
INTRODUCTION
Arsenic trioxide (As2O3) has been accepted as a standard treatment for the patients with acute promyelocytic leukemia (APL) in China [1], which has also been applied for the treatment of other forms of cancers [2–3]. Although, As2O3 has been studied in vivo and in vitro for many years, however, there is a little information regarding the anticancer effect of the intermediate metabolites of As2O3, namely, monomethylarsonous acid (MMAIII) and dimethylarsinous acid (DMAIII) in APL patients receiving As2O3 treatment.
Generally speaking, liver is the major site for arsenic methylation, where As2O3 is metabolically transformed into trivalent mono- and di-methylated metabolites (i.e., MMAIII and DMAIII) by arsenicmethyltransferase (AS3MT) [4]. Finally, it is excreted into urine mostly in the form of pentavalent methylated metabolites such as monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV) [5–6]. In fact, methylated pentavalent arsenic species; MMAV and DMAV could frequently be found in the bloodstream and urine of APL patients after injection of As2O3 [7–8]. Wang et al. (2004) has reported that the highly toxic trivalent arsenic metabolite, MMAIII was found in the urine of APL patients receiving As2O3 injection [8]. Likewise, it was also found in human saliva and urine following exposure to iAsIII in Inner Mongolia [9].
Toxicological studies have recently indicated that trivalent arsenic intermediate metabolites (i.e., MMAIII and DMAIII) are more toxic as compared to their precursor; arsenite (iAsIII) [10], and these trivalent arsenicals have also shown to display a much higher degree of cytotoxicity than the corresponding pentavalent species. However, little is known about the molecular role of these active trivalent intermediate arsenicals in the clinical remission of APL patients. Chen et al. (2003) has reported that methylated MMAIII may contribute to arsenic-induced apoptosis in leukemia and lymphoma cells [11], however, no detailed mechanism has been investigated so far. Based on such observations, it can be suggested that the trivalent methylated arsenicals may contribute to the therapeutic effects in APL.
APL is characterized by a reciprocal translocation between chromosomes 15 and 17, t(15;17), expressing the fusion of promyelocytic leukemia (PML) gene to the retinoic acid receptor α (RARα) gene resulting in the production of a PML-RARα fusion protein [12], this fusion protein may block the differentiation of hematopoietic progenitor cells [13–14]. On the other hand, PML-RARα regulated adapter molecule-1 (PRAM-1), an adaptor protein which is expressed and regulated during normal myelopoiesis is found to be down-regulated by PML-RARα fusion protein in NB4 cells, suggesting important contribution of this protein in signaling pathway involved in differentiation of leukemia promyelocytes [15].
Successful clinical remission in APL patients has been obtained with all-trans retinoic acid (ATRA) and As2O3 treatment [16–17]. Zhang et al. has reported that As2O3 may directly bind to cysteine residues in the RING finger-B Box-Coiled Coil (RBCC) domain of PML-RARα fusion protein, which results in enhanced SUMOylation and degradation of PML-RARα fusion protein, promoting cell differentiation leading to clinical remission [18]. Moreover, binding of trivalent arsenicals to the zinc finger domains of DNA repair proteins and cysteine rich metallothionein (MT) has extensively been studied recently [19]. Although, at normal physiological conditions, trivalent arsenicals have shown to provoke zinc release from the zinc finger domain of the xeroderma pigmentosum group A protein (XPA), the methylated metabolites; MMAIII and DMAIII were found to have stronger binding affinity to zinc finger proteins as compared to iAsIII [20]. In fact, the studies that have shown binding of iAsIII to zinc finger peptides were mostly performed using apo-Zn finger proteins [18–19], thus, more studies are needed to evaluate that whether iAsIII can also bind to Zn finger peptides or proteins (i.e., with Zn2+) even under normal physiological conditions.
Scientists have mainly focused on the effect of As2O3 on APL [21–22], showing a minimal concern to reveal the effects of the intermediates MMAIII and DMAIII on treatment of APL patients. Based on the above observations, we are interested in whether the degradation of PML-RARα fusion is related to the binding of As2O3 and its intermediate metabolites. Thereby, in the current investigation we opt to determine the effect of the three arsenicals, including iAsIII (represent the As2O3) and its two intermediate metabolites (i.e., MMAIII/DMAIII) on induction of cellular differentiation and apoptosis. In addition, we further compared the binding affinity of iAsIII and its intermediate metabolites to the recombinant PML-zinc finger protein to reveal the probable association between the arsenic binding to protein and PML degradation. Interestingly, our present work demonstrated that iAsIII predominantly activates cells differentiation, while its intermediate MMAIII and DMAIII specifically induce apoptosis. Moreover, it was found that binding of arsenic intermediate metabolites to PML-zinc finger protein do not induce the PML-protein degradation.
RESULTS
The proliferation of NB4 cells after exposure to iAsIII and MMAIII and DMAIII
As2O3 in body is commonly present in its hydrolyzed form (e.g., As2O3 → iAsIII) (Fig. 1A), thereby, in current study, iAsIII will represent As2O3. In humans, iAsIII has found to be rapidly methylated to trivalent mono- and di-methylated metabolites (i.e., MMAIII and DMAIII) by arsenic methyltransferse (AS3MT) in liver (Fig. 1B), however, little is known about the therapeutic effects of intermediate metabolites of iAsIII on APL patients receiving As2O3 treatment. Here, we determined the effects of iAsIII, MMAIII and DMAIII on the NB4 cell proliferation, where MMAIII and DMAIII were found to be more cytotoxic to NB4 cells as compared to iAsIII (Fig. 1C). In addition, we also determined the cell viability by trypan blue exclusion assay, the data of cell viability were consistent with MTT assay (data not shown). On the other hand, according to the dosage of As2O3 (0.15 mgAs/kg) in clinical treatment [1, 16–18] as well as concentration of arsenic species in blood samples [23], we selected 1 μM dose of arsenic compounds for our subsequent experiments.
