
INTRODUCTION
Substantial clinical progress in immunosuppression and anticancer chemotherapy has improved the overall survival of patients. Unfortunately, the survival benefits conferred by some long-term treatments such as thiopurine are also associated with an increased risk of developing therapy-related cancers. Thiopurine drugs comprise azathioprine (Aza) and its metabolite 6-mercaptopurine. Aza is widely used as an immunosuppressive agent for the prevention of transplant graft rejection and for autoimmune diseases such as inflammatory bowel disease (IBD) and autoimmune hepatitis. 6-mercaptopurine is commonly used as part of maintenance therapy in childhood acute lymphoblastic leukemia. Aza is classified as a human carcinogen but the specific mechanisms that underlie its in vivo carcinogenicity following thiopurine therapy remain unclear.
Interestingly, several clinical studies have reported an association between the use of prolonged thiopurine regimens and the occurrence of tumors displaying microsatellite instability (MSI). The MSI tumor phenotype is the hallmark of a defective DNA mismatch repair (MMR) system, the major function of which is to edit errors produced during replication. Inactivation of the MMR system greatly increases spontaneous mutation rates. This is observed as MSI and manifests as frequent deletion mutations in DNA regions comprising mostly of long mono- or dinucleotide tracts. In vitro studies have demonstrated that MMR also plays an important role in thiopurine-mediated cytotoxicity, as evidenced by the increased tolerance of MMR-deficient cells to thiopurine exposure. The major cytotoxic effects of thiopurine drugs occur following incorporation of thio-guanine (TG) into DNA and subsequent misincorporation of thymidine opposite TG during DNA replication. The aberrant processing of these DNA base pairs by MMR generates potentially lethal DNA lesions. MMR–deficient cells are unable to initiate lethal processing of the TG-containing base pairs and escape cytotoxicity. Owing to this resistance conferred by defective MMR, known as thiopurine tolerance, it has been suggested that thiopurines may constitute a risk factor for development of human cancer via selection of MMR-defective cells that subsequently undergo neoplastic cell transformation following the accumulation of genetic mutations caused by MSI (for review [1]).
Despite in vitro functional data suggesting a putative causal role for thiopurine therapy in the development of MSI cancers, the clinical evidence to date is mixed and hence this remains to be established conclusively. An MSI phenotype was observed in acute myeloid leukemia/myelodysplastic syndromes that developed after organ transplantation and were associated with immunosuppression by Aza therapy [2]. Previous studies from our group [3, 4] also reported a low frequency of the MSI phenotype in immunodeficiency-related non-Hodgkin lymphomas arising in transplant patients who received Aza as part of their immunosuppressive regimen (13/143 vs 0/500 in immunocompetent patients). However, some patients with MSI tumors of suspected iatrogenic origin did not receive thiopurine therapy [4]. Interpretation of these findings is further complicated by the fact that immunosuppressive regimens in transplant patients often combine Aza with other drugs such as steroids and ciclosporin A (CsA). Additionally, the MSI phenotype was not detected in immunodeficiency-related skin cancers and no significant association was observed between Aza intake and MSI in intestinal neoplasias arising in patients with IBD [5–8]. Thus, the clinical association between thiopurine treatment-related cancer and MMR defects remains uncertain and requires further investigation.
Here we describe results using an animal model to address whether thiopurine tolerance is the putative oncogenic mechanism that underlies the emergence of MSI tumors. In an earlier proof of concept study we demonstrated that Aza induced massive and early development of MSI lymphomas in Msh2+/– mice carrying a germline mutation in the MMR gene Msh2 [9]. In contrast, untreated Msh2+/– mice developed MSI lymphomas at a very low frequency. This work established the Msh2 mouse model, the murine equivalent of Lynch syndrome in humans, as relevant for the investigation of thiopurine tolerance as a causal mechanism for MSI cancers. However, the substantial toxicity of Aza in Msh2 wild type (Msh2wt) mice prevented us from investigating the ability of this agent to induce MSI-driven malignancy in mice without a genetic predisposition to MMR defects. Moreover, we did not investigate other immunosuppressants whose action is independent of MMR. In the present work we have therefore performed a dose effect study using wild type, null or heterozygous mice for the Msh2 gene and identified appropriate long-term treatment conditions for Aza, thus avoiding the major cytotoxic effects encountered previously. We hypothesized that an Aza dosage that enables a substantial survival would allow the development of lymphomas, a small proportion of which would be of MSI phenotype, as observed in humans. To evaluate whether the observed effects could be attributed specifically to Aza, we also investigated CsA, another frequently prescribed immunosuppressant that inhibits the transcription of cytokines, leading to reduced functional ability of effector T-cells.
