
Introduction
During the last years the role of vitamin D, primarily vitamin D3, in tumor suppression has attracted considerable attention (reviewed in [1-4]). There is now increasing evidence strongly suggesting that vitamin D3 reduces the risk of developing cancer and may be applied for the treatment of cancer as it was shown to induce differentiation and apoptosis, and inhibit proliferation and angiogenesis (reviewed in [5]). A tumor suppressive effect of vitamin D3 has also been shown in vivo in animal models of colon, breast and prostate cancer [6-11]. In addition, several epidemiological studies have implicated vitamin D3 in reducing cancer risk, progression and mortality with promising results especially in colon cancer [12-20]. Several large-scale clinical trials aiming to determine the therapeutic value of vitamin D3 are expected to be finalized within the next five years [21].
Vitamin D3, the precursor of the hormonally active calcitriol, is mainly synthesized in the skin after exposure to sunlight but is also supplied by nutritional sources [22]. Two hydroxylation steps mediated by the hydroxylases CYP27A1 and CYP27B1 in the liver and in the kidney, respectively, lead to the active vitamin D3 metabolite calcitriol (1,25(OH)2D3) [23]. Interestingly, CYP27B1 is also expressed at extra-renal sites including brain, colon, pancreas or skin and mediates local conversion of the intermediate metabolite 25-hydroxy-vitamin D3 (25(OH)D3) to calcitriol [24]. Calcitriol binds to the vitamin D receptor (VDR), which regulates the expression of several genes involved in colorectal cancer, such as BIRC5, CDKN1A, CDH1 or HIF1α (reviewed in [5]). Recently, calcitriol-induced expression of cystatin D (CST5) was shown to inhibit proliferation, migration, anchorage-independent growth as well as tumor formation of xenografted colorectal cancer cell lines [25]. Moreover, poor differentiation of colorectal tumors correlates with decreased CST5 expression [25].
Cystatin D is an endogenous inhibitor of cystein proteases, such as the cathepsins S, H and L [26, 27]. Cysteine cathepsins are lysosomal proteases and are often upregulated in different types of cancers. During tumorigenesis cathepsins are translocated from intracellular compartments, typically lysosomes, to the outer cell-membrane or secreted into the extracellular space. Cathepsins promote tumor progression via degradation of components of the basement membrane, extracellular matrix or cleavage of the adhesion molecule E-cadherin (reviewed in [28]).
The p53 transcription factor is one of the most important tumor suppressors as also evidenced by its common inactivation in human tumors [29]. Several different stress signals including oncogene activation, oxidative stress or DNA damage lead to its posttranslational stabilization and subsequent association with binding sites within promoters [30], which are composed of two half sites of RRRCWWGYYY (R: purine, W: adenine or thymine, Y: pyrimidine) divided by a spacer of 0 - 13 bp [31, 32]. Once activated, p53 exerts diverse tumor suppressive effects through a large number of target genes [33]. Interestingly, p53 directly induces the expression of the VDR [34, 35]. Moreover, the CDKN1A promoter harbors p53 and VDR binding sites suggesting the possibility of coordinated regulations after p53 and VDR activation [36]. Furthermore, calcitriol induces MDM2 expression in a p53-dependent manner therefore limiting p53 activation [37]. In addition, mutant p53 may convert the effect of vitamin D3 by modulating VDR-mediated transcription [38].
Here, we show that p53 directly induces CST5 expression by binding to a p53 response element upstream of the CST5 promoter. We present evidence that a combination of p53 activation and calcitriol treatment results in enhanced CST5 expression and suppression of the EMT transcription factor SNAIL. Furthermore, we show that upregulation of CST5 by p53 contributes to the induction of mesenchymal-epithelial transition. In addition, we demonstrate that SNAIL directly represses CST5 expression and that calcitriol treatment reverses this effect.
Results
p53 induces CST5 expression
We had previously observed a ~6-fold increase of CST5 protein by pulsed SILAC and a ~9-fold induction of CST5 mRNA expression by RNA-Seq analysis after addition of doxycycline (DOX) to activate expression of ectopic p53 in the colorectal cancer cell line SW480 harboring a pRTR-p53-VSV vector for 48 and 40 hours, respectively (Hünten et al., submitted). Here, we confirmed the upregulation of the CST5 protein by p53 by Western blot analysis (Figure 1A). As expected, p53 expression also resulted in the induction of the known p53 target p21. The kinetics of induction by p53 were similar for p21 and CST5: at 24 hours an induction was detectable and by 72 hours both proteins were still elevated. After p53 activation, CST5 mRNA expression displayed a similar induction as p21 (Figure 1B). Recently, CST5 was shown to be induced by vitamin D3 via direct binding of the vitamin D receptor (VDR) to its promoter in human colon cancer cell lines [25]. In addition, the VDR gene is a known p53 target [34]. To determine whether p53 induces CST5 in a VDR-independent manner, VDR-deficient HEK293T cells were transfected with a DOX-inducible pRTR-p53-VSV vector (Figure 1C). Activation of p53 expression by treatment with DOX resulted in a significant induction of CST5 and, as a control, p21 mRNA. These results show that p53 induces CST5 expression in a VDR-independent manner. An immunofluorescence analysis confirmed increased CST5 protein levels after ectopic p53 expression and showed its predominant cytoplasmic localization after DOX treatment in SW480/pRTR-p53-VSV cells (Figure 1D).
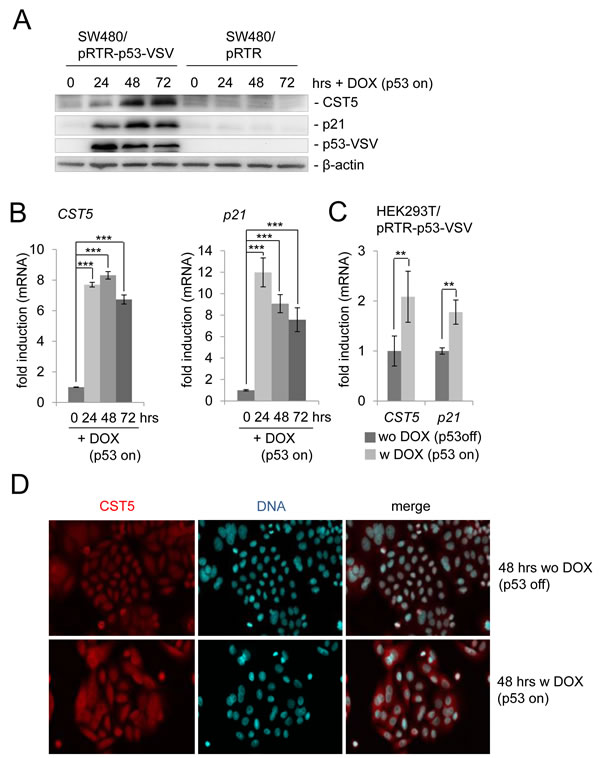
Figure 1: Regulation of CST5 by p53 in SW480 colorectal cancer cells. SW480 cells harboring a pRTR-p53-VSV vector or the empty vector pRTR were treated with DOX for the indicated periods of time to activate p53-VSV expression. A. Western blot analysis of the indicated proteins in SW480/pRTR-p53-VSV. β-actin served as a loading control. B. The expression of the CST5- and p21-mRNA was determined by qPCR analysis. Fold changes represent mean values of triplicate analyses of DOX- versus un-treated cells normalized to β-actin expression. Standard deviations are represented by error bars. C. HEK293T cells were transfected with the pRTR-p53-VSV vector and p53 expression was induced by adding DOX for 24 hours. The expression of CST5- and p21-mRNA levels was measured by qPCR analysis. Fold changes represent mean values of triplicate analyses of DOX-treatment versus untreated cells normalized to β-actin expression. Standard deviations are represented by error bars. D. Analysis of CST5 expression and localization in SW480/pRTR-p53-VSV cells by confocal immunofluorescence microscopy after 48 hours with and without DOX treatment. Nuclear DNA was visualized by DAPI staining. 200x magnification.
