these observations, HER-2 expression is increased during cancerogenesis of PCa and leads to elevation of expression of the AR downstream gene PSA [95, 96]. In addition, compensatory upregulation of the Etx/BMX tyrosine kinase was observed in CRPC cells [97].
MAPK are implicated in activation of the AR by interleukin-6, a cytokine whose expression is elevated in human PCa [98]. It was also demonstrated that interleukin-6 activation of the AR occurs through activation of MAPK and phosphorylation of N-terminal amino acids [99]. Notably, the tyrosine kinase Src and FAK are implicated in regulation of growth and migration of PCa cells by interleukin-6 [100]. Interleukin-6 is a positive growth factor being responsible for inhibition of apoptosis and angiogenesis in several human cancers, including PCa. Similarly, tyrosine kinase Pim1 and Etk are required for AR activation by interleukin-6 [101]. AR coactivators p300 and SRC-1 are particularly important for AR activation by IL-6 [102, 103]. Both coactivators are highly expressed in prostate cancer and are identified as targets for therapy [104-106]. After IL-6 binding to the receptor, signal transducer and activator of transcription (STAT)3 factor is translocated to the nucleus and phosphorylated. Although in LNCaP cells phosphorylation of STAT3 may be associated with either growth inhibition or stimulation, there is evidence obtained with other models according to which STAT3 is a valid target for therapy [107]. AR activation by IL-6 is potentiated by STAT3 and MAPK pathways [108] as evidenced by association of STAT3 with the AR, which occurs in an IL-6-dependent manner [109].
Taken together, the results of several studies mentioned above have indicated that tyrosine kinases are particularly important in AR activation in conditions in which the levels of circulating androgen are diminished during therapy, indicating an adaptation response.
There is also a link between AR mutations and growth factor receptors. Androgens and hormones that activate mutated AR in LNCaP cells increase expression of epidermal growth factor receptor expression [110]. These findings could be explained by association between AR and epidermal growth factor receptor [111]. Proliferative effect of androgens and epidermal growth factor could also be explained by their down-regulation of the cell cycle inhibitor p27 [112].
Because of these findings one can propose the development of preclinical and clinical inhibitors of tyrosine kinases to inhibit the crosstalk between growth factor receptor and AR signaling pathways. Treatment of cells with an anti-androgen and the anti-HER2 receptor monoclonal antibody herceptin could open the way for novel PCa therapies. In line with these observations, the dual ErbB1/ErbB2 tyrosine kinase inhibitor PKI-166 was tested in series of human PCa xenografts [113]. The effects of the inhibitor could be abolished by androgenic administration [113].
In context of crosstalk between signaling pathways, further aspects of inhibition of tyrosine kinases by sorafenib, a multikinase inhibitor, have to be discussed. Sorafenib inhibits proliferation and induced apoptosis in several PCa cell lines [114]. Sorafenib is a multikinase inhibitor that is approved for therapy of renal and liver cancer. Androgen-sensitive PCa cells were inhibited to a higher extent compared to androgen-insensitive ones. It was demonstrated that several targets of sorafenib, which were identified previously, are also inhibited in PCa cells. It was also demonstrated that cell lines that become resistant to endocrine and chemotherapy in PCa are at least partially responsive to sorafenib. A higher sensitivity of AR-positive cell lines to sorafenib could be explained by down-regulation of AR expression.
These in vitro findings could also have clinical implications. Although PCa is a heterogenous disease, patients with higher expression of AR and several other sorafenib targets may be good candidates for sorafenib treatment. Thus, preclinical results obtained with sorafenib may serve as a basis for the development of a more personalized approach for PCa patients.
In summary, there are multiple interactions between signaling pathways that include tyrosine kinase receptors and the AR. These interactions are basis for a rational therapeutic targeting of these interactions in clinics.
Adaptive responses in AR signaling through the activation of the Src-Akt - and MAPK- pathways inactivate AR-corepressors and lead to enhanced cancer progression
The AR signaling is not only controlled by the ligands of AR but also by signal transduction pathways including the MAPK and non-receptor tyrosine kinases. Among tyrosine kinases, Src is particularly important for AR phosphorylation [115]. In PCa, a correlation between AR tyrosine phosphorylation and Src tyrosine kinase activity was observed. Importantly, AR activation by Src was confirmed in CRPC cells [116, 117].
