
INTRODUCTION
Mitochondria represent a vital signaling hub in cellular activities since they produce ATP, reactive oxygen species (ROS) and have the ability to shape intracellular Ca2+ signals [1, 2, 3]. Re-programming of mitochondrial metabolism and intracellular Ca2+ distribution are crucial for cells with high proliferative potential such as cancer cells and activated T lymphocytes. There are three main distinct features of mitochondrial metabolism common for tumor and activated T cells, which allow them to respond rapidly to the requirements associated with biomass increase:
A switch from preferentially mitochondrial ATP production to aerobic glycolysis. This phenomenon was first described by Otto von Warburg in 1956 for tumor cells and named after him as the Warburg effect. The Warburg effect was recently applied to explain metabolic reprogramming in activated T cells [4, 5, 6].
Increased, but not excessive mitochondrial ROS (mitoROS) production that is beneficial for tumor cells proliferation as well as for activation and proliferation of T lymphocytes [7–10, 11].
Common metabolic pathways. Similarly to tumor cells, activated T cells increase glutamine and glucose uptake while down-regulating free fatty acid beta-oxidation, a metabolic pathway important for resting and memory T cells. Glycolytic and glutaminolytic enzymes are key factors of the pentose-phosphate pathway involved in nucleotide synthesis. Further, glutamine catabolism fuels the tricarbonic cycle (TCA) via conversion into α-ketoglutarate as well as synthesis of various amino acids [6].
Therefore, mutations or altered levels of tumor suppressors or oncogenes that control mitochondrial homeostasis could adversely affect both proliferation of tumor cells and regulation of immune responses. In turn, impaired immune responses can promote cancer growth since the latter is preceded and maintained by chronic inflammation [12].
In this review, we will focus on the role of mitochondrial Ca2+ transport in the activation of T lymphocytes and proliferation of cancer cells. We will highlight the role of the mitochondrial tumor suppressor Fus1/Tusc2 that we recently characterized as a novel regulator of Ca2+ ion handling in mitochondria [12]. Using current knowledge about Fus1 functions, we will try to decipher how loss of Fus1 activities could promote development of autoimmune disorders and tumors observed in Fus1 knockout mice [13]. We believe that the described concept of a critical link between Ca2+ dynamics, mitochondrial metabolism and development of cancer, inflammation, and autoimmunity may be applied to other tumor suppressors and oncogenes providing new avenues for therapeutic interventions.
FUS1 ACTIVITY AT THE CELLULAR AND WHOLE-ORGANISM LEVELS
Fus1 as a tumor suppressor
Fus1, also known as Tusc2 (tumor suppressor candidate 2), is a small (110 a.a.), highly conserved, ubiquitously expressed mitochondrial protein. Loss of the Fus1-harboring 3p21.3 chromosomal region or decrease in the Fus1 mRNA and protein levels have been reported in the majority of lung cancers [13, 14], mesotheliomas [15], bone and soft tissue sarcomas [16], and many head–and–neck cancers [17]. Noteworthy, Fus1 levels are decreased in bronchial squamous metaplastic and dysplastic lesions and even in normal epithelium of smokers [13, 14] suggesting that Fus1 deficiency in bronchial epithelial cells occurs at early stages of lung carcinogenesis and is associated with tobacco smoke. Tumor suppressor properties of Fus1 were established in vitro in lung cancer cell lines and in vivo in mouse lung cancer xenografts [18–20]. At the molecular level, the tumor suppressor activity of Fus1 is associated with the inhibition of tyrosine kinase c-Abl and activation of the Apaf-1 apoptotic pathway [21, 22, 23].
Recent data revealed multiple levels and mechanisms of Fus1 regulation. Suppression of Fus1 protein levels via miRNAs miR-93, miR-98, and miR-197 was suggested as one of potential mechanisms of malignization of bronchial epithelial cells [24]. Certain mRNA sequence elements in the 5′- and 3′-untranslated regions were identified as regulatory for Fus1 protein and mRNA expression [24, 25]. Partial methylation of Fus1 promoter region was identified in head and neck tumors and normal salivary rinses as compared to normal mucosa [17]. Remarkably, two Fus1/TUSC2 pseudogenes (TUSC2P) found on chromosomes X and Y that are homologous to the 3′-UTR of TUSC2 were described recently as regulators of Fus1 activities. [26].