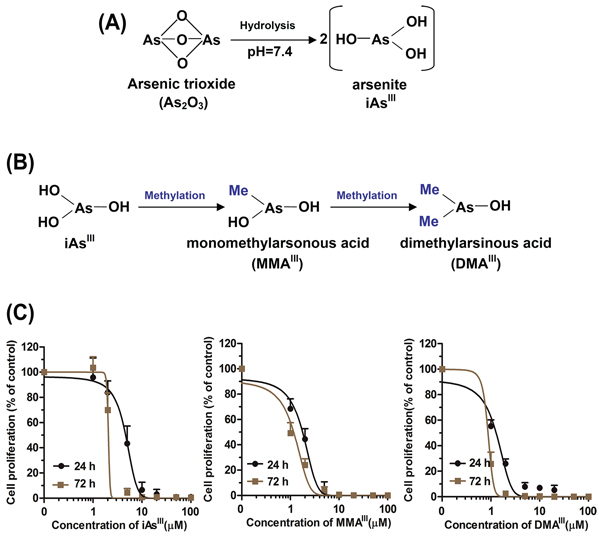
Figure 1: Proposed metabolic pathway for arsenic trioxide in vivo and proliferation of NB4 cells after exposure to arsenicals. Arsenic trioxide is commonly hydrolyzed into arsenite (iAsIII) at the physiological conditions A. iAsIII can immediately be methylated to MMAIII and DMAIII in body B. Cells were exposed to various concentrations of arsenicals for 24 and 72 h, and then viability was determined by MTT assay C. Data are expressed as mean values ± standard deviation (n = 4).
Effects of arsenicals on NB4 cells differentiation or on PML-RARα fusion protein degradation as well as PML nuclear bodies (PML-NBs) formations
In order to better understand the role of three arsenic species in NB4 cells, cellular differentiation and PML-RARα fusion protein degradation were determined at 24 or 72 h following exposure to arsenicals (Fig. 2).
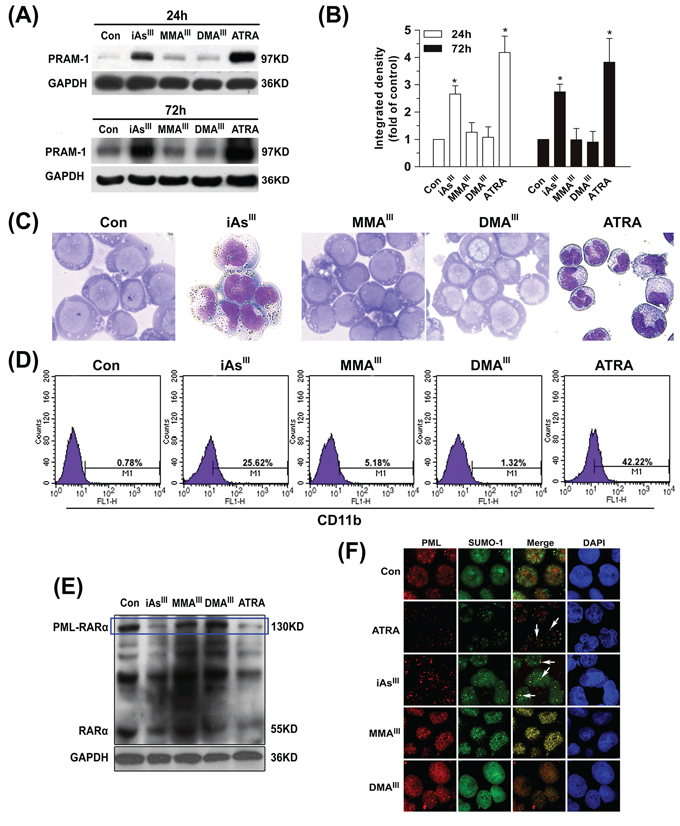
Figure 2: Effects of arsenicals on differentiation of NB4 cells and PML-RARα fusion protein degradation. NB4 cells were exposed to 1 μM of arsenicals for 24 and 72 h to determine the changes in PRAM-1 protein A. and quantified by Image J public domain software B. Additionally, NB4 cells were stained with Wright-Giemsa C. and the expression of CD11b in NB4 cells were determined by flow cytometry D. Degradation of PML-RARα fusion protein in NB4 cells were determined by western blot with RARα antibody E. For immunofluorescence, NB4 cells were double-labeled with PML (red) and SUMO-1 (green) after exposure to 1 μM arsenicals for 12 h. Blue fluorescence indicates cell nucleus. Cells were imaged with a laser scanning confocal microscope F. ATRA was used as positive control in the present study. Asterisks indicate a significant difference from the untreated control group at *p < 0.05. Arrow indicates the PML-NBs.
The expression of PRAM-1 was initially found to be very low in NB4 cells, however, its expression markedly increased as early as 6 h after exposure to iAsIII (Fig. S1A). Moreover, expression of PRAM-1 was observed to be further increased after 24/72 h of exposure to iAsIII and/or ATRA. However, there were no appreciable changes observed after exposure to either MMAIII or DMAIII (Fig. 2A–2B). Moreover, the results of Wright-Giemsa stain and CD11b clearly showed cellular differentiation induced by both iAsIII and ATRA, however, no cellular differentiation in NB4 cells was observed after exposure to MMAIII or DMAIII at 1 μM (Fig. 2C, 2D). Likewise, iAsIII (or ATRA)-treated cells displayed typical nuclear morphology of differentiation (i.e., polylobular nuclei) as compared to control, while MMAIII and DMAIII-treated cells have shown apoptotic chromosome condensation and fragmented nuclei (Fig. S1B).
PML-RARα fusion protein degradation in NB4 cells was observed to occur as early as 6 h after exposure to iAsIII and ATRA (Fig. S1C). However, MMAIII or DMAIII could not degrade PML-RARα fusion protein at any time point (Fig. 2E), suggesting that these metabolites may not have any effect on NB4 cell differentiation and PML-RARα protein degradation. In order to confirm whether massive apoptosis might inhibit their (i.e., MMAIII and DMAIII) potential ability of promoting cellular differentiation, we further used lower doses of methylated arsenicals (e.g., 0.1 and 0.5 μM) to determine NB4 cell differentiation (Fig. S1). Our results clearly found that although both methylated MMAIII and DMAIII did not induce apoptosis significantly at lower doses, (Fig. S1E), they were also unable to induce cell differentiation (Fig. 1D).
It is known that iAsIII and ATRA are able to redirect wild-type PML from aberrant subnuclear distribution in APL cells to its normal localization [24]. Thereby, trying to understand whether the methylated arsenic species could induce PML-NBs formation/SUMOylation, the co-localization of PML with small ubiquitin-like modifier 1 (SUMO-1) in NB4 cells were examined after exposure to three arsenic species. We found that iAsIII and ATRA remarkably induced a characteristic redistribution of PML together with SUMO-1 to form the nuclear speckled structures as compared to control, while the treatment with either MMAIII or DMAIII showed no effect (Fig. 2F), suggesting that the methylated trivalent arsenic species can not trigger the relocalization of the PML-NBs in NB4 cells.