RESULTS
Impact of azathioprine on Msh2+/– and Msh2ko mice
We first specified the way Aza treatment could trigger MSI lymphomagenesis in the context of a genetic predisposition to MSI tumor development (i.e. Msh2+/–). A dose-response relationship was observed for Msh2+/– mice, with median survival of 164, 220 and 575 days for Aza 50, 30 and 10 mg/L treatment, respectively (P < .0001; Log-rank test) (Figure 1A). Aza50-treated Msh2+/– mice systematically developed early onset, MSI lymphomas (22/22 vs 1/10 for untreated Msh2+/– animals) (Figure 1B). The onset of tumors was significantly delayed at lower Aza doses, with a mean age of onset of 154, 255 and 539 days for Aza50, Aza30 and Aza10, respectively (P < .0001; ANOVA test). The incidence of the MSI phenotype was also significantly reduced with the lowest Aza dose (21/23 and 6/18 for Aza30 and Aza10, respectively P < .0001; Fisher's exact test) (Figure 1C). Compared to untreated Msh2+/– mice, Aza10-treated mice showed a slightly shorter median survival (575 vs 625 days, respectively; P = .020; Log-rank test) but comparable median age of lymphoma onset (539 vs 576 days, respectively; P = .59; Student's t test).
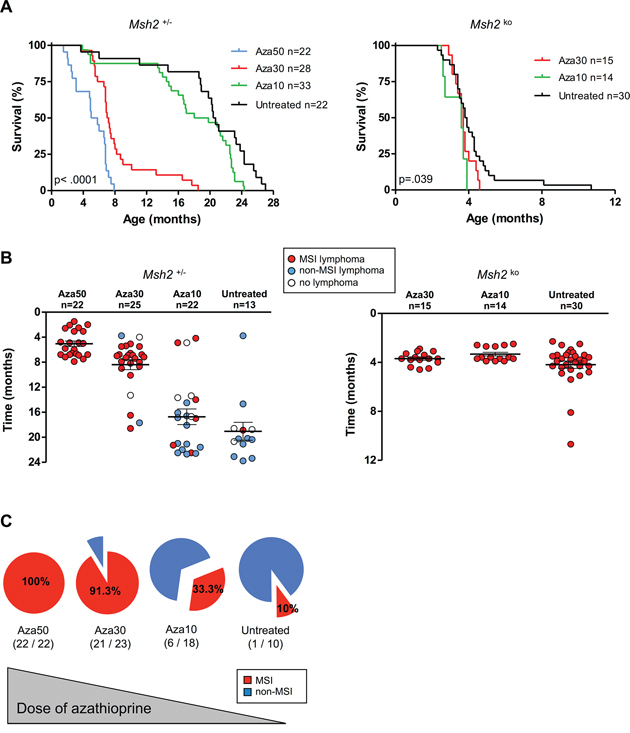
Figure 1: Effect of azathioprine treatment on survival, lymphoma incidence and MSI tumor phenotype in Msh2+/– and Msh2ko mice. A. Evidence for a dose-response relationship between azathioprine treatment and the survival of Msh2+/– mice: Kaplan–Meier survival analysis of untreated and azathioprine-treated Msh2+/– mice (left) and Msh2ko mice (right). Survival curves for Msh2+/– mice after Aza50 treatment are from our previous study [9]. All P values are two-sided. Log-rank test. B. Lymphoma occurrence and delay of onset in the different mice cohorts according to Msh2+/– (left) or Msh2ko (right) genotype. Mean age of lymphoma onset and standard error of the mean are indicated. C. Evidence for a dose-response relationship between azathioprine treatment and incidence of MSI phenotype in lymphomas of Msh2+/– mice. Aza10 = azathioprine 10 mg/L; Aza30 = azathioprine 30 mg/L; Aza50 = azathioprine 50 mg/L.