Intracellular kinases that mediate the signal transduction of membrane associated receptor tyrosine kinases are often linked to cellular growth and seem to play a critical role in cancer development and progression. The family of non-receptor tyrosine kinases, including Src, are activated by and interact with various cellular pathways and regulate a plethora of different pathways including cell proliferation, cell motility, invasion, epithelial-to-mesenchymal transition, resistance to apoptosis, and metastatic spread (reviewed in: [118, 119]. Src expression is upregulated in human CRPC cells [116, 118, 120]. Several lines of evidence indicate that the expression of Src and Src kinase family members can drive the formation of PCa or the progression to CRPC [118]. Interestingly the interaction of AR with Src has been shown to lead to AR activation by phosphorylation of AR [115].
Another underlying molecular basis to activate the AR by the Src family has been suggested to be mediated through inactivation of corepressors. Corepressors are transcriptional regulators that interact with DNA-bound transcription factors and lead to inhibition of their transactivation. Some corepressors interact with the amino-terminus of AR and some with the DBD, which may be used to reduce the transcriptional activity of AR point mutations and AR∆DBD isoforms [121].
In the presence of AR antagonists (anti-androgens), the corepressor recruitment leads to recruitment of chromatin repressor complexes to AR target genes or to coactivator displacement on the AR. These mechanisms are the molecular basis for AR inactivation mediated by some AR antagonists [121-123]. In addition to antagonist induced corepressor binding to AR, LCoR, a ligand-dependent corepressor for AR, reduces AR activity in the presence of androgens including dihydrotestosterone [117]. In contrast to non-PCa cells, LCoR has only weak repression function in CRPC cells indicating an adaptive response in CRPC to inactivate corepressor function. Therefore, combination therapy of inhibitors against Src and Src family members and against the AR may be fruitful.
To analyze the underlying molecular mechanism, a battery of various inhibitors of signaling pathways were used to treat CRPC cells. The results indicated that the LCoR silencing function is repressed by the Src-Akt pathway [117]. Inhibition of Src signaling represses AR target gene expression indicating that the Src family members enhance the AR transactivation at chromatin level. In the presence of a Src inhibitor, LCoR is more potently recruited to AR target genes and reduced the expression such as that of PSA. The Src-LCoR-AR pathway was confirmed by LCoR expression and inactivation of Src to regulate CRPC tumor growth of human xenografts in mice. Thus, the Src-Akt signaling inactivates LCoR, which subsequently activates AR signaling in human CRPC cells in culture and in mouse xenograft model [117].
These findings and those from other groups strongly suggest that inhibitors of members of the Src tyrosine kinase family in combination therapy with antagonists may be a very useful tool to inactivate AR signaling and progression of CRPC, which has been nicely reviewed recently [124]. In line with this combinatorial treatment idea, the use of PI3K inhibitors is currently under clinical investigation in combination with AR antagonists such as enzalutamide (UKCRN Study ID: 16580).
Similarly to LCoR, the corepressor Silencing Mediator for Retinoid and Thyroid hormone receptors (SMRT) seems also to be inactivated by signaling pathways in PCa. An activated MAPK signaling was shown to inhibit the SMRT corepressor function [125]. SMRT binds to the AR in the presence of AR antagonists and inactivates the AR-mediated transactivation. SMRT is recruited to AR binding sites at chromatin and recruits the SAP30-SIN3A-HDAC repressor complex [126]. In addition SMRT competes with the coactivator SRC1 for the binding to AR [127]. However in PCa cells, SMRT exhibited only little silencing activity. Analyses of various signaling pathways suggest that the MAPK is one major pathway that inactivates SMRT repressor function. Using an ERK1/2 specific inhibitor, the binding of SMRT to AR was enhanced as well as the chromatin recruitment of SMRT to the PSA gene was increased. Furthermore, a synergy by the co-treatment of the AR antagonist and ERK1/2 inhibitor revealed a potent inhibition of PCa cell growth and colony formation [125]. These findings strongly suggest that the MAPK inactivates AR corepressors and thereby activates the AR signaling as an adaptive response. These observations also suggest that inhibitors of the MAPK pathway [128] in combination therapy with AR antagonists may be a very useful tool to inactivate AR signaling and progression of PCa.