Stage I clinical trials of lipoparticles that deliver Fus1 transgene to compensate for Fus1 deficiency in tumors of patients with chemotherapy refractory stage IV lung cancer [27] showed no significant side effects, resulted in the uptake and expression of the gene by primary and metastatic tumors, and produced anti-tumor effects [28]. Introduction of cationic liposomes with Fus1/hIL-12 co-expression plasmid led to reduction of lung tumor growth in mice [29]. Based on these trials, a new strategy that combines Fus1 lipoparticles and other agents (LKB1 plasmid, AKT inhibitor MK2206) or established chemotherapy agents have been proposed [30, 31, 23, 31].
Thus, Fus1 protein is a classical tumor suppressor that is decreased in tumor cells via multiple molecular mechanisms and, if restored, could produce a potent anti-tumor effect.
Fus1 has a potential to control cancer and other pathologies via regulation of immune response and inflammation
It is commonly accepted that initial tumor growth often originates at sites with long-lasting chronic inflammation. Infiltration of immune cells to such sites creates specific inflammatory milieu (tumor-promoting cytokines, elevated ROS, etc.), which predisposes tissue cells to malignization and tumor growth [12, 32]. Chronic inflammatory processes affect all stages of tumor development [33]. Fus1-deficient mice display signs of chronic inflammation such as altered basal production of cytokines, NF-κB activation, and increased ROS levels in immune and epithelial cells [34]. Peritoneal exposure to asbestos, a carcinogenic mineral strongly linked to mesothelioma and lung cancer, resulted in a skewed cytokine response and more robust immune infiltration into the peritoneum of Fus1 KO mice [34]. Interestingly, in WT mice asbestos exposure led to down-regulation of Fus1 expression in cells of inflammatory milieu [34]. Our experiments with normal mesothelial cells of human origin exposed to asbestos revealed a ROS-dependent nature of Fus1 regulation [35] suggesting that ROS produced due to constant inflammation or environmental stress may keep Fus1 in epithelial and immune cells at a persistently low level. We also found that Fus1 knockdown in cancer cells confers resistance to oxidative stress [34].
Our in silico analysis of NCBI database GeoProfiles (http://www.ncbi.nlm.nih.gov/geoprofiles/) provided data on Fus1 expression in immune cells from different physiological and pathological states. We found that Fus1 is down-regulated in PBMC from multiple sclerosis patients (Ref # GDS3920) and in PBMC monocytes after interaction with bacteria Francisella tularensis (Ref # GDS3298). We also found that Fus1 level is decreasing during monocyte to macrophage differentiation. Interestingly, during subsequent macrophage polarization to M1 and M2, M1 macrophages showed 2-fold lower Fus1 level than M2 macrophages (Ref # GDS2429). At the same time, leukocytes and alveolar macrophages demonstrated up-regulation of Fus1 RNA upon activation with G-CSF (granulocyte colony-stimulating factor) (Ref # GDF2959) and LPS, respectively (Ref # GDS4419). Future experiments should further address dynamic changes of Fus1 expression in innate immune cells under polarizing conditions, during CD3/CD28 stimulation of T cells and Th1/Th2/Th17 polarization, during CD4+CD25+ Treg formation, etc.
At systemic level, Fus1 inactivation in mice led to development of multiple signs of autoimmunity reminiscent of systemic lupus erythematosus (SLE) [36]. Interestingly, adult Fus1 KO mice spontaneously developed tumors at the sites of chronic vessel inflammation, thus linking Fus1 loss to inflammation, autoimmunity, and tumorigenesis [36]. Thus, Fus1 as a suppressor of inflammation and carcinogenesis may serve as a therapeutic for treatment of inflammation-associated cancers, autoimmune and other inflammatory disorders.
Putative molecular structure and cellular functions of Fus1
The molecular structure and functions of Fus1 protein are still not well established. Fus1 is a basic, soluble, globular protein residing in the mitochondria [36]. Early in silico studies of the Fus1 structure predicted a protein kinase A and C (PKA and PKC) phosphorylation sites (a.a. 43–57) as well as a site for ERK kinase binding (a.a. 33–47) [19, 37]. Also, computational and protein chip analyses revealed in Fus1 protein a C-terminally-located A kinase anchor protein (AKAP) interface, a PDZ (PSD95/Dlg1/ZO-1) class II domain, and interaction with PDZ class I of the pro-apoptotic protein Apaf-1 [19, 21, 38, 39]. We identified similarity of Fus1 with the DNA-binding domain of the IRF7 (Interferon Regulatory transcription Factor 7) [35]. Uno et al. demonstrated that Fus1 possesses a site for N-terminal myristoylation, which is crucial for tumor suppressor activities of Fus1 [19]. Finally, our recent pairwise alignment analysis revealed an EF-hand Ca2+-binding domain and a hydrophobic pocket with a potential capacity to bind the myristoyl tail in the Fus1 protein [40]. These features allowed to classify Fus1/Tusc2 as a member of Ca2+/myristoyl switch protein family [41].