As expected, all control Msh2ko mice, whether treated or not, were sacrificed before the age of 11 months. The median survival of Msh2ko mice was 117 days for untreated animals, 113 days with Aza30 and 109 days with Aza10 (P = .039; Log-rank test) (Figure 1A). As expected, all Msh2ko mice developed patent tumor syndromes, i.e. abnormal enlargement of the thymus, spleen or liver and massive and diffuse proliferation of lymphoma cells, that displayed MSI (Figure 1B).
Impact of azathioprine and ciclosporin A on Msh2wt mice
Similar to Msh2+/– mice, a dose-response relationship was observed between Aza treatment and the survival of Msh2wt mice. The lowest Aza dose was associated with a significantly longer lifespan, with median survival of 78,123 and 568 days for Aza50, Aza30, and Aza10, respectively (P < .0001; Log-rank test) (Figure 2A). Whereas Aza10 was the only dose compatible with a substantial survival of animals, it still demonstrated a significant impact upon lifespan (568 vs 652 days for untreated mice; P = .007; Log-rank test). A patent tumor syndrome was observed in a substantial proportion of the Aza10 cohort (7/23, 30.5%) (Figures 2B and 2C). Msh2wt mice exposed to higher Aza doses (Aza30 and Aza50) were sacrificed within several months of birth due to poor tolerance to therapy. Consistent with our previous results [9], micro-lymphoproliferations were frequently observed during histological examination of these animals (Figures 2B, 2C). Microscopic foci comprised of medium-to-large neoplastic lymphoid cells were restricted to the spleen area located under the capsule.
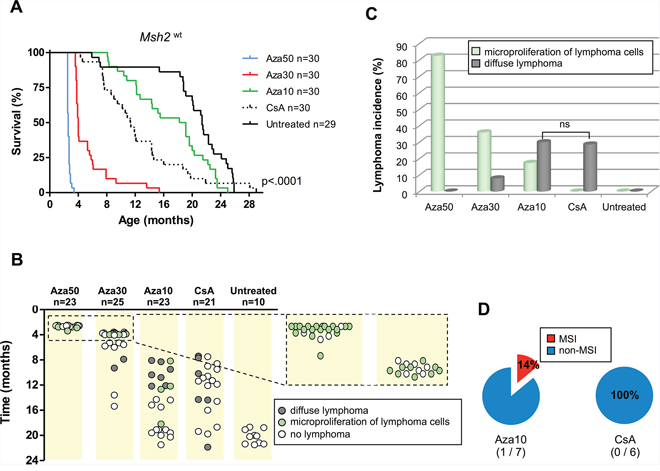
Figure 2: Effect of azathioprine and ciclosporin A treatments on survival, lymphoma incidence and MSI tumor phenotype in Msh2wt mice. A. Evidence for a dose-response relationship between immunosuppressive treatment and the survival of Msh2wt mice: Kaplan–Meier survival analysis of untreated, azathioprine- and ciclosporin A-treated Msh2wt mice. All P values are two-sided. Log-rank test. B. Occurrence of lymphoproliferations and delay of onset in the different cohorts of Msh2wt mice. C. Lymphoma incidence. Fisher's exact test; ns=not significant. D. Incidence of MSI phenotype in lymphomas. Aza10 = azathioprine 10 mg/L; Aza30 = azathioprine 30 mg/L; Aza50 = azathioprine 50 mg/L; CsA = ciclosporin A 300 mg/L.
CsA-treated Msh2wt mice also showed significantly shorter survival compared to untreated mice (345 vs 652 days, respectively; P = .002; Log-rank test) (Figure 2A). Aza10- and CsA-treated Msh2wt mice showed comparable survival (median of 568 vs 345 days, respectively; P = .045; Log-rank test) and both groups showed the presence of lymphomas in about one third of cases (7/23 vs 6/21, respectively; P = .9; Chi2 test) (Figures 2B and 2C).
MSI status of azathioprine- and ciclosporin A-induced lymphomas in Msh2wt mice
Microsatellite genotyping was performed in all Msh2wt mice that developed patent tumor syndrome. As described above, these had mostly been treated with Aza10 or CsA. A diffuse MMR-deficient lymphoma due to proliferation of MSI tumor cells was found in 1/7 Aza10-treated Msh2wt mice. At contrast, all tumor DNAs from the 6 CsA-treated mice with lymphoma were non-MSI (Figure 2D). Two Aza30-treated mice that had not been sacrificed at a young age, showed a late onset lymphoma syndrome that was also non-MSI. Tumor cells obtained by laser microdissection from splenic microproliferations in four Aza50-treated Msh2wt mice also failed to show MSI. Results of microsatellite instability analysis are detailed in the Supplementary Table S1.