MMAIII or DMAIII have strong effect on induction of apoptosis in NB4 cells
Based on the above results, we hypothesized that MMAIII and DMAIII may contribute to induce apoptosis in NB4 cells. As anticipated, we found that MMAIII and DMAIII (1 μM) significantly induced apoptosis in NB4 cells, while no appreciable effect was observed after exposure to iAsIII at 1 μM (Fig. 3A–3D), where the toxicity of DMAIII was observed to be greater than MMAIII.
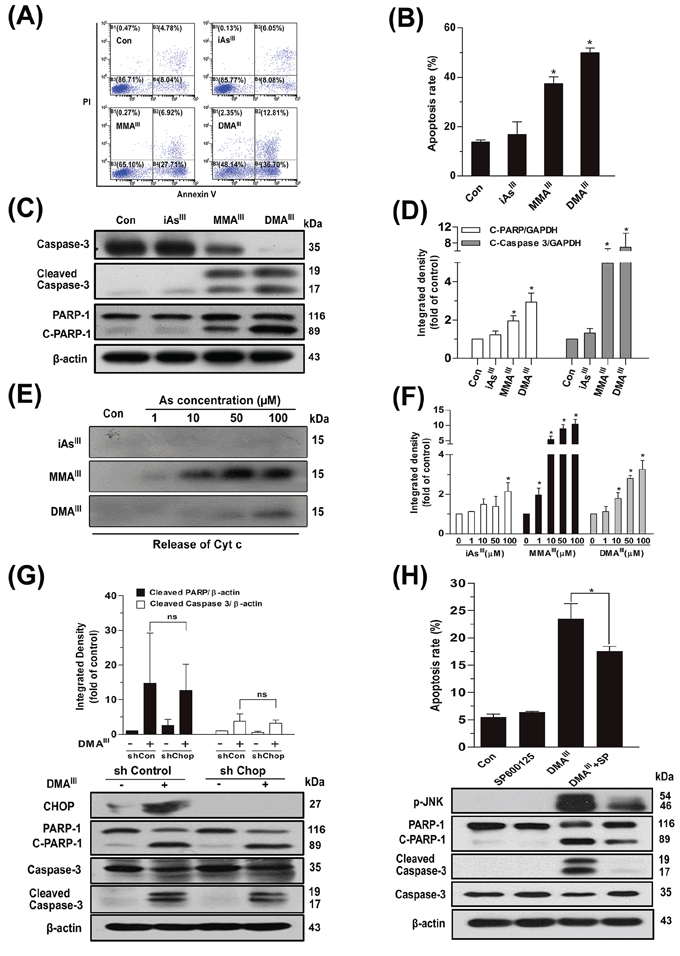
Figure 3: Effect of arsenicals on induction of apoptosis in NB4 cells or release of Cyt c from intact mitochondria. Apoptosis in NB4 cells was determined by flow cytometry following exposure to 1 μM of arsenicals for 24 h A-B. Apoptosis-related proteins were determined by western blot C. and quantified by Image J public domain software D. Isolated pure rat liver mitochondria (200 μg protein/ml) were incubated with three arsenicals with indicated concentration for 30 min, and the release of Cyt c in supernatant was determined by western blot E, F. Apoptosis was determined by knockdown of CHOP by shRNA G. or inhibition of p-JNK using its specific inhibitor SP600125 H. in NB4 cells following exposure to DMAIII. n.s. (not significant).
On the other hand, apoptosis related proteins such as cleaved caspase-3 and poly (ADP-ribose) polymerase (PARP) were clearly observed at early time following exposure to MMAIII and DMAIII, which were found to increase in a time- and dose-dependent manner (Fig. S2). Similarly, proapoptotic protein, Bcl-2-associated X protein (Bax) and/or anti-apoptotic protein, B-cell lymphoma 2 (Bcl-2), both were found to significantly decrease in cytoplasm and mitochondria as early as 6 to 12 h after exposure to MMAIII and DMAIII, and continuously decreased till 24 h (Fig. S3A/C). Especially, Cyt c was markedly found to be released from mitochondria in the cytoplasm of NB4 cells, and increased with exposure time of MMAIII and DMAIII as compared to iAsIII (Fig. S3B/D). Likewise, the mitochondrial membrane potential (ΔΨm) was also significantly reduced by MMAIII and DMAIII, suggesting that these trivalent intermediates indeed contributed in the induction of apoptotic cell death (Fig. S3E).
To reveal the mechanism underlying the release of Cyt c, pure rat liver mitochondria were incubated with three arsenic species for 30 min. Surprisingly, iAsIII was unable to release Cyt c from intact mitochondria (even at 100 μM), while methylated MMAIII showed to have potential effect on the release of Cyt c from intact mitochondria, which was observed to be much stronger than DMAIII (Fig. 3E–3F). Similarly, MMAIII can induce opening of mPTP even at low concentrations; however, DMAIII only showed effect at high concentration, while no effects were observed with iAsIII (data not shown). However, we found that DMAIII–induced increase in cytoplasmic Cyt c (Fig. S3D), was not consistent with the data of intact mitochondria (Fig. 3F). Thereby, these apparently contradictory results call into question the signaling pathway that could be involved in DMAIII–induced apoptosis.
We hypothesized that DMAIII may target endoplasmic reticulum and may show its effect via ER-related pathways. Interestingly, we found that the phosphorylated protein kinase-like endoplasmic reticulum kinase (PERK) was significantly activated in NB4 cells after exposure to DMAIII as compared to the other two arsenic species; iAsIII and MMAIII (Fig. S4A), suggesting DMAIII as a potent inducer of ER stress. Similarly, a significant increase in the expressions of p-PERK and α submits of eukaryotic translation initiation factor-2 (elF2α), or downstream activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) were observed following exposure to DMAIII (Fig. S4B). Likewise, DMAIII was also capable of inducing JUN N-terminal kinase (JNK) phosphorylation in NB4 cells and triggering JNK dependent ER-stress-induced apoptosis (Fig. S4C–S4D). Moreover, after the knockdown of CHOP, no appreciable difference was observed in apoptosis related proteins such as PARP and caspase-3 between the shCHOP and shControl group (Fig. 3G), however, inhibition of p-JNK significantly prevented cell death in NB4 cells after exposure to DMAIII (Fig. 3H).