Endogenous p53 mediates induction of CST5 by DNA damage
To further interrogate the p53-dependency of the CST5 expression, the colorectal cancer cell lines HCT116 and RKO and their isogenic p53-deficient variants, which were generated by homologous recombination [39], were treated with either nutlin-3a, a small-molecule inhibitor of MDM2 [40], or the DNA-damaging agent etoposide (Figure 2). Both treatments resulted in a time-dependent upregulation of CST5 in HCT116 p53+/+ cells (Figure 2A). Since CST5 expression was not induced in HCT116 p53-/- cells, the activation of CST5 transcription was mediated by p53. Also RKO cells showed a p53-dependent induction of CST5 after induction of DNA damage by etoposide (Figure 2B). The p53-dependent induction of p21 mRNA was more pronounced than the induction of CST5 expression in RKO and HCT116 cells (Figure 2C, 2D). Taken together, these results show that p53 mediates the induction of CST5 by DNA damage.
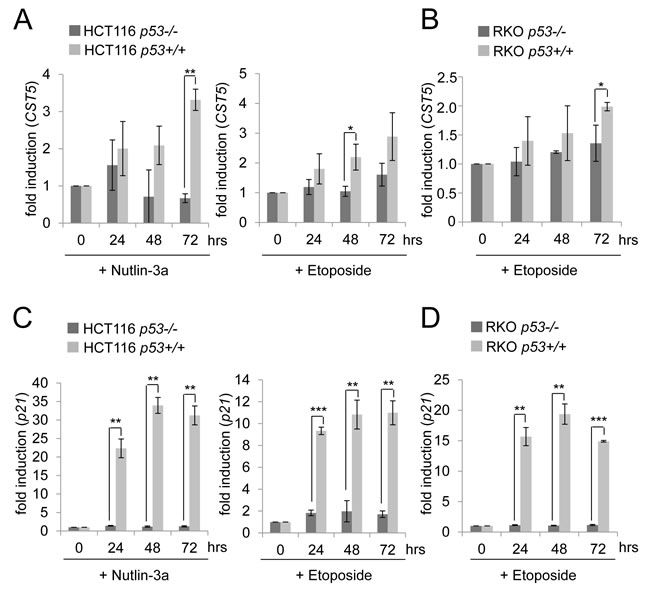
Figure 2: p53-dependent regulation of CST5 in HCT116 and RKO cancer cells. A., B. CST5- and C., D. p21-mRNA levels were measured by qPCR analysis at the indicated time points. A., C. HCT116 p53-/- or p53+/+ and B., D. RKO p53+/+ or p53-/- were treated with nutlin-3a or etoposide or the vehicle DMSO. Fold changes represent mean values of triplicate analyses of nutlin-3a/etoposide versus DMSO treated cells normalized to β-actin expression. Error bars represent standard deviations.
CST5 is a direct p53 target
In a genome-wide p53/ChIP-Seq analysis we had detected ChIP-signals in the vicinity of the CST5 promoter indicating direct p53 binding (Figure 3A; Hünten et al., submitted). A sequence that fits well to the p53 binding consensus sequence [31, 32] was identified 1106 base-pairs upstream of the transcriptional start site (TSS) of CST5 underneath a ChIP-signal with ~10 reads (Figure 3A). This p53 binding element was conserved between human and rat CST5 promoters (Figure 3B). p53 binding at this site was confirmed by qChIP analysis in SW480/pRTR-p53-VSV cells treated with DOX for 16 hours, whereas no enrichment was detected in the control cell line SW480/pRTR (Figure 3C). After DNA damage induced by etoposide, HCT116 p53+/+, but not HCT116 p53-/- cells, displayed increased p53 occupancy at this site (Figure 3D). Therefore, also endogenous p53 occupied these binding sites. Together with the results presented above, these findings establish CST5 as a direct transcriptional target of p53.
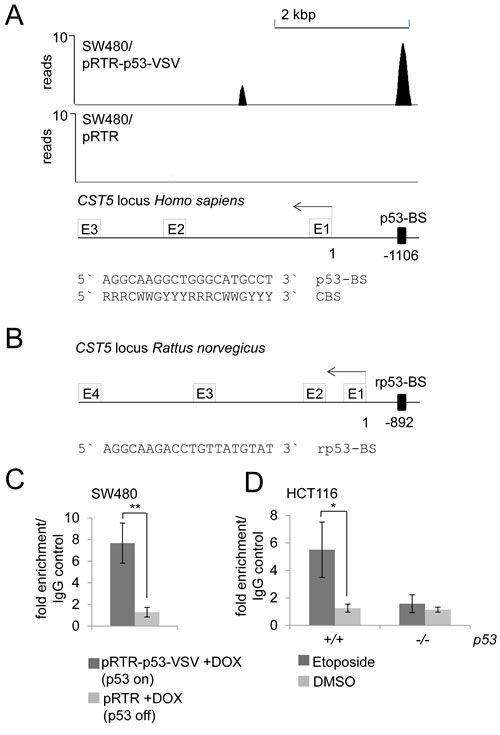
Figure 3: Direct binding of p53 upstream of the CST5 promoter. A. ChIP-Seq analysis was performed with a VSV-specific antibody 16 hours after addition of DOX to SW480/pRTR-p53-VSV and SW480/pRTR cells. The ChIP-Seq results are represented in the UCSC browser showing p53 binding to the indicated p53 binding site in a region 1106 bp upstream of the CST5 promoter. B. p53 binding site upstream of the CST5 locus of Rattus norvegicus. C. qChIP analysis of SW480/pRTR-p53-VSV cells 16 hours after activation of p53-VSV expression versus SW480/pRTR cells using anti-VSV and anti-rabbit IgG for ChIP. Experiments were performed in triplicates. Error bars represent +/- SD (n = 3). D. qChIP analysis of HCT116 p53+/+ versus HCT116 p53-/- after 16 hours etoposide treatment using anti-p53 and anti-mouse IgG for ChIP; this ChIP analysis was performed in unicates and measured in triplicates.