The single Msh2wt mouse that developed an MSI tumor syndrome was sacrificed at the age of 315 days because of posterior leg paralysis. At autopsy, it presented an enlarged spleen and lymph nodes. Histological examination revealed massive (>90%) infiltration of the spleen, thymus and lymph node tissues by medium to large size lymphoid cells. Examination of tumor DNA from the splenic tissue with three microsatellite markers revealed a change in length of all three loci compared with normal tissue DNA from the same mouse (Figures 3A and 3C).
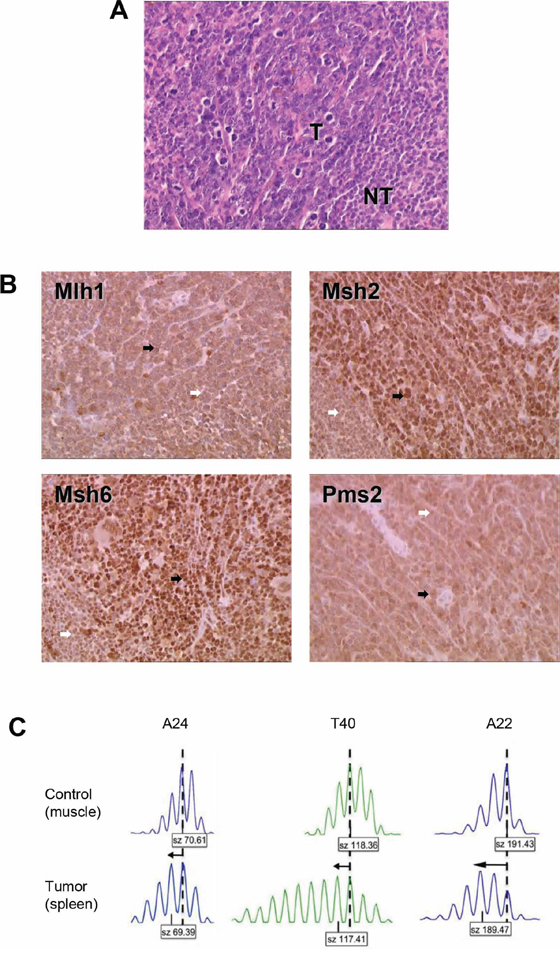
Figure 3: Characterization of the MSI lymphoma syndrome arising in Aza10-treated Msh2wt mouse. A. Histological characterization showing hematoxylin–eosin staining of splenic lymphoma. T indicates tumor cells; NT indicates normal lymphocytes. B. Immunohistochemistry of spleen tissue sections. Tumor cells (black arrows) stained positively. Normal lymphocytes (white arrows) showed less pronounced staining of MMR proteins due to their lower proliferative rate. Antibodies against Mlh1 and Pms2 provided less intense IHC staining however positive staining was retained in tumor cells (black arrows). An IHC picture of a non-MSI/MMR-proficient splenic lymphoma can be seen in the Supplementary Figure S2 to illustrate that technical point. C. Allelic profiles of the three noncoding microsatellite markers (A24, T40 and A22) displaying shorter alleles (arrows) for the three markers in tumor (spleen) compared to control (muscle) tissue. Dashed vertical lines indicate the predominant allele size observed in control DNA. Predominant allele size (in base pair) is indicated in the box below each profile.
Molecular mechanism underlying MMR-deficiency due to thiopurine tolerance in tumor cells
PCR-based genotyping was performed on all MSI tumors that developed in Msh2+/− mice following an immunosuppressive regimen. In the majority of cases (22/26, including 19/21 Aza30- and 3/5 Aza10-treated Msh2+/– mice), inactivation of the MMR system resulted from somatic loss of the remaining wild type Msh2 allele (i.e. loss of heterozygosity), as previously reported [9]. cDNA sequencing of Msh2 was performed in the tumor from the single Msh2wt mouse that displayed an MSI lymphoma following Aza treatment, however no somatic mutation was detected. Immunohistochemical analysis of Msh2, Mlh1, Msh6 and Pms2 expression was then performed to further investigate the mechanism underlying loss of MMR function in the tumor from this Msh2wt mouse. As shown in Figure 3B, the tumor cells stained positively for these four major MMR proteins.