Thus, mitogenic signaling pathways activate the AR signaling. One underlying molecular basis is the inactivation of AR corepressors by the Src kinase family members and/or the MAPK signaling. Co-treatment with signal transduction inhibitors may be a useful tool and therapeutic approach to inhibit CRPC. Presumably tissue specific inhibitors of Src, PI3K, Akt and MAPK are more useful in combination with AR inhibitors to reduce side-effects.
Taken together, multiple adaptation processes seem to exist that allow persistent AR signaling in PCa (Figure 2). The detailed knowledge about possible pathways that lead to the activation of the AR allows to detect individualized adaptation processes in PCa and consequently allows an optimal individualized treatment option.
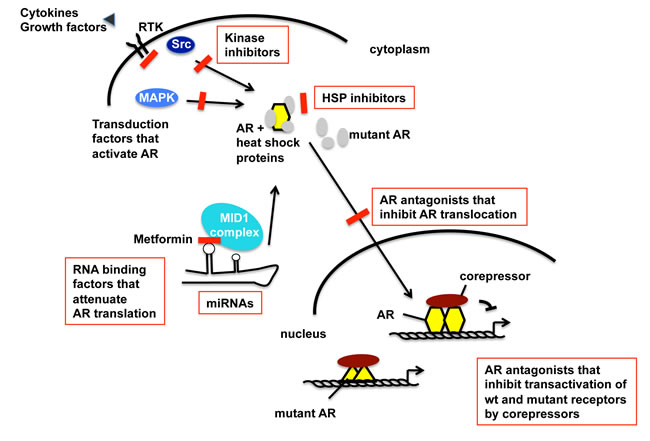
Figure 2: Schematic view of the AR activation pathway that undergoes adaptation during progression towards CRPC and therapy resistance. Summarized targets for future therapeutic interventions of CRPC within the AR signaling pathway and AR adaptive responses. The level of increased AR protein in CRPC is in part regulated at mRNA level. In addition to its cognate hormone, the AR is activated by tyrosine kinase and MAPK signaling, leading to dissociation of heat shock proteins (HSP). HSPs might thus present targets of intervention into AR signaling [23]. Further steps of AR signaling are the translocation to the nucleus, DNA binding and regulation of AR target gene expression. Besides the wild-type (wt) AR, isoforms and mutants of AR exist for which the inhibition is also an important future goal for treatment of CRPC. Detection of activated individualized pathways that activate the AR may allow using highly specific inhibitors in combination therapies. Inhibitors of specific AR activation signaling are highlighted in red color.
Acknowledgments
We are grateful to ISMAR Healthcare NV-SA for editorial support provided by Astellas Pharma GmbH. Independent review article written by investigators where Astellas Germany has only provided funding for editorial support and has had no other involvement in the writing, payment or review of the article. This work was supported by the Rudolf-Becker-Foundation to S.P.
Conflicts of interest
The authors served as advisors for Astellas Pharma GmbH.
Abbreviations
ADT, androgen deprivation therapy; AR, androgen receptor, CRPC, castration resistant prostate cancer; CTC, circulating tumor cells; DBD, DNA binding domain; HR, hinge region; LBD, ligand binding domain; LHRH, luteinizing hormone-releasing hormone; mCRPC, metastatic CRPC; MAPK, Mitogen-activated protein kinases; PCa, prostate cancer; PSA, prostate specific antigen; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor; TAD, transactivation domain, wt, wild-type.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63: 11-30.
2. Welty CJ, Cowan JE, Nguyen H, Shinohara K, Perez N, Greene KL, Chan JM, Meng MV, Simko JP, Cooperberg MR, Carroll PR. Extended followup and risk factors for disease reclassification in a large active surveillance cohort for localized prostate cancer. J Urol. 2015; 193: 807-11.
3. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T, Zattoni F, Mottet N. EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol. 2014; 65: 124-37.
4. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T, Zattoni F, Mottet N. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014; 65: 467-79.
5. Hussain M, Tangen CM, Higano C, Schelhammer PF, Faulkner J, Crawford ED, Wilding G, Akdas A, Small EJ, Donnelly B, MacVicar G, Raghavan D. Absolute prostate-specific antigen value after androgen deprivation is a strong independent predictor of survival in new metastatic prostate cancer: data from Southwest Oncology Group Trial 9346 (INT-0162). J Clin Oncol. 2006; 24: 3984-90.
6. Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009; 6: 76-85.
7. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011; 65: 1180-92.
8. Tombal B. What is the pathophysiology of a hormone-resistant prostate tumour? Eur J Cancer. 2011; 47: S179-S188.
9. Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, Said J, Reiter RE, Sawyers CL. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999; 59: 5030-6.
10. Tso CL, McBride WH, Sun J, Patel B, Tsui KH, Paik SH, Gitlitz B, Caliliw R, van Ophoven A, Wu L, deKernion J, Belldegrun A. Androgen deprivation induces selective outgrowth of aggressive hormone-refractory prostate cancer clones expressing distinct cellular and molecular properties not present in parental androgen-dependent cancer cells. Cancer J. 2000; 6: 220-33.
11. Nadiminty N, Gao AC. Mechanisms of persistent activation of the androgen receptor in CRPC: recent advances and future perspectives. World J Urol. 2012; 30(3): 287-95.
12. Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57: 314-9.
13. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013; 63: 920-6.
14. Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol. 2009; 27: 36-41.
15. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008; 68: 6407-15.
16. Culig Z, Santer FR. Androgen receptor co-activators in the regulation of cellular events in prostate cancer. World J Urol. 2012; 30: 297-302.
17. Cronauer MV, Schulz WA, Burchardt T, Anastasiadis AG, de la Taille A, Ackermann R, Burchardt M. The androgen receptor in hormone-refractory prostate cancer: relevance of different mechanisms of androgen receptor signaling. Int J Oncol. 2003; 23: 1095-102.
18. Toren PJ, Gleave ME. Novel non-AR therapeutic targets in castrate resistant prostate cancer. Transl Androl Urol. 2013; 2: 265-277.
19. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase (PI3K) pathway in cancer. Nat Rev Drug Discov. 2009; 8: 627-44.
20. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012; 72: 5588-99.
21. Lee RJ, Smith MR. Targeting MET and vascular endothelial growth factor receptor signaling in castration-resistant prostate cancer. Cancer J. 2013; 19: 90-8.
22. Varkaris A, Corn PG, Gaur S, Dayyani F, Logothetis CJ, Gallick GE. The role of HGF/c-Met signaling in prostate cancer progression and c-Met inhibitors in clinical trials. Expert Opin Investig Drugs. 2011; 20: 1677-84.
23. Hessenkemper W, Baniahmad A. Targeting heat shock proteins in prostate cancer. Curr Med Chem. 2013; 20: 2731-40.
24. Eisermann K, Wang D, Jing Y, Pascal LE, Wang Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl Androl Urol. 2013; 2: 137-147.
25. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012; 33: 887-94.
26. Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990; 173: 534-40.
27. Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999; 59: 2511-5.
28. Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RT, Kantoff P, Montgomery B, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015; 21: 1273-80.
29. Mostaghel EA. Beyond T and DHT - novel steroid derivatives capable of wild type androgen receptor activation. Int J Biol Sci. 2014; 10: 602-13.
30. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013; 3:1030-43.
31. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013; 3: 1020-9.
32. Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991; 5: 1396-404.
33. Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, Balk SP. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res. 2014; 20:1590-600.
34. Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014; 21: T87-T103.
35. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013; 73: 483-9.
36. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR, Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012; 72: 3457-62.
37. Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, Chi KN. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013; 24: 1802-7.
38. Schrader AJ, Boegemann M, Ohlmann CH, Schnoeller TJ, Krabbe LM, Hajili T, Jentzmik F, Stoeckle M, Schrader M, Herrmann E, Cronauer MV. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014; 65: 30-6.
39. Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010; 70: 7992-8002.
40. Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR, Giannakakou P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014; 74: 2270-82.
41. de Leeuw R, Berman-Booty LD, Schiewer MJ, Ciment SJ, Den RB, Dicker AP, Kelly WK, Trabulsi EJ, Lallas CD, Gomella LG, Knudsen KE. Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clin Cancer Res. 2015; 21: 795-807.
42. van Soest RJ, de Morrée ES, Kweldam CF, de Ridder CM, Wiemer EA, Mathijssen RH, de Wit R, van Weerden WM. Targeting the androgen receptor confers in vivo cross-resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration-resistant prostate cancer. Eur Urol. 2015; 67: 981-5.