Role of CST5 in p53-mediated MET
In order to determine the requirement of CST5 for p53-mediated mesenchymal-epithelial transition (MET) and related processes, such as cellular migration, we silenced CST5 expression in the colorectal cancer cell line SW480/pRTR-p53-VSV using siRNAs (Figure 4A). After treatment with a CST5-directed siRNA CST5 protein was undetectable after induction of ectopic p53 expression (Figure 4A). In the presence of CST5 silencing the p53-mediated repression of SNAIL protein was less pronounced than in cells transfected with control siRNAs (Figure 4A). The same effect was observed on the level of mRNA expression (Figure 4B). Subsequently, we determined whether downregulation of CST5 influences p53-mediated inhibition of cellular migration in a scratch/wound-closure assay and in a Boyden-chamber assay (Figure 4C-4E). When CST5 was silenced by siRNAs, wound closure of SW480/pRTR-p53-VSV cells was generally less suppressed by p53 than in control cells (Figure 4C, 4D). Also in a Boyden-chamber assay a reduction in the inhibition of migration by p53 after treatment with siCST5 was observed (Figure 4E). Taken together, these results show that the induction of CST5 by p53 contributes to the inhibition of migration by p53.
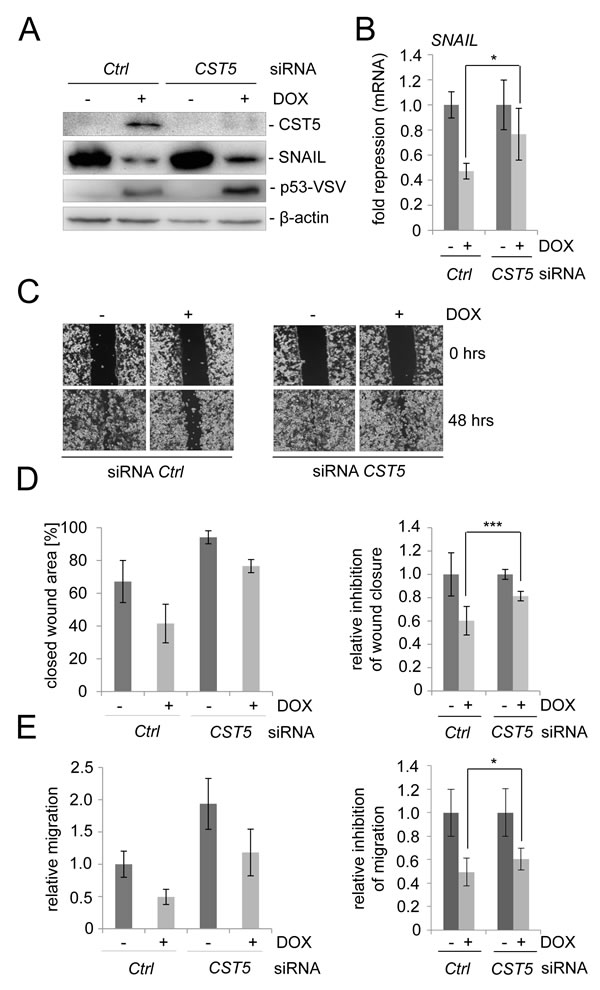
Figure 4: CST5 is relevant for p53-mediated MET. SW480/pRTR-p53-VSV cells were transfected with a CST5-specific siRNA or control oligonucleotide for 72 hours and treated with DOX for 48 hours. A. Western Blot analysis of the indicated proteins. β-actin served as a loading control. B. SNAIL mRNA levels were measured by qPCR analysis after treatment with DOX. Fold changes represent mean values of triplicate analyses of DOX treatment versus vehicle treated cells normalized to β-actin expression. The relative p53-mediated repression of SNAIL is indicated. C., D. Wound-healing assay using Ibidi-Inlets. C. Representative pictures of the wound area obtained 48 hours after scratching. 100 x magnification. D. Left: Average [%] of the closed wound area determined by the final width of the scratch in three independent wells. Right: The relative p53-mediated inhibition of wound-closure. E. Boyden chamber-assay of cellular migration. Left: The relative migration through the filter with the untreated control set as one. Right: The relative p53-mediated inhibition of migration. B. D. E.: Error bars represent +/- SD (n = 3).
Cooperation between p53 and VDR at the CST5 promoter
Recently, the active metabolite of vitamin D3, calcitriol (1,25(OH)2D3), was shown to induce CST5 expression in colorectal cancer cells [25]. To examine whether a combination of p53 activation and treatment with calcitriol may lead to an enhanced CST5 induction, we treated the colorectal cancer cell line SW480/pRTR-p53-VSV with DOX, calcitriol or both agents (Figure 5). As shown before, induction of ectopic p53 expression led to a robust increase in CST5 expression on the mRNA level (Figure 5A). Calcitriol treatment alone resulted in a comparatively minor induction in CST5 expression, whereas the combination of DOX and calcitriol led to significantly higher CST5 mRNA levels when compared to single DOX or calcitriol treatments. Furthermore, CST5 protein induction was more pronounced after combined activation of p53 and the VDR when compared to addition of DOX or calcitriol alone (Figure 5C). In addition, the EMT transcription factor SNAIL displayed a stronger decrease on the mRNA and protein level after a combined treatment with DOX and calcitriol when compared to each stimulus alone (Figure 5B, 5C).
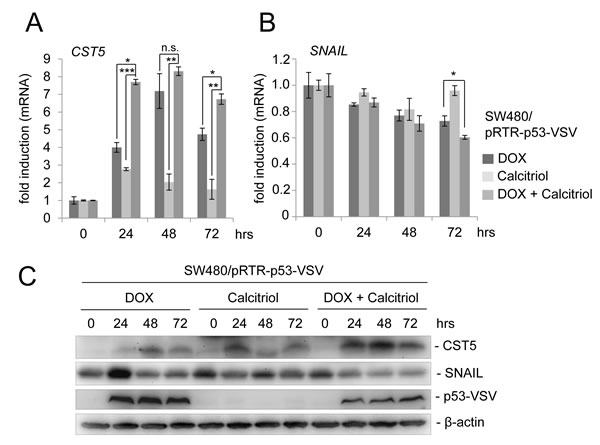
Figure 5: p53 activation in combination with calcitriol treatment enhances CST5 induction. A. CST5- and B. SNAIL mRNA levels were measured by qPCR analysis after treatment with DOX, calcitriol or both agents at the indicated time-points. Fold changes represent mean values of triplicate analyses of DOX / calcitriol / DOX+calcitriol treatment versus vehicle treated cells normalized to β-actin expression. Error bars represent standard deviations. C. Western blot analysis of the indicated proteins in SW480/pRTR-p53-VSV cells. β-actin served as a loading control.