Determination of formation of nuclear bodies in PML and PML-RARα-transfected cells after exposure to arsenicals
As2O3 can induce formation of PML-NBs, SUMOylation of PML-RARα (or PML) protein in APL cells [25]. In the current study, we found that MMAIII and DMAIII could not induce relocalization of PML-NBs, SUMOylation and PML-RARα degradation in NB4 cells (Fig. 2). Thus, HEK293T and HeLa cells were transiently transfected with PML or PML-RARα genes to confirm the effect of the three arsenicals on PML/PML-RARα protein degradation, relocalization of PML-NBs, SUMOylation. We found that the PML and PML-RARα successfully overexpressed in both cell lines; HEK293T and HeLa cells (Fig. S5A–S5B).
Furthermore, methylated MMAIII and DMAIII were found unable to degrade the PML or PML-RARα proteins in HEK293T and HeLa cells, however, iAsIII showed to induce complete degradation of these proteins (Fig. 4A–4B). These results were found be consistent with our above data obtained using NB4 cells (Fig. 2E). Similarly, re-localization of PML-NBs in PML-HEK293T cells or PML-RARα-HeLa cells was examined after exposure to the three arsenicals. The green fluorescence indicates PML, while the red fluorescence indicates SUMO-1. As anticipated, only iAsIII induced a characteristic redistribution of PML together with SUMO-1 to form the nuclear speckled structures as compared to control (Fig. 4C–4D), while the NBs are dispersed as microspeckles in PML-HEK293T cells or PML-RARα-HeLa following exposure to methylated arsenicals (Fig. 4C–4D), suggesting that MMAIII and DMAIII have no effect on PML protein degradation and PML-NBs formation.
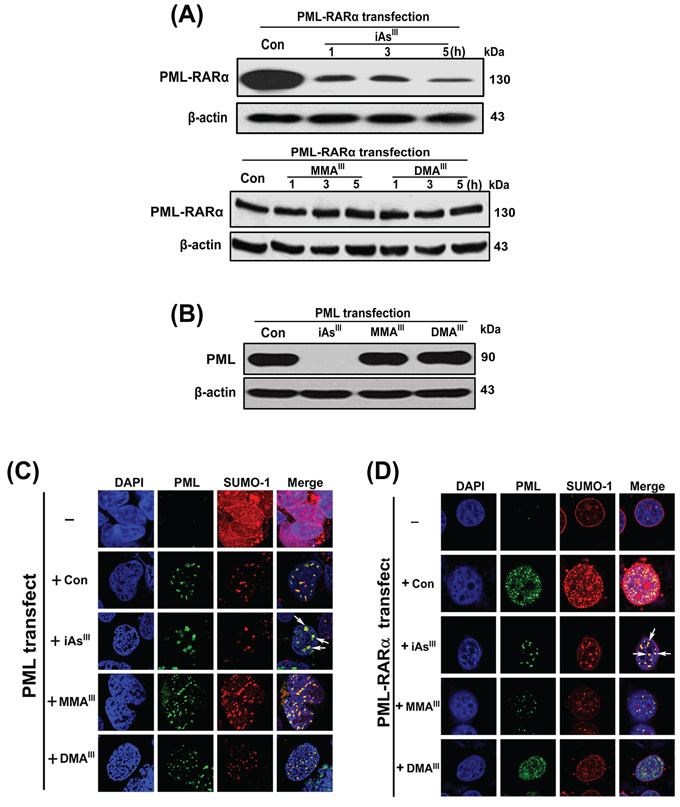
Figure 4: Examination of PML-NBs formation or PML and PML-RARα proteins degradation in HEK293T and HeLa cells after exposure to arsenicals. Degradation of PML-RARα fusion protein was examined in PML-RARα-transfected HeLa cells following exposure to 4 μM of arsenicals at indicated time points A. Likewise, degradation of PML protein was determined in PML-HEK293T cells following exposure to 4 μM of arsenicals for 6 h B. Formation of PML-NBs in PML or PML-RARα overexpressed HEK293T C. or HeLa cells D. were determined after exposure to arsenicals for 6 h. Cells were double-labeled with PML (green) and SUMO-1 (red). Blue fluorescence indicates cell nucleus. Arrow indicates the PML-NBs.
Binding of arsenic species with PML-Zinc finger protein and other proteins in NB4 cells
It has been indicated that iAsIII directly binds to cysteine residues of zinc fingers located within the RBCC domain of PML-RARα resulting in degradation of PML-RARα fusion protein. Thus, to address the binding affinity of the three arsenic species to the cellular proteins, we used a gel filtration GS-220 column and determined the arsenic-binding proteins and unbound arsenicals in NB4 cells after exposure to 1 μM of arsenicals for 24 h.
We detected few arsenic-binding proteins along with a large amount of free iAsIII (unbound-form) in NB4 cells after exposure to iAsIII (Fig. 5A). However, in the MMAIII treated cells, most of the arsenic was detected in protein-binding form and no free MMAIII was observed. Moreover, only a small amount of arsenic was found to bind to proteins, and most of the arsenic was detected as DMAV following exposure to DMAIII (Fig. 5A). Thus, these data indicated that MMAIII has much stronger binding affinity to SH-group of proteins as compared to iAsIII or DMAIII. Additionally, MMAIII and DMAIII were more efficiently taken up by NB4 cells than the precursor iAsIII (Fig. 5B), where, most of MMAIII was found to bind to proteins in whole cell extract (Fig. 5C).