43. Jiang Y, Palma JF, Agus DB, Wang Y, Gross ME. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clin Chem. 2010; 56: 1492-5.
44. Steinestel J, Luedeke M, Arndt A, Schnoeller TJ, Lennerz JK, Wurm C, Maier C, Cronauer MV, Steinestel K, Schrader AJ. Detecting predictive androgen receptor modifications in circulating prostate cancer cells. Oncotarget 2015; in press.
45. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371: 1028-38.
46. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Bañuelos CA, Williams DE, McEwan IJ, Wang Y, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010; 17: 535-46.
47. Zengerling F, Streicher W, Schrader AJ, Schrader M, Nitzsche B, Cronauer MV, Höpfner M. Effects of sorafenib on C-terminally truncated androgen receptor variants in human prostate cancer cells. Int J Mol Sci. 2012; 13: 11530-42.
48. Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, Hessein M, Hsing M, Singh K, LeBlanc E, Dehm S, Tomlinson Guns ES, Cherkasov A, et al.Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014; 289: 33877.
49. Laschak M, Spindler KD, Schrader AJ, Hessenauer A, Streicher W, Schrader M, Cronauer MV. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells. BMC Cancer. 2012; 12: 130.
50. Streicher W, Luedeke M, Azoitei A, Zengerling F, Herweg A, Genze F, Schrader MG, Schrader AJ, Cronauer MV. Stilbene induced inhibition of androgen receptor dimerization: implications for AR and ARΔLBD-signalling in human prostate cancer cells. PLoS One. 2014; 9: e98566.
51. Reeb CA, Gerlach C, Heinssmann M, Prade I, Ceraline J, Roediger J, Roell D, Baniahmad A. A designed cell-permeable aptamer-based corepressor peptide is highly specific for the androgen receptor and inhibits prostate cancer cell growth in a vector-free mode. Endocrinology. 2011; 152: 2174-83.
52. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10: 33-9.
53. Koivisto P, Visakorpi T, Kallioniemi OP. Androgen receptor gene amplification: a novel molecular mechanism for endocrine therapy resistance in human prostate cancer. Scand J Clin Lab Invest Suppl. 1996; 226: 57-63.
54. Wilce JA, Leedman PJ, Wilce MC. RNA-binding proteins that target the androgen receptor mRNA. IUBMB Life. 2002; 54: 345-9.
55. Yeap BB, Voon DC, Vivian JP, McCulloch RK, Thomson AM, Giles KM, Czyzyk-Krzeska MF, Furneaux H, Wilce MC, Wilce JA, Leedman PJ. Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3’-untranslated region of the androgen receptor messenger RNA. J Biol Chem. 2002; 277: 27183-92.
56. Cloke B, Shah K, Kaneda H, Lavery S, Trew G, Fusi L, Higham J, Dina RE, Ghaem-Maghami S, Ellis P, Brosens JJ, Christian M. The poly(c)-binding protein-1 regulates expression of the androgen receptor. Endocrinology. 2010; 151: 3954-64.
57. Zhou H, Mazan-Mamczarz K, Martindale JL, Barker A, Liu Z, Gorospe M, Leedman PJ, Gartenhaus RB, Hamburger AW, Zhang Y Post-transcriptional regulation of androgen receptor mRNA by an ErbB3 binding protein 1 in prostate cancer. Nucleic Acids Res. 2010; 38: 3619-31.
58. Zhang Y, Fondell JD, Wang Q, Xia X, Cheng A, Lu ML, Hamburger AW. Repression of androgen receptor mediated transcription by the ErbB-3 binding protein, Ebp1. Oncogene. 2002; 21: 5609-18.
59. Zhang Y, Wang XW, Jelovac D, Nakanishi T, Yu MH, Akinmade D, Goloubeva O, Ross DD, Brodie A, Hamburger AW. The ErbB3-binding protein Ebp1 suppresses androgen receptor-mediated gene transcription and tumorigenesis of prostate cancer cells. Proc Natl Acad Sci U S A. 2005; 102: 9890-5.