DISCUSSION
In the present study we investigated the ability of Aza, an immunosuppressant from the thiopurine family, to trigger MSI lymphomagenesis in mice. Aza induces apoptosis of MMR-proficient cells via the MMR–dependent signaling of Aza-induced DNA damage. Aza also has immunosuppressive properties due to induction of apoptosis in T cells [10] and inhibition of de novo purine biosynthesis which hampers cell division especially in lymphocytes, which unlike other cell types cannot use the salvage pathway of purine synthesis [11]. Importantly, the two latter effects are not dependent on MMR status. We previously observed considerably more toxicity at the higher dose of Aza (50 mg/L) in Msh2wt mice compared to Msh2+/− mice [9] and confirmed here that heterozygous mice are more tolerant to the cytotoxic effects of Aza (Supplementary Figure S1). These observations are consistent with an earlier report using another MMR murine model (Mlh1 mice) that showed Mlh1+/– mice were less sensitive to the lethal effects of a methylating agent than Mlh1wt control mice (thiopurines and methylating agents share a common MMR processing mechanism for converting damaged DNA bases into lethal lesion) [12]. Our results also concur with in vitro experiments showing that immortalized lymphoblasts from Lynch patients who are heterozygous at the MSH2 locus were more tolerant to the methylating agent temozolomide than MSH2wt cells [13].
Together, these observations suggest the overall clinical impact of Aza results from a combination of factors such as Msh2 genotype, drug dosage and the balance between the cytotoxic and oncogenic properties of this drug. To account for the observed survival and tumor phenotypes in drug-treated mice with different Msh2 genotype status, we propose the following explanations. First, all Msh2ko mice (untreated and Aza-treated) exhibited the characteristic development of MSI diffuse lymphomas that ultimately resulted in their early death. Whereas untreated Msh2+/– mice only rarely develop spontaneous MSI neoplasms, Aza induced a high incidence of MSI lymphomas in a dose-dependent manner in these animals. This is likely to be related to the burden of induced thiopurine DNA lesions and the resulting selective pressure to escape cell apoptosis through MMR inactivation. In most Msh2+/– mice, this inactivation occurs via somatic loss of the remaining wild type Msh2 allele. In Aza-treated Msh2wt mice, a balance was observed between the cytotoxic and oncogenic properties of the drug. Survival and incidence of diffuse lymphomas were inversely proportional to the dose of azathioprine. We hypothesize that the microproliferations of lymphoma cells, which were mostly observed in Aza50- and Aza30-treated Msh2wt mice, did not lead to lymphoma because either their progression has been efficiently prevented by the immunosuppressive effect of Aza treatment or the mice died prematurely from organ failure related to the high drug cytotoxicity at this dosage. Aza10 and CsA treatments resulted in similar survival times and induction of lymphomas in a substantial and comparable proportion of Msh2wt mice. This mirrors the situation in humans where the development of lymphomas is a known adverse effect of immunosuppressive treatment, although the incidence of iatrogenic lymphomas is much higher in mice. An MSI tumor phenotype was observed in one Msh2wt mouse following Aza10 treatment. At contrast, none was observed in the CsA treated Msh2wt mice and we found that CsA treatment did not result in a significant induction of MSI lymphomas in Msh2+/– mice (data not shown). All together, our data support a causal role for Aza treatment, but not CsA treatment, in triggering MSI-driven lymphomagenesis in Msh2wt or Msh2+/– mice.
Considering that the risk of lymphoproliferative disorder (LPD) was five times higher in IBD patients treated with thiopurines than in those never exposed to these drugs [14], we searched for MSI phenotype in a series of 13 IBD-related LPD and found one case, which interestingly developed after thiopurine (imurel) therapy (Supplementary Table S2). Thus, this observation broadens the clinical contexts of thiopurine-associated lymphoproliferative disorders with MSI phenotype. Interestingly, as with the lymphoma that developed in the Aza10-treated Msh2wt mouse, the tumor cells from this MSI case were shown by immunohistochemistry to retain expression of the 4 MMR proteins (Supplementary Figure S3). This was also the case in about one third (3/8) of the immunodeficiency-related non-Hodgkin lymphomas with a MSI phenotype that develop in transplant patients treated with Aza, that we previously reported [4]. These findings suggest that new mechanisms for inactivation of MMR, which remain to be determined, are associated with clinical contexts of thiopurine treatment.