Fus1 activities related to cell functions are still not well characterized. Thus, cell cycle arrest and inhibition of lung cancer cell growth followed by Fus1 overexpression are consistent with the crucial role of Fus1 in cancer cell proliferation [18]. Synergism of Fus1 and p53 in the activation of the Apaf-1 apoptotic pathway suggests overlapping of the Fus1 and p53 pathways in the regulation of apoptosis in cancer cells [22]. Additionally, Fus1 displayed inhibitory effect toward tyrosine kinase c-Abl [21] and tyrosine kinase receptors, such as EGFR, PDGFR, and c-Kit [27], suggesting its crucial role in signal transduction. A recently established link between Fus1 and AMPK/AKT/mTOR signaling in cancer cells suggests involvement of Fus1 in cell survival and energetics [31]. However, molecular mechanisms of Fus1 involvement in these processes are still not determined.
Fus1 and mitochondrial homeostasis
In our earlier work, we established Fus1 as a nucleus-encoded protein that resides in mitochondria [36]. Its mitochondrial residency was confirmed by our recent data on Fus1 loss-mediated mitochondrial dysfunction. Thus, we found that Fus1 loss in immune cells results in elevation of mitochondrial membrane potential (ΔΨm) at resting and asbestos-activated states. Additionally, asbestos-activated Fus1 KO leukocytes showed excessive ROS production and high levels of a mitochondrial protein UCP2 that regulates ΔΨm [34]. Further, depletion of Fus1 in cancer cells led to ΔΨm and ROS increases accompanied by resistance to oxidative stress [34]. Immune response to pulmonary infection by Acinetobacter baumannii in Fus1 KO mice also led to increased ΔΨm, ROS production, and UCP2 expression in innate immune cells that may mediate resistance of Fus1 KO mice to non-lethal bacterial infection [42].
Based on these data, we suggest that Fus1 plays a key role in the control of inflammation and cancer cell growth via maintenance of mitochondrial homeostasis. These Fus1 activities may mediate a crucial link between cancer and inflammation.
MITOCHONDRIA IN CELLULAR CALCIUM HOMEOSTASIS AND CALCIUM SIGNALING
Mitochondrial Ca2+ transport and its effect on mitochondrial homeostasis
Mitochondrial Ca2+ transport consists of two opposing processes: uptake and extrusion. Uptake of the positively charged Ca2+ ions is driven by a negative potential on the inner membrane (IMM), which is formed by pumping protons into the intermembrane space (IMS) driven by the mitochondrial respiratory chain [43] (Fig. 1). Ca2+ ions enter the mitochondrial matrix through a highly selective channels formed by a transmembrane IMM protein MCU (mitochondrial Ca2+ uniporter; also named MCUa) [44, 45] (Fig. 1). Another protein, MICU1 (mitochondrial Ca2+ uniporter 1) and its two paralogs, MICU2 and MICU3, were identified as Ca2+ sensors. They have two EF-hand motifs, which face IMS, interact with MCU, and control Ca2+ threshold and its import [44, 46, 45, 47, 48]. MCUR1 (mitochondrial calcium uniporter regulator 1), a yet another component of Ca2+ import machine, has been described recently as MCU-interacting, but the mechanism of its action remains unknown [49]. Another layer of complexity to the organization of the MCU complex (MCUC) was added after the identification of two novel components - MCUb, an inhibitor of MCUa, and EMRE (Essential MCU REgulator) that connects MCUa with the MICU1/2 regulatory subunits [reviewed in [50]]. Finally, the roles of ryanodine receptor RyR1, uncoupling proteins UCP2/3, and Ca2+/H+ exchanger LETM1 in Ca2+ uptake have also been demonstrated [51, 52, 53].