60. Zhang Y, Linn D, Liu Z, Melamed J, Tavora F, Young CY, Burger AM, Hamburger AW. EBP1, an ErbB3-binding protein, is decreased in prostate cancer and implicated in hormone resistance. Mol Cancer Ther. 2008; 7: 3176-86.
61. Mukhopadhyay NK, Kim J, Cinar B, Ramachandran A, Hager MH, Di Vizio D, Adam RM, Rubin MA, Raychaudhuri P, De Benedetti A, Freeman MR. Heterogeneous nuclear ribonucleoprotein K is a novel regulator of androgen receptor translation.Cancer Res. 2009; 69: 2210-8.
62. Schweiger S, Schneider R. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays. 2003; 25: 356-66.
63. Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997; 17: 285-91.
64. Köhler A, Demir U, Kickstein E, Krauss S, Aigner J, Aranda-Orgillés B, Karagiannidis AI, Achmüller C, Bu H, Wunderlich A, Schweiger MR, Schaefer G, Schweiger S, et al. A hormone-dependent feedback-loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol Cancer. 2014; 13: 146.
65. Aranda-Orgillés B, Trockenbacher A, Winter J, Aigner J, Köhler A, Jastrzebska E, Stahl J, Müller EC, Otto A, Wanker EE, Schneider R, Schweiger S. The Opitz syndrome gene product MID1 assembles a microtubule-associated ribonucleoprotein complex. Hum Genet. 2008; 123: 163-76.
66. Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, Schneider R, Schweiger S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 2001; 29: 287-94.
67. Bhardwaj A, Singh S, Srivastava SK, Honkanen RE, Reed E, Singh AP. Modulation of protein phosphatase 2A activity alters androgen-independent growth of prostate cancer cells: therapeutic implications. Mol Cancer Ther. 2011; 10: 720-31.
68. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Köhler A, Glossmann H, Schneider R, Sutherland C, Schweiger S. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A. 2010; 107: 21830-5.
69. Colquhoun AJ, Venier NA, Vandersluis AD, Besla R, Sugar LM, Kiss A, Fleshner NE, Pollak M, Klotz LH, Venkateswaran V. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012; 15: 346-52.
70. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27: 3576-86.
71. Preston MA, Riis AH, Ehrenstein V, Breau RH, Batista JL, Olumi AF, Mucci LA, Adami HO, Sørensen HT. Metformin use and prostate cancer risk. Eur Urol. 2014; 66: 1012-20.
72. Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013; 8: e71583.
73. Tseng CH. Metformin significantly reduces incident prostate cancer risk in Taiwanese men with type 2 diabetes mellitus. Eur J Cancer. 2014; 50: 2831-7.
74. Anwar MA, Kheir WA, Eid S, Fares J, Liu X, Eid AH, Eid AA. Colorectal and Prostate Cancer Risk in Diabetes: Metformin, an Actor behind the Scene. J Cancer. 2014; 5: 736-44.
75. Demir U, Koehler A, Schneider R, Schweiger S, Klocker H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer. 2014; 14: 52.
76. Rothermundt C, Hayoz S, Templeton AJ, Winterhalder R, Strebel RT, Bärtschi D, Pollak M, Lui L, Endt K, Schiess R, Rüschoff JH, Cathomas R, Gillessen S. Metformin in chemotherapy-naive castration-resistant prostate cancer: a multicenter phase 2 trial (SAKK 08/09). Eur Urol. 2014; 66: 468-74.
77. Spratt DE, Zhang C, Zumsteg ZS, Pei X, Zhang Z, Zelefsky MJ. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol. 2013; 63: 709-16.
78. Bensimon L, Yin H, Suissa S, Pollak MN, Azoulay L. The use of metformin in patients with prostate cancer and the risk of death Cancer Epidemiol Biomarkers Prev. 2014; 23: 2111-8.
79. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5: 522-31.
80. Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008; 455: 64-71.
81. Sikand K, Slaibi JE, Singh R, Slane SD, Shukla GC. miR 488* inhibits androgen receptor expression in prostate carcinoma cells. Int J Cancer. 2011; 129: 810-9.
82. Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E, Alves P, Tewari AK, Gerstein MB, Beltran H, Melnick AM, Elemento O, Demichelis F, et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer Res. 2013; 73: 1232-44.