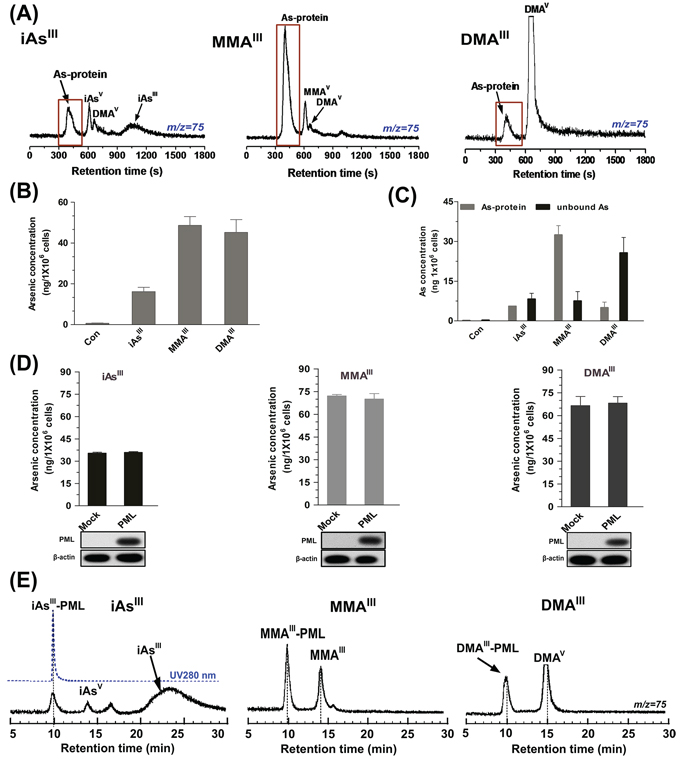
Figure 5: Analysis of arsenic distribution and concentration in NB4 cells after exposure to three arsenic compounds. NB4 cells were treated with 1 μM of arsenicals for 24 h. Arsenic species in supernatant were determined by ICP-MS on GS-220 column with 50 mM ammonium acetate (pH6.5) at the flow rate of 0.6 mL/min A. Arsenic accumulation in NB4 cells B. or concentration of arsenic-binding proteins and free arsenic (unbound) C. were measure by ICP-MS after washing with HNO3 and H2O2. D. Arsenic accumulation in Mock or PML transfected HEK293T cells after exposure to 2 μM arsenicals for 6 h was measure by ICP-MS after washing with HNO3 and H2O2. E. Binding of three arsenic species to recombinant PML-R protein (25 μg/mL) was determined by ICP-MS on GS-220 column with 50 mM ammonium acetate (pH7.0) at the flow rate of 0.8 mL/min. Arsenic was monitored at m/z 75.
We were further interested in detecting that whether overexpression of PML in HEK293T cells can also increase arsenic accumulations, vector or PML transfected HEK293T cells were exposed to 2 μM of three arsenicals for 6 h. Interestingly, no significant differences were noted in the results between the PML and Mock-transfected HEK293T cells after exposure to the three arsenicals (Fig. 5D). Furthermore, we evaluated the binding affinity of the three arsenicals to the zinc fingers in the RBCC domain of PML-RARα fusion protein. For this, the recombinant zinc finger protein PML-Ring (containing Zn2+) was incubated with the three arsenicals for 30 min in Tris-HNO3 buffer (pH7.4). Surprisingly, inorganic iAsIII was found unable to bind to PML-R at physiological condition (pH7.4), while MMAIII exhibited high binding affinity for recombinant PML-R (Fig. 5E). Although DMAIII was capable of binding to PML-R, it has exhibited less efficiently as compared to MMAIII. Here, we concluded that although intermediate metabolites have a high binding affinity for zinc finger proteins, they could not degrade PML or PML-RARα proteins in NB4 cells or PML-transfected HEK293T cells, suggesting that binding to zinc finger proteins may not be a requirement for PML-RARα degradation.
Determination of PML proteins SUMOylation and ubiquitination after exposure to arsenicals
In order to confirm the effect of three arsenicals on modification of PML proteins, PML-HEK293T cells were exposed to three arsenicals in a concentration and time dependent manner. Cells were lysed with RIPA buffer, and then separated into soluble (Supernatant) and insoluble fractions (Pellet). Interestingly, iAsIII was observed to induce a shift of PML protein from supernatants to insoluble fractions in a concentration-dependent manner, while no changes were observed following exposure to MMAIII and DMAIII (Fig. 6A). Meanwhile, this shift of PML was found to occur as early as 30 min by iAsIII and then increased with exposure time. However, the result of methylated metabolites was found to be in contrast to this (Fig. 6B/Fig. S5C) i.e., having no effect even with the increase in the exposure time. These data indicated that only iAsIII can induce PML modification.
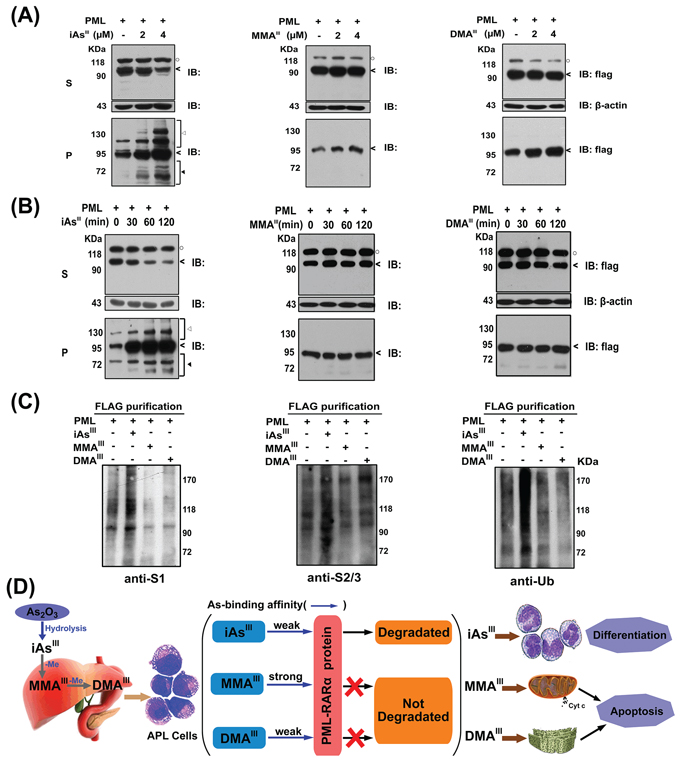
Figure 6: Changes in PML protein in soluble (S) and insoluble fractions (P) of PML-HEK293T cells by exposure to arsenicals in time and dose-dependent manners. PML overexpressed-HEK293T cells were exposed to 2 μM of arsenicals in time A. or dose-dependent B. manners. Arsenic-treated cells were lysed in RIPA buffer, centrifuged into supernatant (S) and pellet (P) fractions, and then determine the PML proteins (<), modified PML-proteins (◄) and degraded PML-protein (◄) by western blot analysis. (○) indicates a non-specific protein in supernatant. C. PML protein was immunoprecipitated from extract of PML overexpressed-HEK293T cells by anti-FLAG-M2 beads after exposure to 2 μM three arsenic species for 2 h. SUMO-1, 2/3 and ubiquitin antibodies were used to determine the modification of PML proteins (anti-S1: SUMO-1; anti-S2/3: SUMO-2/3; anti-Ub: ubiquitin). D. Proposed mechanism underlying the arsenic trioxide and its methylated metabolites-induced cell differentiation and apoptosis in NB4 cells.