In summary, our study allows an estimation of the clinical risk associated with prolonged Aza intake based upon the development of iatrogenic tumors with MSI in a mouse model. Our data clearly demonstrate the ability of Aza to trigger lymphomagenesis through an MSI-driven process in vivo in the absence of genetic predisposition to MMR defects. The limitation of this study is the small number of Msh2wt mice that developed iatrogenic lymphoma with MSI but increasing the number of treated Msh2wt mice was not feasible from an ethical point of view. However, based upon post-transplant lymphoproliferative disorders in humans, such event was expected to be rare (9%) [3, 4]. Together, our findings could have important clinical implications for patients receiving azathioprine therapy and more broadly thiopurines. Therefore, we recommend systematic screening for MSI in lymphomas and other neoplasms that arise in patients treated with thiopurine drugs. Such screening could be particularly important because tumors with MSI usually have a different prognosis and sensitivity to chemotherapeutic agents compared with tumors that lack MSI, as has been shown for colorectal carcinomas [15].
MATERIALS AND METHODS
Mice and treatments
Msh2+/– mice on an FVB background [16] were intercrossed to obtain animals that were null, heterozygous, or wild type for the Msh2 gene. Mice genotypes were determined by PCR analysis [17]. Aza was given orally via the drinking water using Imurel 50 mg (GlaxoSmithKline, Marly-le-Roi, France) to prepare a 50 mg/L, 30 mg/L or 10 mg/L drinking water solution (referred to as Aza50, Aza30 and Aza10 treatment in the text). The dose of azathioprine administered through the Aza50 treatment was estimated at 6–20 mg/kg body weight per day which corresponds to a human dose equivalent of 0.5–1.6 mg/kg body weight per day [18] and lies within the therapeutic range [19, 20]. Groups of mice received Aza10 (14 Msh2ko, 33 Msh2+/– and 30 Msh2wt) or Aza30 (15 Msh2ko, 28 Msh2+/– and 30 Msh2wt) and a group of 30 Msh2wt mice also received Aza50. Control mice (30 Msh2ko, 22 Msh2+/– and 29 Msh2wt mice) received only water to drink. A final group of 30 Msh2wt mice received drinking water that contained 300 mg/L ciclosporin A (CsA) (Neoral 100 mg/mL, Novartis Pharma SAS, Rueil-Malmaison, France). The dose of CsA administered to mice was estimated at 40–120 mg/kg body weight per day i.e. a human dose equivalent of 6.48–9.72 mg/kg body weight per day, which lies in the upper limit of the maintenance treatment administered to human transplant patients (i.e. 2 to 6 mg/kg according to Novartis recommendations). Food and drinking were ad libitum. All treatments were administered from the age of 6 weeks. Mice were kept in a pathogen-free environment and health was monitored daily. Animals that displayed signs of poor health, including breath insufficiency and posterior leg paralysis were sacrificed by cervical dislocation. The spleen, liver, thymus, lymph nodes and gut were removed for examination together with muscle (as source of germinal DNA). Since Msh2wt mice on FVB background are prone to develop lymphomas of late occurrence, analysis was not continued beyond 24 months. All animal experiments were conducted in accordance with the regulations controlling procedures in live animals (approval Ce5/2010/065 of the Ethical Committee for Laboratory Animal Care Charles Darwin France).
Histopathological studies
Tissues were formalin fixed, embedded in paraffin and sectioned for histological analysis with hematoxylin–eosin staining. Mice were categorized into three classes: (i) with patent diffuse lymphoma, (ii) with microproliferation of lymphoma cells limited to the spleen, and (iii) with no histological evidence of lymphoma.