Opposing regulation of CST5 by SNAIL and VDR activation
Since we had observed that VDR activation by exposure to calcitriol results in the induction of CST5 expression and in the repression of SNAIL, we asked whether conversely SNAIL may directly or indirectly lead to repression of CST5 as part of a SNAIL-induced EMT program. Indeed, CST5 mRNA levels decreased by up to 80% after induction of SNAIL by DOX treatment in SW480/pRTR-SNAIL-VSV cells (Figure 6A). To determine whether this repression is mediated directly by SNAIL occupancy at the CST5 promoter, we inspected this region for E-boxes, which may mediate SNAIL binding. We identified several E-boxes upstream of the CST5 promoter and could confirm the direct binding of SNAIL to an E-box, which is located 50 bp upstream of the CST5 promoter, using qChIP analysis (Figure 6B, 6C). The SNAIL binding site in the E-cadherin/CDH1 promoter served as a positive control. Next, we assessed the combined effect of SNAIL activation with calcitriol treatment in SW480/pRTR-SNAIL-VSV cells (Figure 6D). When ectopic SNAIL expression was combined with calcitriol treatment, CST5 was no longer repressed by SNAIL but showed a five-fold induction. Treatment with calcitriol alone led to a robust induction of CST5, whereas SNAIL mRNA levels were not affected. Therefore, induction of CST5 expression by VDR activation is dominant over its repression by SNAIL. As discussed below, this may at least partially explain the effect of vitamin D3 in the treatment of cancer.
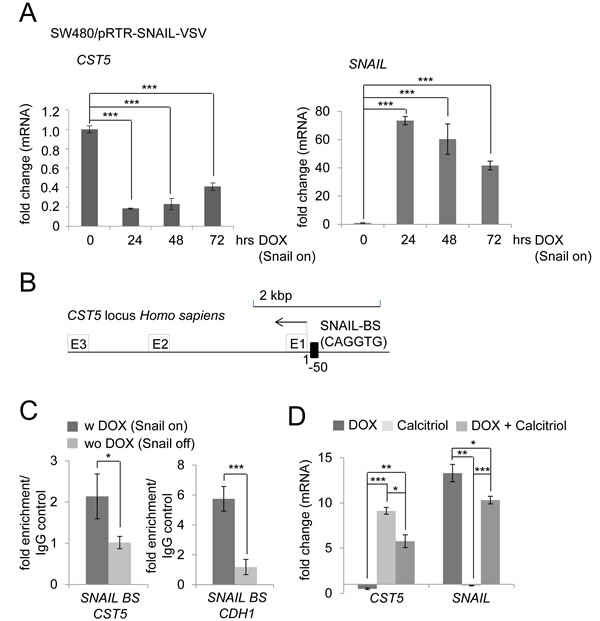
Figure 6: Calcitriol treatment prevents SNAIL-mediated repression of CST5. A. CST5 and SNAIL mRNA levels were measured by qPCR analysis after treatment with DOX at the indicated time-points in SW480/pRTR-SNAIL-VSV cells. Fold changes represent mean values of triplicate analyses after DOX treatment versus vehicle treated cells normalized to β-actin expression. B. Schematic SNAIL binding site 50 bp upstream of the CST5 transcriptional start site. C. qChIP analysis of DLD1/pRTR-SNAIL-VSV cells 24 hours after activation of SNAIL-VSV expression by addition of DOX using anti-VSV and anti-rabbit IgG for ChIP; this ChIP analysis was performed in unicates and measured in triplicates. D. CST5 and SNAIL mRNA levels were measured by qPCR analysis after treatment with DOX, calcitriol or both agents at the indicated time-points in SW480/pRTR-SNAIL-VSV cells. Fold changes represent mean values of triplicate analyses of DOX / calcitriol / DOX+calcitriol treatment versus vehicle treated cells normalized to β-actin expression. Error bars represent standard deviations.
Discussion
Here we identified CST5, which encodes cystatin D, an inhibitor of cysteine cathepsins, as a new p53 target. Furthermore, we showed that CST5 mediates, at least in part, some of the tumor suppressive functions of p53 in colorectal cancer cells (summarized in Figure 7). We showed that CST5 is directly induced by p53, which occupies a canonical p53 binding site upstream of the CST5 promoter. Furthermore, we identified SNAIL as a direct repressor of CST5.
Recently, individual cysteine cathepsins were shown to have diverse roles during tumorigenesis [28, 41]. A study in a mouse model of pancreatic islet cell tumors suggested a multistep process integrating several members of the cysteine cathepsin family that induce angiogenesis, degradation of the basement membrane, tumor growth and invasion [41, 42]. Different members of the cystatin family of protease inhibitors are known to inhibit the protumorigenic actions of cathepsins. For example, cystatin M exerts tumor suppressive effects by inhibiting cell proliferation, migration and invasion in breast cancer cells and is epigenetically silenced in breast cancer [43, 44]. Cystatin D presumably plays an important role as a mediator of tumor suppression in colon cancer cells and is directly induced by the VDR [25] (see also Figure 7). Furthermore, CST5 expression negatively correlates with human colon cancer progression emphasizing its potential tumor suppressive role for colorectal cancer [25]. By showing that p53 directly induces the expression of CST5, we uncovered a new mechanism as to how p53 inhibits tumor progression and growth.
Vitamin D3 is a prohormone, which mainly controls bone mineralization by regulating calcium and phosphate metabolism. During the last years there has been increasing evidence that vitamin D3 deficiency enhances colorectal cancer incidence and mortality [5]. In addition to convincing tumor suppressive effects of vitamin D3 in in vivo and in vitro studies, meta-analyses of clinical trial results also provided evidence for a tumor suppressive effect of vitamin D3 (reviewed in [1]). Furthermore, several studies showed a negative correlation between 25-hydroxy-vitamin D3 (25(OH)D3) serum levels, a marker allowing an overall estimation of the vitamin D3 status, and cancer incidence [45] or overall survival [46], especially for colorectal cancer [20, 47]. Notably, a 30 - 40% reduction of colon cancer risk in patients with high 25(OH)D3 serum levels was observed [14]. This is in line with the notion that the risk of colorectal cancer can be strongly influenced by primary prevention including physical activity or diet [48]. Given the strong evidence that vitamin D3 may decrease the risk for colorectal cancer and the world-wide shortage of vitamin D3 alimentation [49], a feasible approach for reducing colorectal cancer incidence and risk would be to increase vitamin D3 uptake by nutritional sources or its synthesis in the skin by exposure to sunlight. Notably, the VDR is highly expressed in early stages of colorectal tumorigenesis but decreases in advanced stages [50-54]. Also p53 mutation is a late event in colorectal cancer development that mostly occurs during the transition from adenoma to carcinoma [55]. Therefore, a combination of p53-activation and calcitriol treatment to enhance CST5 induction might be more effective in early tumor stages. In case p53 is mutated, a combination of small molecules reactivating mutant p53, such as PRIMA or MIRA-1 [56], together with calcitriol (or an analog to avoid hypercalcemia by high vitamin D3 doses) may lead to the inhibition of cancer progression. Similar approaches combining calcitriol with different known anti-cancer agents, such as doxorubicin or cisplatin, to achieve an enhanced anti-tumor effect have been conducted in several in vitro and in vivo analyses [57-64]. This provides an opportunity for successful anticancer treatment if chemotherapy alone is not effective. Furthermore, we found that the direct induction of CST5 by p53 is, at least in part, required for the induction of MET by p53. Several other p53 target genes, among them the miR-34, miR-200 and miR-15a/16-1 genes, are likely to also contribute to the anti-migratory effects of p53 [65-68].