Based on above observation, we hypothesize that methylated arsenic species probably have no effect on induction of SUMOylation/ubiquitination of PML proteins. Thus, SUMOylation/ubiquitination of PML proteins was determined in PML-HEK293T cells by immunoprecipitation using specific antibodies after exposure to three arsenic species (Fig. 6C). As anticipated, PML proteins were modified to SUMO-1, SUMO-2/3, and ubiquitin conjugations after exposure to iAsIII, but no changes were observed following exposure to both MMAIII and DMAIII, suggesting that methylated arsenic species could not induce PML proteins SUMOylation as well as ubiquitination.
DISCUSSION
In the present study, we clearly demonstrated that iAsIII and its intermediate metabolites exert different roles in NB4 cells; especially MMAIII and DMAIII have shown to have no effect on NB4 cell differentiation, however, they showed to have strong effect on inducing NB4 cell apoptosis compared with iAsIII. This clearly suggests that the formation of these methylated metabolites may probably contribute to the antileukemic effect in APL patient after As2O3 treatment through other pathways. In fact, liver is considered to be the major organ for arsenic methylation, where As2O3 is rapidly methylated to different intermediates such as trivalent mono- and dimethylated arsenic species by AS3MT, and then redistributed to body fluids and other organs [6]. However, Khaleghian et al. (2014) has recently reported that NB4 cells are capable of metabolizing As2O3 into methylated metabolites, suggesting that the production of these methylated metabolites may contribute to the therapeutic effect of As2O3 in APL treatment [26].
In fact, DMAIII showed higher cytotoxic activity against NB4 cells than MMAIII (Fig. 3), and induced abundance of Cyt c release into the cytosolic (Fig. S3), suggesting that DMAIII may also target cellular mitochondria for the induction of apoptosis. Contrary to our hypothesis, MMAIII was shown to have more potent ability to release Cyt c from intact mitochondria, while only a small amount of Cyt c was released by DMAIII at high dose (Fig. 3E), indicating that MMAIII can directly attack mitochondria. Thus, we hypothesize that the DMAIII-induced release of Cyt c in NB4 cells could have probably occurred through other signaling pathways.
Expectedly, ER-stress can be specifically induced by DMAIII, however, little changes were observed following exposure to iAsIII and MMAIII (Fig. S4A–S4B). Additionally, activation of JNK is a mediator of ER stress-induced apoptosis [27], and ER stress is majorly responsible for JNK activation [28]. Here, DMAIII significantly induced apoptosis signal regulating kinase 1 (ASK1)-JNK activation (Fig. S4C–S4D), and inhibition of p-JNK could prevent DMAIII-induced apoptosis (Fig. 3H). These results suggest that the activation of ASK1-JNK is involved in the induction of apoptosis via ER stress. Our results demonstrated that MMAIII directly attacks the mitochondria to induce apoptosis, while DMAIII–induced apoptosis is through induction of ER stress (Fig. S6).
Trivalent arsenicals are known to be highly reactive towards the free-cysteine residues in proteins [29]. Especially, MMAIII have shown strong ability to release zinc ion from the zinc finger protein, and binds to the protein [20]. In NB4 cells, MMAIII showed stronger binding affinity to proteins as compared to iAsIII and DMAIII (Fig. 5A). Correspondingly, MMAIII is able to release Zn2+ ions and bind to Ring domain of PML-R recombinant protein at physical condition (pH7.4), but DMAIII exhibited less efficiently as compared to MMAIII (Fig. 5E). Furthermore, little amount of iAsIII was also observed to bind to PML-R protein. This is probably because a small amount of zinc might be released from the PML-R during the purification process providing free cysteine residues for iAsIII binding. In addition, our results are consistent with previous studies; proposing that iAsIII could not bind to zinc finger protein XPAzf (containing Zn2+) in ammonium acetate buffer (pH7.4) as determined by ESI-MS, while MMAIII exhibits potent binding affinity for XPAzf [20]. Collectively, it seems that at physiological condition, binding of iAsIII to Zinc finger proteins (except to the apo-Zinc finger protein; without Zn2+ ions) is difficult however, the methylated arsenic compounds demonstrate to have strong binding affinity for PML or other thio-containing proteins.
Notably, although MMAIII was able to bind to ring domain of PML protein, the methylated arsenicals could not induce NB4 cell differentiation or PML-RARα protein degradation (Fig. 2). Similarly, we also further confirmed that PML proteins could not be degraded by exposure to both MMAIII and DMAIII in PML or PML-RARα overexpressed HEK293T or HeLa cells, however, these proteins were completely degraded by iAsIII, which was found to be consistent with previous reports [30–31]. Additionally, there was no re-localization of PML-NBs found in HEK293T or HeLa cells after exposure to the two methylated arsenic species (Fig. 4C–4D), indicating that the arsenic binding to PML protein actually does not correlate with PML protein degradation. Moreover, our results of immunoprecipitation have clearly shown that the two methylated arsenic species are incapable of inducing PML protein SUMOylation and ubiquitination (Fig. 6C), which clearly indicates that iAsIII once methylate to organic compounds, it will lose its ability to induce cell differentiation or PML protein degradation.
In summary, we suggest that the three different arsenic species exert different roles in NB4 cells; where, iAsIII can predominantly induce cellular differentiation, while the two intermediate metabolites; MMAIII and DMAIII can induce cellular apoptosis as schematically drawn in Figure 6D. However, further studies are required to confirm whether the formation of the arsenic intermediate metabolites (i.e., induction of apoptosis) in APL patients may help increase the therapeutic efficacy of As2O3 treatment.
MATERIALS AND METHODS
Reagents
All reagents were of analytical grade. Milli-Q water (Millipore) was used throughout the experiment. Trizma® HCl and Trizma® Base were purchased from Sigma (St. Louis, MO, USA). Nitric acid, ammonium acetate, acetic acid, 28% ammonia solution, L-cysteine, sodium arsenite (iAsIII), sodium arsenate (iAsV), and dimethylarsinic acid [(CH3)2AsO(OH)] (DMAV) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Monomethylarsonic acid (MMAV) was obtained from Tri Chemicals (Yamanashi, Japan). The arsenic standard solution (1, 000 μg/mL) for ICP-MS was purchased from SPEX CentiPrep (Metuchen, NJ, USA). Stock solutions of all arsenic compounds (10 mmol/L) were prepared from the respective standard compounds. All stock solutions were stored in the dark at 4°C. Diluted standard solutions for analysis were prepared daily prior to use.