83. Shi XB, Xue L, Ma AH, Tepper CG, Gandour-Edwards R, Kung HJ, deVere White RW. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene. 2013; 32: 4130-8.
84. Hagman Z, Haflidadóttir BS, Ceder JA, Larne O, Bjartell A, Lilja H, Edsjö A, Ceder Y. miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br J Cancer. 2013; 108: 1668-76.
85. Stope MB, Bradl J, Peters S, Streitbörger A, Weiss M, Zimmermann U, Walther R, Lillig CH, Burchardt M. Shortened isoforms of the androgen receptor are regulated by the cytoprotective heat-shock protein HSPB1 and the tumor-suppressive microRNA miR-1 in prostate cancer cells. Anticancer Res. 2013; 33: 4921-6.
86. Qu F, Cui X, Hong Y, Wang J, Li Y, Chen L, Liu Y, Gao Y, Xu D, Wang Q. MicroRNA-185 suppresses proliferation, invasion, migration, and tumorigenicity of human prostate cancer cells through targeting androgen receptor. Mol Cell Biochem. 2013; 377: 121-30.
87. Östling P, Leivonen SK, Aakula A, Kohonen P, Mäkelä R, Hagman Z, Edsjö A, Kangaspeska S, Edgren H, Nicorici D, Bjartell A, Ceder Y, Perälä M, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells.Cancer Res. 2011; 71: 1956-67.
88. Ikonen T, Palvimo JJ, Kallio PJ, Reinikainen P, Jänne OA. Stimulation of androgen-regulated transactivation by modulators of protein phosphorylation. Endocrinology. 1994; 135: 1359-66.
89. Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994; 54: 5474-8.
90. Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci U S A. 1999; 96: 5458-63.
91. Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999; 5: 280-5.
92. Mellinghoff IK1, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004; 6: 517-27.
93. Gregory CW, Whang YE, McCall W, Fei X, Liu Y, Ponguta LA, French FS, Wilson EM, Earp HS 3rd. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005; 11: 1704-12.
94. Berger R, Lin DI, Nieto M, Sicinska E, Garraway LA, Adams H, Signoretti S, Hahn WC, Loda M. Androgen-dependent regulation of Her-2/neu in prostate cancer cells. Cancer Res. 2006; 66: 5723-8.
95. Lee MS, Igawa T, Yuan TC, Zhang XQ, Lin FF, Lin MF. ErbB-2 signaling is involved in regulating PSA secretion in androgen-independent human prostate cancer LNCaP C-81 cells. Oncogene. 2003; 22: 781-96.
96. Neto AS, Tobias-Machado M, Wroclawski ML, Fonseca FL, Teixeira GK, Amarante RD, Wroclawski ER, Del Giglio A. Her-2/neu expression in prostate adenocarcinoma: a systematic review and meta-analysis. J Urol. 2010; 184: 842-50.
97. Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, Li W, Guo Z, Xu K, Kim O, Kong X, Melamed J, Qiu S, et al. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010; 70: 5587-96.
98. Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, Culig Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998; 58: 4640-5.
99. Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002; 277: 7076-85.
100. Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, Kung HJ. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene. 2004; 23: 2197-205.
101. Kim O, Jiang T, Xie Y, Guo Z, Chen H, Qiu Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2004; 23: 1838-44.
102. Debes JD, Schmidt LJ, Huang H, Tindall DJ. P300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002; 62: 5632-6.
103. Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator in prostate cancer cells. J Biol Chem. 2002; 277: 38087-94.
104. Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. P300 in rostate cancer proliferation and progression. Cancer Res. 2003; 63: 7638-40.
105. Agoulnik IU, Vaid A, Bingman WE 3rd, Erdeme H, Frolov A, Smith CL, Ayala G, Ittmann MM, Weigel NL. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005; 65: 7959-67.
106. Santer FR, Höschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011; 10: 1644-55.
107. Sun M, Liu C, Nadiminty N, Lou W, Zhu Y, Yang J, Evans CP, Zhou Q, gao AC. Inhibition of Stat3 activation by sanguinarine suppresses prostate cancer cell growth and invasion. Prostate. 2012; 72: 82-9.
108. Yang L, Wang L, Lin HK, Kann PY, Xie S, Tsai MY, Wang PH, Chen YT, Chang C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3-K, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem Biophys Res Commun. 2003; 305: 462-9.
109. Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000; 60: 2132-5.
110. Schuurmans AL, Bolt J, Voorhorst MM, Blankenstein RA, Mulder E. Regulation of growth and epidermal growth factor receptor levels of LNCaP prostate tumor cells by different steroids. Int J Cancer. 1988; 42: 917-22.
111. Bonaccorsi L, Carloni V, Muratori M, Formigli L, Zecchi S, Forti G, Baldi E. EGF receptor (EGFR) signaling promoting invasion is disrupted in androgen-sensitive prostate cancer cells by an interaction between EGFR and androgen receptor (AR). Int J Cancer. 2004; 112: 78-86.
112. Ye D, Mendelsohn J, Fan Z. Androgen and epidermal growth factor down-regulate cyclin-dependent kinase inhibitor p27Kip1 and costimulate proliferation of MDA PCa 2a and MDA PCa 2b prostate cancer cells. Clin Cancer Res. 1999; 5: 2171-7.
113. Mellinghoff IK, Tran C, Sawyers CL. Growth inhibitory effects of the dual ErbB1/ErbB2 tyrosine kinase inhibitor PKI-166 on human prostate cancer xenografts. Res. 2002; 62: 5254-9.
114. Oh SJ, Erb HH, Hobisch A, Santer FR, Culig Z. Sorafenib decreases proliferation and induces apoptosis of prostate cancer cells by inhibition of the androgen receptor and Akt signaling pathways. Endocr Relat Cancer. 2012; 19: 305-19.
115. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006; 10: 309-19.
116. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008; 27: 3596-604.
117. Asim M, Hafeez BB, Siddiqui IA, Gerlach C, Patz M, Mukhtar H, Baniahmad A. Ligand-dependent corepressor acts as a novel androgen receptor corepressor, inhibits prostate cancer growth, and is functionally inactivated by the Src protein kinase. J Biol Chem. 2011; 286: 37108-17.
118. Gelman IH. Androgen receptor activation in castration-recurrent prostate cancer: the role of Src-family and Ack1 tyrosine kinases. Int J Biol Sci. 2014; 10: 620-6.
119. Vlaeminck-Guillem V, Gillet G, Rimokh R. SRC: marker or actor in prostate cancer aggressiveness. Front Oncol. 2014; 4: 222.
120. Qin J, Lee HJ, Wu SP, Lin SC, Lanz RB, Creighton CJ, DeMayo FJ, Tsai SY, Tsai MJ. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J Clin Invest. 2014; 124: 5013-26.
121. Dotzlaw H, Moehren U, Mink S, Cato AC, Iñiguez Lluhí JA, Baniahmad A. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002; 16: 661-73.
122. Wang L, Hsu CL, Chang C. Androgen receptor corepressors: an overview. Prostate. 2005; 63(2): 117-30.
123. Moehren U, Papaioannou M, Reeb CA, Hong W, Baniahmad A. Alien interacts with the human androgen receptor and inhibits prostate cancer cell growth. Mol Endocrinol. 2007; 21: 1039-48.
124. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013; 20: R83-R99.
125. Eisold M, Asim M, Eskelinen H, Linke T, Baniahmad A. Inhibition of MAPK-signaling pathway promotes the interaction of the corepressor SMRT with the human androgen receptor and mediates repression of prostate cancer cell growth in the presence of antiandrogens. J Mol Endocrinol. 2009; 42: 429-35.
126. Vermeulen M, Walter W, Le Guezennec X, Kim J, Edayathumangalam RS, Lasonder E, Luger K, Roeder RG, Logie C, Berger SL, Stunnenberg HG. A feed-forward repression mechanism anchors the Sin3/histone deacetylase and N-CoR/SMRT corepressors on chromatin. Mol Cell Biol. 2006; 26: 5226-36.
127. Dotzlaw H, Papaioannou M, Moehren U, Claessens F, Baniahmad A. Agonist-antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol Cell Endocrinol. 2003; 213: 79-85.
128. Choudhury Y, Yang Z, Ahmad I, Nixon C, Salt IP, Leung HY. AMP-activated protein kinase (AMPK) as a potential therapeutic target independent of PI3K/Akt signaling in prostate cancer. Oncoscience. 2014, 1: 446-56.