It has been shown that ectopic expression of CST5 indirectly leads to the repression of SNAIL presumably via the inhibition of the transcriptional activity of β-catenin/TCF complexes [25]. In our study we could show that SNAIL binds upstream of the CST5 promoter to suppress CST5 expression and that this repression can be suppressed by treating the cells with calcitriol. It has been shown before that SNAIL can directly repress VDR which might also contribute to the inhibition of CST5 by SNAIL [69]. However, direct binding of SNAIL to the CST5 promoter has not been demonstrated until now. Since SNAIL-mediated repression of CST5 is abrogated by calcitriol treatment, calcitriol may inhibit tumor progression even in cells that show high SNAIL expression and therefore have a high tumorigenic potential. Others and we have previously shown that SNAIL expression is downregulated by p53 via the induction of miR-34a [65, 66]. Alternatively, p53 may indirectly suppress SNAIL via MDM2 [70]. Therefore, p53 presumably activates CST5 expression by direct and SNAIL-mediated indirect mechanisms, which together form a feed-forward loop.
Taken together, our findings reveal that CST5 represents an important mediator of p53 tumor suppression by mediating MET. Notably, CST5 expression decreases during human colon cancer progression [25]. The possibility to reactivate CST5 expression by dietary vitamin D3 or/and p53 activation suggests that our findings may have potential therapeutic implications.
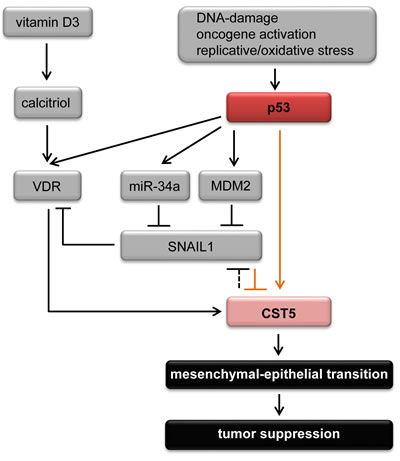
Figure 7: The p53 - vitamin D3 - CST5 regulatory network. Schematic model of CST5 regulation integrating the results of this study (marked with orange arrows) with previous findings (marked with black arrows). Vitamin D3, that is converted to its active metabolite calcitriol, induces CST5 via the vitamin D receptor (VDR) [25]. In addition, p53 can positively influence CST5 expression via different pathways. p53 directly induces VDR which might lead to the induction of CST5 [34]. Furthermore, p53 directly binds to a p53 BS upstream of the CST5 promoter. And third, p53 can indirectly repress SNAIL expression [65, 66, 70] which results in the induction of CST5. Moreover, CST5 expression indirectly leads to the repression of SNAIL by inhibiting the transcriptional activity of β-catenin/TCF complexes [25]. Upon induction, CST5 promotes mesenchymal-epithelial transition to suppress tumor progression and metastasis.
Materials and Methods
Cell culture, conditional expression and treatment
The colorectal cancer cell line SW480 was kept in DMEM and the cell lines DLD1, HCT116, and RKO were cultured in McCoys medium supplemented with 10% FCS (Invitrogen) and 1% Penicillin/Streptavidin at 5% CO2. For conditional expression of p53, SW480 cells were stably transfected with the episomal expression vector pRTR-p53-VSV [66, 71] or the control vector pRTR using Fugene 6 (Roche) and subsequently selected with 2 µg/ml Puromycin (Sigma; stock solution 2 mg/ml in water) for 10 days. Ectopic expression of p53 was induced for the indicated time-points using 100 ng/ml doxycycline (DOX). HEK293T cells were transfected with the pRTR-p53-VSV vector by calcium-phosphate transfection. siRNA against CST5 and the respective control (Applied Biosystems) were transfected at a final concentration of 10 nM using HiPerfect (Qiagen). Nutlin-3a (Sigma) and etoposide were both dissolved in DMSO and used at a final concentration of 10 µM and 20 µM, respectively. Calcitriol (Sigma) was dissolved in EtOH and used at a final concentration of 0.1 µM.
Western blot analysis
SDS-PAGE and Western blotting were performed according to standard protocols. Cells were lysed in RIPA lysis buffer (50 mM Tris/HCl, pH 8.0, 250 mM NaCl, 1% NP40, 0.5% (w/v) Sodium Deoxycholate, 0.1% Sodium Dodecylsulfate, complete mini protease inhibitor tablets (Roche)). Lysates were sonicated and centrifuged at 16.060 g for 15 min, 4°C. 100 µg of whole cell lysate per lane was separated using 10% SDS-Acrylamide gels and transferred on Immobilon PVDF membranes (Millipore). ECL signals were recorded using a CF440 Imager (Kodak). Antibodies used: CST5 (Santa Cruz, sc-46890, 1:200), p21 (Neomarkers, CP-74, 1:1000), SNAIL (R&D Systems, AF3639, 1:250), β-actin (Sigma, A2066, 1:1000).
Quantitative PCR
Total RNA was isolated using the High Pure RNA Isolation Kit (Roche) according to the manufacturer´s instructions. 1 µg of total RNA per sample was used for cDNA-generation using anchored oligo(dT) primers (Ambion Verso Kit). Quantitative PCR (qPCR) was performed by using the Fast SYBR Green Master Mix (Applied Biosystems) and the LightCycler 480 (Roche). Only oligonucleotide pairs resulting in a single peak in the melting curve analysis were used. For sequences of the oligonucleotides used for qPCR see Table 1.