Cell culture
NB4 cells were kindly provided by Dr. Qiaojun He (Zhejiang University, China) in September 2012 and no authentication was done by the author. HEK293T and HeLa cells were purchased from Cell Bank of China Science in April 2013. Following receipt, cells were grown and frozen as a seed stock as they were available. Cells were passaged for a maximum of 3 months, after which new seed stocks were thawed. Two cell lines were authenticated using DNA fingerprinting (variable number of tandem repeats), confirming that no cross-contamination occurred during this study. Two cell lines were tested for mycoplasma contamination at least two times per year.
Cells (1.0 × 106) were cultured in T25 flask, and were maintained in logarithmic growth phase using RPMI-1640 or DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin, at 37°C in 5% CO2 atmosphere. After 24 h of seeding, cultures were washed with PBS, fresh medium was added, and the cells were treated with indicated doses of arsenicals and all-trans retinoic acid for indicated time.
HPLC-ICP MS analysis
A polymer-based gel filtration column (Shodex Asahipak GS-220 HQ, 300 mm 7.6 mm i.d., Showa Denko, Tokyo) with an exclusion limit of 3000Da was used to separate unbound arsenic species from protein-bound arsenicals. A 20 μL aliquot of a sample solution was applied to the column, and then the column was eluted with 50 mM ammonium acetate buffers (pH6.5/7.0), at a flow rate of 0.6 mL/min or 0.8 ml/min, respectively. Arsenic species and concentrations were monitored with an Agilent 7500ce ICP-MS (Agilent Technologies, Tokyo, Japan). Arsenic (As) and Zinc (Zn) were monitored at m/z 75 and 66 (respectively). Arsenic concentration was determined by ICP MS after wet-digestion with concentrated nitric acid (HNO3) and 30% H2O2 (v/v = 1/1) at 135°C for 2 days.
Antibodies
Primary antibodies RARα, PRAM-1, rabbit anti-Poly (ADP-ribose) polymerase (PARP) polyclonal antibody, Cyt c, JNK, P-JNK, P-PERK, eif2α, rabbit anti-human PML, anti-human SUMO-1, Ub Antibody were purchased from Santa Cruz Biotechnology (CA, USA). Primary antibodies; Bax, BCL-2, β-actin, anti-cleaved caspase-3 antibody, caspase-3, Perk, P-ASK1, ASK, P-eif2α, GAPDH were purchased from Cell Signaling Technology (Danvers, MA). Anti-FLAG mouse monoclonal antibody was purchased from Sigma.
MTT assay for cell proliferation
NB4 cells were seeded at a density of 2 × 104 cells/100 μL/well in 96-well microtiter plates (Promega Corporation). Twenty-four hours post-seeding, the cultures were washed twice with PBS and then exposed to various concentrations of arsenicals. Then, 20 μL of an MTT solution was added to each well, and the plates were incubated for an additional 3 h at 37ºC. Afterward, cell cultures were washed with PBS, and DMSO was added to each well. Cell proliferation was measured as absorbance at 570 nm with a microplate reader.
Assay for CD11b and apoptosis
For CD11b determination, arsenicals (or ATRA)-treated NB4 cells were washed twice with PBS before incubation with FITC-conjugated anti-CD11b antibody, and then analyzed on flow cytometer (Beckmancoulter). For apoptosis, cells were stained with AnnexinV-FITC and PI for analysis of cellular apoptosis.
Western blot analysis
NB4 cells were washed twice with D-hanks solution, followed by the lysis of cell pellets using RIPA lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.2 mM PMSF, and a complete mini protease inhibitor tablet) with 8M urea. Whole cell lysates were incubated on ice for 30 min and centrifuged for 30 min at 13,000g at 4°C to obtain the supernatant for western blot analysis. Notably, HEK 293T and HeLa cells were lysed in RIPA buffer without 8 M urea, and then incubated on ice and centrifuged as mentioned above to obtain the supernatant (S) and pellet (P) fractions. The supernatant was transferred and the pellet was washed with PBS, lysed in SDS buffer (1 × TBS, 10% glycerol, 0.015% EDTA, 50 mM DTT, and 2% SDS), boiled for 10 min at 95°C. Twenty-five microgram of each protein sample was resolved by 10~12% SDS-PAGE and electroblotted onto nitrocellulose membranes. The membranes were blocked with non-fat milk and incubated overnight with different primary antibodies at 4ºC, followed by incubation with HRP-linked secondary antibodies for 1 h at room temperature and then the proteins were visualized by enhanced chemiluminescence.
Immunoprecipitation assay
Immunoprecipitation was performed as described by Zhang et al. [18]. Briefly, HEK293T cells were transfected with Flag-PML using Lipofectamine 2000 (Invitrogen). After 24 h of post-transfection, the cells were treated with arsenicals and then lysed in IP buffer in an ice bath, followed by centrifugation at 14000g to obtain the supernatant. In addition, appropriate specific antibody, normal IgG and proteinA/G-beads were added to supernatant and incubated at 4°C for overnight. Then, removing the supernatant after centrifugation, the SDS loading buffer was added to the pellets and boiled for 10 min at 95°C. The samples were further centrifuged to obtain the supernatant for western blot analysis.
Immunofluorescence microscopy
Arsenic-treated NB4 cells were transferred onto glass slides using a Shandon cytospin (Runcorn, UK), while FLAG-PML (or FLAG-PML-RARα) transfected HEK293T cells and HeLa cells were cultured on Chamber slides and then exposed to arsenicals. After washing with PBS twice, the slides were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100, blocked with PBST with 10% fetal bovine serum. Further, the cells were then blocked with 1% BSA in PBS, followed by incubation with primary antibodies overnight at 4°C. Later they were tagged with fluorescent secondary antibodies for 2 h. Slides were mounted using Vectashield mounting liquid (Vector Labs) and sealed, stored in dark at 4°C. The cells were examined under a Zeiss (Göttingen, Germany) 510 confocal microscope.
Statistical analysis
Each proliferation value represents the mean ± S.D. from four determinations, and IC50 values were calculated from the log-log plot between the percentages of viable cells. Subsequently, each experiment was performed at least three times. Statistical analysis of data was carried out using a one-way ANOVA followed by Holm-Sidak pairwise multiple comparison test (Sigmaplot, Systat Software Inc), and a probability value of less than 0.05 (*p < 0.05) was accepted as a significant difference.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGMENTS AND GRANT SUPPORT
This work is supported by National Natural Science Foundation of China No: 81274138 and 81473289 (to H. Naranmandura), Zhejiang Provincial Natural Science Foundation of China No. R2110231 (to H. Naranmandura), Zhejiang University K.P. Chao’s High Technology Development Foundation No.2013RC026 (to H. Naranmandura), Scientific and Technological of Zhejiang Province No. 2015C33154 (to H. Naranmandura), Health Bureau of Zhejiang Province Foundation, China No.WKJ-ZJ-1512 (to H. Naranmandura), Zhejiang Provincial Program for the Cultivation of Medical Rookies and National High-tech Research and Development Program of China No.SS2012AA020203 (to J. Zhou).