Immunohistochemical studies
Immunohistochemical staining for Mlh1, Msh2, Msh6 and Pms2 proteins was performed in lymphoma cells from the single Msh2wt mouse that displayed an MSI lymphoma following Aza treatment. Briefly, 4 μm sections were incubated with monoclonal antibodies against MLH1 (1/20 dilution, clone G168–728, Pharmingen, San Diego, CA), MSH2 (1/25 dilution, clone FE11, Calbiochem, Oncogene Research Products, Cambridge, MA), MSH6 (1/25 dilution, clone 44, Becton Dickinson, Lexington, NC) and PMS2 (1/20 dilution, clone A16–4, BD PharMingen, Le Pont de Claix, France).
Loss of heterozygosity (LOH) analysis
Normal (muscle) and tumor DNA were extracted for Msh2 genotype analysis (DNA extraction kit Qiagen, Courtaboeuf, France). PCR products were resolved on agarose gels to assess loss of heterozygosity of the Msh2 allele [9].
Microdissection of mouse tumor tissue
The tumor cell component from four Aza50-treated Msh2wt mice was collected from 10 hematoxylin-eosin-stained 7-μm tissue sections with a laser-capture microdissection system (PALM Laser, Zeiss, LePecq, France).
Msh2 gene sequencing
Total RNA from the single Aza-treated Msh2wt that displayed an MSI lymphoma tissue sample (two tumor sites and the corresponding normal tissue) was analyzed. Reverse transcription–PCR and sequencing were performed as previously reported [9], except that electrophoresis was performed for each amplified fragment and this was extracted specifically with a Gel Extraction Kit (Qiagen, Redwood City, USA). A second amplification was performed on these fragments according to the same PCR protocol in order to magnify the sequence signal and reduce noise.
MSI testing
Polymerase chain reaction analysis of three noncoding mononucleotide repeats (A22, A24, T40) was used to determine the MSI status of tumor tissues [9]. Amplified PCR products were separated by capillary electrophoresis on an ABI PRISM 3100 Genetic Analyzer and analyzed using GeneMapper software (version 3.7, Aplera). Tumors were scored as MSI if the length of at least one mononucleotide repeat marker in tumor DNA differed from that observed in normal tissue DNA obtained from the same animal.
Kaplan-Meier survival curves and statistical analysis
Time of death was recorded for each mouse that was found dead or had been sacrificed. Kaplan-Meier survival curves were generated using Graphpad Prism software (version 5, San Diego, CA). All statistical tests were two-sided.
Characterization of lymphoproliferative disorders (LPD) in patients with inflammatory bowel disease (IBD)
A total of 19,486 patients were enrolled in the CESAME cohort, a French nationwide prospective observational cohort spanning May 2004 to June 2005 to assess the risk of LPD in patients with IBD treated with thiopurines [14]. MSI status was evaluated in 13 cases of LPD from paraffin material using a fluorescent multiplex system comprising 5 quasimonomorphic mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and NR-27) [21] and expression of MMR proteins was evaluated by immunohistochemistry [4]. LPD were classified according to the WHO classification 2001 and tested for Epstein Barr virus by immunohistochemistry or in situ hybridization.
FUNDING
This work was partly supported by grants to Martine Muleris from the “Association pour la Recherche contre le Cancer” (ARC 5079). Sahra Bodo is a recipient of the “Ministère de l'Enseignement Supérieur et de la Recherche“ and the ”Association pour la Recherche contre le Cancer” fellowship.
CONFLICTS OF INTEREST
The authors disclose no conflicts.
REFERENCES
1. Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. British medical bulletin. 2006; 79-80:153–170.
2. Offman J, Opelz G, Doehler B, Cummins D, Halil O, Banner NR, Burke MM, Sullivan D, Macpherson P, Karran P. Defective DNA mismatch repair in acute myeloid leukemia/myelodysplastic syndrome after organ transplantation. Blood. 2004; 104:822–828.
3. Duval A, Raphael M, Brennetot C, Poirel H, Buhard O, Aubry A, Martin A, Krimi A, Leblond V, Gabarre J, Davi F, Charlotte F, Berger F, Gaidano G, Capello D, Canioni D, et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci U S A. 2004; 101:5002–5007.
4. Borie C, Colas C, Dartigues P, Lazure T, Rince P, Buhard O, Folliot P, Chalastanis A, Muleris M, Hamelin R, Mercier D, Oliveira C, Seruca R, Chadburn A, Leblond V, Barete S, et al. The mechanisms underlying MMR deficiency in immunodeficiency-related non-Hodgkin lymphomas are different from those in other sporadic microsatellite instable neoplasms. Int J Cancer. 2009; 125:2360–2366.