Table 1: Oligonucleotides used for qPCR
Gene |
Fwd 5`- 3` |
Rev 5`- 3` |
CST5 |
CCTCTGCAGGTGATGGCTG [25] |
GGACTTGGTGCATGTGGTTC [25] |
p21 |
GGCGGCAGACCAGCATGACAGATT |
GCAGGGGGCGGCCAGGGTAT |
SNAIL |
GCACATCCGAAGCCACAC |
GGAGAAGGTCCGAGCACA |
VDR |
TTGCCATACTGCTGGACGC [69] |
GGCTCCCTCCACCATCATT [69] |
β-Actin |
TGACATTAAGGAGAAGCTGTGCTAC |
GAGTTGAAGGTAGTTTCGTGGATG |
Immunofluorescence and confocal laser-scanning microscopy
For immunofluorescence analysis SW480/pRTR-p53-VSV cells treated with and without DOX were cultivated on glass cover-slides and fixed in 4% paraformaldehyde/PBS for 10 minutes, permeabilized in 0.2% Triton X 100 for 20 minutes and blocked with 100% FBS for 30 minutes. CST5 (Santa Cruz, sc-46890, dilution 1:50) was used as a primary antibody. α-goat Cy3 (Jackson ImmunoResearch, dilution 1:400) served as secondary antibody. Slides were covered with ProLong Gold antifade (Invitrogen). All stainings were performed without primary antibody as a negative control.
LSM (laser scanning microscopy) images were captured with a confocal microscope (LSM 700, Zeiss) using a Plan Apochromat 20x/0.8 M27 objective, ZEN 2009 software (Zeiss) and the following settings: image size 2048x2048 and 16 bit; Pixel/dwell of 25.2 µs; Pixel Size 0.31 µm; laser power 2%; Master gain 600-1000.
Wound healing assay
SW480/pRTR-p53-VSV cells were seeded into culture inserts (Ibidi, 80241) and transfected with a siRNA against CST5 or the respective control for 48 hours. After 42 hours, p53 expression was induced by adding DOX to the indicated samples. Mitomycin C [10 ng/ml] was added for 2 hours before the scratch was applied. To remove mitomycin C and detached cells, cells were washed twice in HBSS +/+ and medium with or without DOX was added. Cells were allowed to close the wound for 48 hours and images were captured on an Axiovert Observer Z.1 microscope connected to an AxioCam MRm camera using the Axiovision software (Zeiss).
Migration analysis in boyden-chambers
SW480/pRTR-p53-VSV cells were seeded in triplicates into 6-well plates and transfected with siRNA against CST5 or the respective control for 48 hours. For the last 24 hours previous to the analysis, cells were serum deprived (0.1% serum). To analyse migration, 3x105 cells were seeded in the upper chamber of a Boyden Chamber (8 µm pore size; Corning) in serum free medium. After 42 hours, p53 expression was induced by adding 100 ng/ml DOX to the indicated samples. 10% FCS was used as a chemo-attractant and placed in the lower chamber. Cultures were maintained for 48 hours, then non-motile cells at the top of the filter were removed and the cells in the bottom chamber were fixed with methanol and stained with DAPI. Cell numbers in three different fields per condition were enumerated by immunofluorescence microscopy. The average number of cells in three fields per membrane was counted in triplicates. The relative migration is expressed as the value of treated cells to control cells with the control set as one.
Chromatin immunoprecipitation (ChIP) assay
SW480/pRTR-p53-VSV, DLD1/pRTR-SNAIL-VSV and HCT116 cells were cultured as described above. Before cross-linking, SW480/pRTR-p53-VSV and DLD1/pRTR-SNAIL-VSV cells were treated with DOX for 24 hours to induce ectopic expression of p53 or SNAIL, respectively, and HCT116 cells were treated with etoposide for 16 hours to induce endogenous p53 expression. Cross-linking was performed with formaldehyde (Merck) at a final concentration of 1% and terminated for SW480/pRTR-p53-VSV and DLD1/pRTR-SNAIL-VSV after five minutes, for HCT116 cells after ten minutes by addition of glycine at a final concentration of 0.125 M. Cells were harvested using SDS buffer (50 mM Tris pH 8.1, 0.5% SDS, 100 mM NaCl, 5 mM EDTA) and after pelleting resuspended in IP buffer (2 parts of SDS buffer and 1 part Triton dilution buffer (100 mM Tris-HCl pH 8.6, 100 mM NaCl, 5 mM EDTA, pH 8.0, 0.2% NaN3, 5.0% Triton X-100)). Chromatin was sheared by sonication (HTU SONI 130, G. Heinemann) to generate DNA fragments with an average size of 500 bp. Preclearing and incubation was performed with polyclonal VSV (V4888, Sigma) antibody or IgG control (M-7023, Sigma) using A sepharose beads for SW480/pRTR-p53-VSV and DLD1/pRTR-SNAIL-VSV cells for 16 hours as previously described [72]. The monoclonal DO-1 antibody (Santa Cruz, sc-126) in combination with sepharose G beads was used for the HCT116 cells. Washing and reversal of cross-linking was performed as described in [73]. Immunoprecipitated DNA was analyzed by qRT-PCR. For oligonucleotides used for qChIP see Table 2.
Table 2: Oligonucleotides used for qChIP
Fwd 5`- 3` |
Rev 5`- 3` |
|
CST5 p53 BS |
TAAGAGACCGGAAAGGTTGAGA |
AGGGCCTTTGCACTGACTATT |
CST5 SNAIL BS |
GGGGACACCCAAGTAGGATAA |
GGAGCTGGATCTCCCAGAG |
CDH1 SNAIL BS |
TAGAGGGTCACCGCGTCTAT |
TCACAGGTGCTTTGCAGTTC |
ELF1α |
CACACGGCTCACATTGCAT |
CACGAACAGCAAAGCGACC |
BS: binding site
ChIP-sequencing
Chromatin immunoprecipitation was performed as described above. The immunoprecipitated DNA-fragments were quantified using a Bioanalyzer (Agilent Technologies). Libraries were generated using a ChIP-Seq Sample Prep Kit (Illumina Part # 11257047) according to the manufacturer´s instructions and sequenced on a HiSeq 2000 device (Illumina). 101-nt sequence reads were aligned to the hg19 genome assembly. The complete ChIP-Seq results analysis will be published elsewhere (Hünten et al., submitted).
Statistical analysis
A Student´s t-test (unpaired, two-tailed) was used to determine significant differences between two groups of samples. p-values < 0.05 were considered as significant (*: p < 0.05; **: p < 0.01; ***: p < 0.001).
Acknowledgments
We thank Bert Vogelstein (Johns Hopkins Medical School, Baltimore) for providing cell lines. We also thank Helge Siemens and René Jackstadt for kindly providing RNA and ChIP samples obtained after ectopic SNAIL expression. This study was supported by grants to HH from the German-Israeli Science Foundation (GIF), the German Cancer Consortium (DKTK) and the Rudolf-Bartling-Stiftung.
References
1. Feldman D, Krishnan AV, Swami S, Giovannucci E and Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nature reviews cancer. 2014; 14:342-357.