Abbreviations
ATRA, all-trans retinoic acid; AS3MT, arsenic (+3 oxidation state) methyltransferase; As2O3, arsenic trioxide; iAsIII, arsenite; iAsV, arsenate; APL, acute promyelocytic leukemia, Cyt c, cytochrome c; DMAIII, dimethylarsinous acid; DMAV, dimethylarsinic acid; MMAIII, monomethylarsonous acid; MMAV, monomethylarsonic acid; mPTP, mitochondrial permeability transition pore; HPLC, high performance liquid chromatography; ICP MS, inductively coupled argon plasma mass spectrometry; PML-RARα, Promyelocytic leukemia-Retinoic acid receptor; PML-NBs, PML-nuclear bodies.
REFERENCES
1. Lallemand-Breitenbach V, Zhu J, Chen Z, de Thé H. Curing APL through PML/RARA degradation by As2O3. Trends Mol Med. 2012; 18:36–42.
2. de Thé H, Chen Z. Acute promyelocytic leukemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010; 10:775–783.
3. Ahn RW, Chen F, Chen H, Stern ST, Clogston JD, Patri AK, Raja MR, Swindell EP, Parimi V, Cryns VL, O’Halloran TV. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer. Clin Cancer Res. 2010; 16:3607–3617.
4. Vahter M, Marafante E. Intracellular interaction and metabolic fate of arsenite and arsenate in mice and rabbits. Chem Biol Interact. 1983; 47:29–44.
5. Kenyon EM, Del Razo LM, Hughes MF. Tissue distribution and urinary excretion of inorganic arsenic and its methylated metabolites in mice following acute oral administration of arsenate. Toxicol Sci. 2005; 85:468–475.
6. Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Davis KH, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, Thomas DJ. Disruption of the arsenic (+ 3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem Res Toxicol. 2009; 22:1713–1720.
7. Yoshino Y, Yuan B, Miyashita S, Iriyama N, Horikoshi A, Shikino O, Toyoda H, Kaise T. Speciation of arsenic trioxide metabolites in blood cells and plasma of a patient with acute promyelocytic leukemia. Anal Bioanal Chem. 2009; 393:689–697.
8. Wang Z, Zhou J, Lu X, Gong Z, Le XC. Arsenic speciation in urine from acute promyelocytic leukemia patients undergoing arsenic trioxide treatment. Chem Res Toxicol. 2004; 17:95–103.
9. Yuan C, Lu X, Oro N, Wang Z, Xia Y, Wade TJ, Mumford J, Le XC. Arsenic speciation analysis in human saliva. Clin Chem. 2008; 54:163–171.
10. Naranmandura H, Carew MW, Xu S, Lee J, Leslie EM, Weinfeld M, Le XC. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells. Chem Res Toxicol. 2011; 24:1586–1596.
11. Chen GQ, Zhou L, Styblo M, Walton F, Jing Y, Weinberg R, Chen Z, Waxman S. Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res. 2003; 63:1853–1859.
12. de Thé H, Chomienne C, Lanotte M, Degos L, Dejean A. The t (15, 17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature. 1990; 347:558–561.
13. Lallemand-Breitenbach V. A new oncoprotein catabolism pathway. Blood. 2010; 116:2200–2201.
14. Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. 2001; 90:105–156.
15. Moog-Lutz C, Peterson EJ, Lutz PG, Eliason S, Cavé-Riant F, Singer A, Di Gioia Y, Dmowski S, Kamens J, Cayre YE, Koretzky G. PRAM-1 is a novel adaptor protein regulated by retinoic acid (RA) and promyelocytic leukemia (PML)-RA receptor α in acute promyelocytic leukemia cells. J Biol Chem. 2001; 276:22375–22381.
16. Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM, Li JM, Tang W, Zhao WL, Wu W, Sun HP, Chen QS, Chen B, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2009; 106:3342–3347.
17. Zhou J, Zhang Y, Li J, Li X, Hou J, Zhao Y, Liu X, Han X, Hu L, Wang S, Zhao Y, Zhang Y, Fan S, et al. Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood. 2010; 115:1697–1702.
18. Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, et al. Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science. 2010; 328:240–243.
19. Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, Hudson LG. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J Biol Chem. 2011; 286:22855–22863.
20. Piatek K, Schwerdtle T, Hartwig A, Bal W. Monomethylarsonous acid destroys a tetrathiolate zinc finger much more efficiently than inorganic arsenite: mechanistic considerations and consequences for DNA repair inhibition. Chem Res Toxicol. 2008; 21:600–606.
21. Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010; 116:2324–2331.
22. Miller WH, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002; 62:3893–3903.
23. Kiguchi T, Yoshino Y, Yuan B, Yoshizawa S, Kitahara T, Akahane D, Gotoh M, Kaise T, Toyoda H, Ohyashiki K. Speciation of arsenic trioxide penetrates into cerebrospinal fluid in patients with acute promyelocytic leukemia. Leuk Res. 2010; 34:403–405.
24. Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P, de Thé H. Role of promyelocytic leukemia (PML) sumoylation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor α degradation. J Exp Med. 2001; 193:1361–1372.
25. Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, Zhou J, Zhu J, Raught B, de Thé H. Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol. 2008; 10:547–555.
26. Khaleghian A, Ghaffari S H, Ahmadian S, Alimoghaddam K, Ghavamzadeh A. Metabolism of arsenic trioxide in acute promyelocytic leukemia cells. J Cell Biochem. 2014; 115:1729–1739.
27. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007; 8:519–529.
28. Urano F, Wang XZ, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000; 287:664–666.
29. Naranmandura H, Suzuki N, Suzuki KT. Trivalent arsenicals are bound to proteins during reductive methylation. Chem Res Toxicol. 2006; 19:1010–1018.
30. Goto E, Tomita A, Hayakawa F, Atsumi A, Kiyoi H, Naoe T. Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood. 2011; 118:1600–1609.
31. Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Bras ML, Duffort S, Peres L, Berthier C, Soilihi H, Raught B, de Thé H. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer cell. 2010; 18:88–98.