5. Borie C, Euvrard S, Verola O, Buhard O, Barete S, Molina JM, Kanitakis J, Kerob D, Lebbe C, Duval A. No evidence for microsatellite instability in immunodeficiency-related skin cancers. Am J Transplant. 2010; 10:192–193.
6. Perrett CM, Harwood CA, McGregor JM, Warwick J, Cerio R, Karran P. Expression of DNA mismatch repair proteins and MSH2 polymorphisms in nonmelanoma skin cancers of organ transplant recipients. The British journal of dermatology. 2010; 162:732–742.
7. Wisgerhof HC, Hameetman L, Tensen CP, Morreau H, de Fijter JW, Bouwes Bavinck JN, Willemze R, de Gruijl FR. Azathioprine-induced microsatellite instability is not observed in skin carcinomas of organ transplant recipients. The Journal of investigative dermatology. 2009; 129:1307–1309.
8. Svrcek M, El-Bchiri J, Chalastanis A, Capel E, Dumont S, Buhard O, Oliveira C, Seruca R, Bossard C, Mosnier JF, Berger F, Leteurtre E, Lavergne-Slove A, Chenard MP, Hamelin R, Cosnes J, et al. Specific clinical and biological features characterize inflammatory bowel disease associated colorectal cancers showing microsatellite instability. J Clin Oncol. 2007; 25:4231–4238.
9. Chalastanis A, Penard-Lacronique V, Svrcek M, Defaweux V, Antoine N, Buhard O, Dumont S, Fabiani B, Renault I, Tubacher E, Flejou JF, Te Riele H, Duval A, Muleris M. Azathioprine-induced carcinogenesis in mice according to Msh2 genotype. J Natl Cancer Inst. 2010; 102:1731–1740.
10. Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, Lehr HA, Wirtz S, Becker C, Atreya R, Mudter J, Hildner K, Bartsch B, Holtmann M, Blumberg R, Walczak H, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. The Journal of clinical investigation. 2003; 111:1133–1145.
11. Quemeneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003; 170:4986–4995.
12. Kawate H, Itoh R, Sakumi K, Nakabeppu Y, Tsuzuki T, Ide F, Ishikawa T, Noda T, Nawata H, Sekiguchi M. A defect in a single allele of the Mlh1 gene causes dissociation of the killing and tumorigenic actions of an alkylating carcinogen in methyltransferase-deficient mice. Carcinogenesis. 2000; 21:301–305.
13. Marra G, D’Atri S, Corti C, Bonmassar L, Cattaruzza MS, Schweizer P, Heinimann K, Bartosova Z, Nystrom-Lahti M, Jiricny J. Tolerance of human MSH2+/– lymphoblastoid cells to the methylating agent temozolomide. Proc Natl Acad Sci U S A. 2001; 98:7164–7169.
14. Beaugerie L, Brousse N, Bouvier AM, Colombel JF, Lemann M, Cosnes J, Hebuterne X, Cortot A, Bouhnik Y, Gendre JP, Simon T, Maynadie M, Hermine O, Faivre J, Carrat F. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet. 2009; 374:1617–1625.
15. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005; 23:609–618.
16. de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995; 82:321–330.
17. Toft NJ, Winton DJ, Kelly J, Howard LA, Dekker M, te Riele H, Arends MJ, Wyllie AH, Margison GP, Clarke AR. Msh2 status modulates both apoptosis and mutation frequency in the murine small intestine. Proc Natl Acad Sci U S A. 1999; 96:3911–3915.
18. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2008; 22:659–661.
19. Yip JS, Woodward M, Abreu MT, Sparrow MP. How are Azathioprine and 6-mercaptopurine dosed by gastroenterologists? Results of a survey of clinical practice. Inflammatory bowel diseases. 2008; 14:514–518.
20. Opelz G, Dohler B. Critical threshold of azathioprine dosage for maintenance immunosuppression in kidney graft recipients. Collaborative Transplant Study. Transplantation. 2000; 69:818–821.
21. Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, Seruca R, Iacopetta B, Hamelin R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002; 123:1804–1811.