2. Krishnan AV and Feldman D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annual review of pharmacology and toxicology. 2011; 51:311-336.
3. Deeb KK, Trump DL and Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nature reviews cancer. 2007; 7:684-700.
4. Lamprecht SA and Lipkin M. Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nature reviews cancer. 2003; 3:601-614.
5. Pereira F, Larriba MJ and Munoz A. Vitamin D and colon cancer. Endocrine-related cancer. 2012; 19:R51-71.
6. Hummel DM, Thiem U, Hobaus J, Mesteri I, Gober L, Stremnitzer C, Graca J, Obermayer-Pietsch B and Kallay E. Prevention of preneoplastic lesions by dietary vitamin D in a mouse model of colorectal carcinogenesis. The Journal of steroid biochemistry and molecular biology. 2013; 136:284-288.
7. Kovalenko PL, Zhang Z, Yu JG, Li Y, Clinton SK and Fleet JC. Dietary vitamin D and vitamin D receptor level modulate epithelial cell proliferation and apoptosis in the prostate. Cancer prevention research. 2011; 4:1617-1625.
8. Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH and Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009; 30:88-92.
9. Ray R, Banks M, Abuzahra H, Eddy VJ, Persons KS, Lucia MS, Lambert JR and Holick MF. Effect of dietary vitamin D and calcium on the growth of androgen-insensitive human prostate tumor in a murine model. Anticancer research. 2012; 32:727-731.
10. Swami S, Krishnan AV, Wang JY, Jensen K, Horst R, Albertelli MA and Feldman D. Dietary vitamin D(3) and 1,25-dihydroxyvitamin D(3) (calcitriol) exhibit equivalent anticancer activity in mouse xenograft models of breast and prostate cancer. Endocrinology. 2012; 153:2576-2587.
11. Zinser GM, Sundberg JP and Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002; 23:2103-2109.
12. Chung M, Lee J, Terasawa T, Lau J and Trikalinos TA. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Annals of internal medicine. 2011; 155:827-838.
13. Fedirko V, Riboli E, Tjonneland A, Ferrari P, Olsen A, Bueno-de-Mesquita HB, van Duijnhoven FJ, Norat T, Jansen EH, Dahm CC, Overvad K, Boutron-Ruault MC, Clavel-Chapelon F, Racine A, Lukanova A, Teucher B, et al. Prediagnostic 25-hydroxyvitamin D, VDR and CASR polymorphisms, and survival in patients with colorectal cancer in western European ppulations. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2012; 21:582-593.
14. Lee JE, Li H, Chan AT, Hollis BW, Lee IM, Stampfer MJ, Wu K, Giovannucci E and Ma J. Circulating levels of vitamin D and colon and rectal cancer: the Physicians’ Health Study and a meta-analysis of prospective studies. Cancer prevention research. 2011; 4:735-743.
15. Yin L, Ordonez-Mena JM, Chen T, Schottker B, Arndt V and Brenner H. Circulating 25-hydroxyvitamin D serum concentration and total cancer incidence and mortality: a systematic review and meta-analysis. Preventive medicine. 2013; 57:753-764.
16. Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Krstic G, Wetterslev J and Gluud C. Vitamin D supplementation for prevention of cancer in adults. The Cochrane database of systematic reviews. 2014; 6:CD007469.
17. Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M and Gluud C. Vitamin D supplementation for prevention of mortality in adults. The Cochrane database of systematic reviews. 2014; 1:CD007470.
18. Maalmi H, Ordonez-Mena JM, Schottker B and Brenner H. Serum 25-hydroxyvitamin D levels and survival in colorectal and breast cancer patients: systematic review and meta-analysis of prospective cohort studies. European journal of cancer. 2014; 50:1510-1521.
19. Mohr SB, Gorham ED, Kim J, Hofflich H and Garland CF. Meta-analysis of vitamin D sufficiency for improving survival of patients with breast cancer. Anticancer research. 2014; 34:1163-1166.
20. Wei MY, Garland CF, Gorham ED, Mohr SB and Giovannucci E. Vitamin D and prevention of colorectal adenoma: a meta-analysis. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2008; 17:2958-2969.
21. Kupferschmidt K. Uncertain verdict as vitamin D goes on trial. Science. 2012; 337:1476-1478.
22. Holick MF. Vitamin D: A millenium perspective. Journal of cellular biochemistry. 2003; 88:296-307.
23. Jones G, Prosser DE and Kaufmann M. Cytochrome P450-mediated metabolism of vitamin D. Journal of lipid research. 2014; 55:13-31.
24. Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM and Hewison M. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. The Journal of clinical endocrinology and metabolism. 2001; 86:888-894.
25. Alvarez-Diaz S, Valle N, Garcia JM, Pena C, Freije JM, Quesada V, Astudillo A, Bonilla F, Lopez-Otin C and Munoz A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. The Journal of clinical investigation. 2009; 119:2343-2358.
26. Balbin M, Hall A, Grubb A, Mason RW, Lopez-Otin C and Abrahamson M. Structural and functional characterization of two allelic variants of human cystatin D sharing a characteristic inhibition spectrum against mammalian cysteine proteinases. The Journal of biological chemistry. 1994; 269:23156-23162.
27. Freije JP, Abrahamson M, Olafsson I, Velasco G, Grubb A and Lopez-Otin C. Structure and expression of the gene encoding cystatin D, a novel human cysteine proteinase inhibitor. The Journal of biological chemistry. 1991; 266:20538-20543.
28. Gocheva V and Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell cycle. 2007; 6:60-64.
29. Hollstein M, Sidransky D, Vogelstein B and Harris CC. p53 mutations in human cancers. Science. 1991; 253:49-53.
30. Vogelstein B, Lane D and Levine AJ. Surfing the p53 network. Nature. 2000; 408:307-310.
31. el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW and Vogelstein B. Definition of a consensus binding site for p53. Nature genetics. 1992; 1:45-49.
32. Funk WD, Pak DT, Karas RH, Wright WE and Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Molecular and cellular biology. 1992; 12:2866-2871.
33. Menendez D, Inga A and Resnick MA. The expanding universe of p53 targets. Nature reviews Cancer. 2009; 9:724-737.
34. Maruyama R, Aoki F, Toyota M, Sasaki Y, Akashi H, Mita H, Suzuki H, Akino K, Ohe-Toyota M, Maruyama Y, Tatsumi H, Imai K, Shinomura Y and Tokino T. Comparative genome analysis identifies the vitamin D receptor gene as a direct target of p53-mediated transcriptional activation. Cancer research. 2006; 66:4574-4583.
35. Kommagani R, Payal V and Kadakia MP. Differential regulation of vitamin D receptor (VDR) by the p53 Family: p73-dependent induction of VDR upon DNA damage. The Journal of biological chemistry. 2007; 282:29847-29854.
36. Saramaki A, Banwell CM, Campbell MJ and Carlberg C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic acids research. 2006; 34:543-554.
37. Chen H, Reed G, Guardia J, Lakhan S, Couture O, Hays E and Chandar N. Vitamin D directly regulates Mdm2 gene expression in osteoblasts. Biochemical and biophysical research communications. 2013; 430:370-374.
38. Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, Shiff I, Kogan I, Shay M, Kalo E, Blandino G, Simon I, Oren M and Rotter V. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer cell. 2010; 17:273-285.
39. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998; 282:1497-1501.
40. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N and Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844-848.
41. Joyce JA and Hanahan D. Multiple roles for cysteine cathepsins in cancer. Cell cycle. 2004; 3:1516-1619.
42. Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M and Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer cell. 2004; 5:443-453.
43. Schagdarsurengin U, Pfeifer GP and Dammann R. Frequent epigenetic inactivation of cystatin M in breast carcinoma. Oncogene. 2007; 26:3089-3094.
44. Zhang J, Shridhar R, Dai Q, Song J, Barlow SC, Yin L, Sloane BF, Miller FR, Meschonat C, Li BD, Abreo F and Keppler D. Cystatin m: a novel candidate tumor suppressor gene for breast cancer. Cancer research. 2004; 64:6957-6964.
45. Giovannucci E. Vitamin D status and cancer incidence and mortality. Advances in experimental medicine and biology. 2008; 624:31-42.
46. Ng K, Meyerhardt JA, Wu K, Feskanich D, Hollis BW, Giovannucci EL and Fuchs CS. Circulating 25-hydroxyvitamin d levels and survival in patients with colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008; 26:2984-2991.
47. Garland CF, Garland FC, Gorham ED, Lipkin M, Newmark H, Mohr SB and Holick MF. The role of vitamin D in cancer prevention. American journal of public health. 2006; 96:252-261.
48. Tomasetti C and Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347:78-81.
49. Holick MF and Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. The American journal of clinical nutrition. 2008; 87:1080S-1086S.
50. Anderson MG, Nakane M, Ruan X, Kroeger PE and Wu-Wong JR. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer chemotherapy and pharmacology. 2006; 57:234-240.
51. Cross HS, Bareis P, Hofer H, Bischof MG, Bajna E, Kriwanek S, Bonner E and Peterlik M. 25-Hydroxyvitamin D(3)-1alpha-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids. 2001; 66:287-292.
52. Matusiak D, Murillo G, Carroll RE, Mehta RG and Benya RV. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1{alpha}-hydroxylase in normal and malignant human colon. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005; 14:2370-2376.
53. Sheinin Y, Kaserer K, Wrba F, Wenzl E, Kriwanek S, Peterlik M and Cross HS. In situ mRNA hybridization analysis and immunolocalization of the vitamin D receptor in normal and carcinomatous human colonic mucosa: relation to epidermal growth factor receptor expression. Virchows Archiv : an international journal of pathology. 2000; 437:501-507.
54. Larriba MJ and Munoz A. SNAIL vs vitamin D receptor expression in colon cancer: therapeutics implications. British journal of cancer. 2005; 92:985-989.
55. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759-767.
56. Selivanova G and Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007; 26:2243-2254.
57. Abe J, Nakano T, Nishii Y, Matsumoto T, Ogata E and Ikeda K. A novel vitamin D3 analog, 22-oxa-1,25-dihydroxyvitamin D3, inhibits the growth of human breast cancer in vitro and in vivo without causing hypercalcemia. Endocrinology. 1991; 129:832-837.
58. Abe-Hashimoto J, Kikuchi T, Matsumoto T, Nishii Y, Ogata E and Ikeda K. Antitumor effect of 22-oxa-calcitriol, a noncalcemic analogue of calcitriol, in athymic mice implanted with human breast carcinoma and its synergism with tamoxifen. Cancer research. 1993; 53:2534-2537.
59. Cho YL, Christensen C, Saunders DE, Lawrence WD, Deppe G, Malviya VK and Malone JM. Combined effects of 1,25-dihydroxyvitamin D3 and platinum drugs on the growth of MCF-7 cells. Cancer research. 1991; 51:2848-2853.
60. Kota BP, Allen JD and Roufogalis BD. The effect of vitamin D3 and ketoconazole combination on VDR-mediated P-gp expression and function in human colon adenocarcinoma cells: implications in drug disposition and resistance. Basic & clinical pharmacology & toxicology. 2011; 109:97-102.
61. Opolski A, Wietrzyk J, Siwinska A, Marcinkowska E, Chrobak A, Radzikowski C and Kutner A. Biological activity in vitro of side-chain modified analogues of calcitriol. Current pharmaceutical design. 2000; 6:755-765.
62. Pelczynska M, Switalska M, Maciejewska M, Jaroszewicz I, Kutner A and Opolski A. Antiproliferative activity of vitamin D compounds in combination with cytostatics. Anticancer research. 2006; 26:2701-2705.
63. Ravid A, Rocker D, Machlenkin A, Rotem C, Hochman A, Kessler-Icekson G, Liberman UA and Koren R. 1,25-Dihydroxyvitamin D3 enhances the susceptibility of breast cancer cells to doxorubicin-induced oxidative damage. Cancer research. 1999; 59:862-867.
64. Siwinska A, Opolski A, Chrobak A, Wietrzyk J, Wojdat E, Kutner A, Szelejewski W and Radzikowski C. Potentiation of the antiproliferative effect in vitro of doxorubicin, cisplatin and genistein by new analogues of vitamin D. Anticancer research. 2001; 21:1925-1929.
65. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ and Yook JI. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. The Journal of cell biology. 2011; 195:417-433.
66. Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U and Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell cycle. 2011; 10:4256-4271.
67. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, Liu M, Chen CT, Yu D and Hung MC. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nature cell biology. 2011; 13:317-323.
68. Shi L, Jackstadt R, Siemens H, Li H, Kirchner T and Hermeking H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer research. 2014; 74:532-542.
69. Palmer HG, Larriba MJ, Garcia JM, Ordonez-Moran P, Pena C, Peiro S, Puig I, Rodriguez R, de la Fuente R, Bernad A, Pollan M, Bonilla F, Gamallo C, de Herreros AG and Munoz A. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nature medicine. 2004; 10:917-919.
70. Lim SO, Kim H and Jung G. p53 inhibits tumor cell invasion via the degradation of snail protein in hepatocellular carcinoma. FEBS letters. 2010; 584:2231-2236.
71. Jackstadt R, Roh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A and Hermeking H. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. The Journal of experimental medicine. 2013; 210:1331-1350.
72. Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G and Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell cycle. 2007; 6:1586-1593.
73. Amati B, Frank SR, Donjerkovic D and Taubert S. Function of the c-Myc oncoprotein in chromatin remodeling and transcription. Biochimica et biophysica acta. 2001; 1471:M135-145.