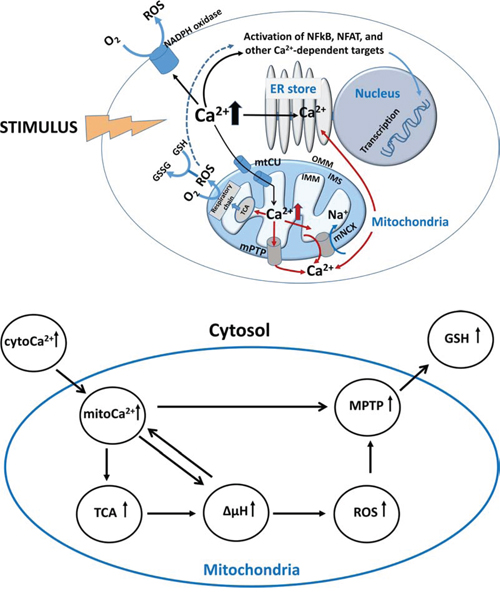
Figure 1: Ca2+/metabolic coupling in a cell. A. Ca2+ metabolism in response to cell stimulation. Stimulus (TCR triggering, cytokines, mediators, hormones, etc.) leads to Ca2+ elevation in cytosol and activation of Ca2+-dependent targets including transcriptional factors NF-κB and NFAT, NADPH oxidase and other proteins. Important intracellular store for Ca2+ accumulation is endoplasmic reticulum (ER). After reaching threshold for Ca2+ concentration in cytosol, mitochondria begins taking up Ca2+ ions via Ca2+ uniport (mtCU) located in the inner mitochondrial membrane (IMM). Inside mitochondria, Ca2+ stimulates dehydrogenases of tricarbonic cycle (TCA) and H+ transport into intermembrane space (IMS) resulting in the increased respiratory chain activities, rise of electrochemical membrane potential (ΔΨm), and production of reactive oxygen species (ROS). In turn, ROS alter redox potential in mitochondria and other intracellular compartments by metabolizing reduced glutathione (GSH) to its oxidized form (GSSG). Finally, mitochondria use two basic mechanisms to extrude Ca2+: mitochondrial Na+/Ca2+ exchanger (mNCX) and mitochondrial permeability transition pore (mPTP). Lower panel. B. Generalized scheme of Ca2+/metabolic coupling in mitochondria.
The directionality of Ca2+ ion transport, from the matrix towards the cytosol and vice versa, is maintained by Na+-dependent and -independent mechanisms. Na+-dependent export of Ca2+ is mediated via a mitochondrial Na+/Ca2+ exchanger (mNCX) represented by NCLX (alternative name is SLC8B1), which returns one Ca2+ ion back to the cytosol in exchange for three Na+ ions [54] (Fig. 1). mNCX is regulated by interactions with the number of proteins including protein kinases PKC and PINK1, SLP-2 and Bcl-2 [54, 55]. Finally, when Ca2+ concentration reaches the threshold, permeability transition pore (mPTP) provides mitochondria with Na+-independent Ca2+ extrusion from the matrix (Fig. 1). Since mitochondria possess high buffering capacity for Ca2+, mPTP prevents the formation of Ca2+ phosphate precipitates, which might destroy mitochondrial integrity. Thus, mitochondria employ several mechanisms for controlling Ca2+ levels, which are translated into signals for proliferation, differentiation, activation, etc.
Metabolic coupling in mitochondria
Different metabolic processes are tightly coordinated and cross-regulated in mitochondria (Fig. 1A and 1B). After Ca2+ enters mitochondria, it activates key enzymes of the tricarbonic (TCA) cycle (pyruvate, isocitrate and alpha-ketoglutarate dehydrogenases) fueling the respiratory chain with protons and thereby increasing ΔΨm [56]. With the ΔΨm elevation, levels of ROS production in mitochondria are increased in a non-linear mode [57, 58]. Single-electron reduction of oxygen by complex I and III leads to the formation of superoxide anion followed by its conversion to hydrogen peroxide (H2O2) driven by superoxide dismutase 2 (MnSOD) [59]. Further, H2O2 is transported into the cytosol where it gets involved in redox signaling or used by the mitochondrial catalase in metabolic reaction leading to production of water. Glutathione peroxidase (GPX) supplies the catalase reaction with reductive equivalents in expense of reduced glutathione (GSH), which, in turn, forms oxidized dimers (GSSG). During the reverse reaction, a reduction of GSSG to GSH, glutathione reductase (GR) uses NADPH as a source of reductive equivalents. It leads to the oxidation of NADH catalyzed by transhydrogenase (TH) resulting in decrease of charge on IMM (ΔΨm depolarization) since protons are directed towards the reduction of NAD+ for TH reaction but not for the formation of ΔΨm [60]. mNCX becomes activated after primary accumulation of Ca2+ in the mitochondrial matrix to prevent Ca2+ overload and the induction of apoptosis [61, 62, 63]. Thus, mNCX limits histamine-induced alterations in mitoCa2+ accumulation and production of NADP(H) by extrusion of Ca2+ from mitochondria [64]. However, excessive mNCX-mediated Ca2+ efflux could lead to deficiency in the activation of respiratory chain complexes, NAD/NADH disbalance, and drop in ATP synthesis [65, 66]. Due to inappropriate TCA cycle activation and diminished NADPH formation, it results in ROS overproduction [67, 68]. A generalized scheme of Ca2+/metabolic coupling is presented in Fig. 2B.
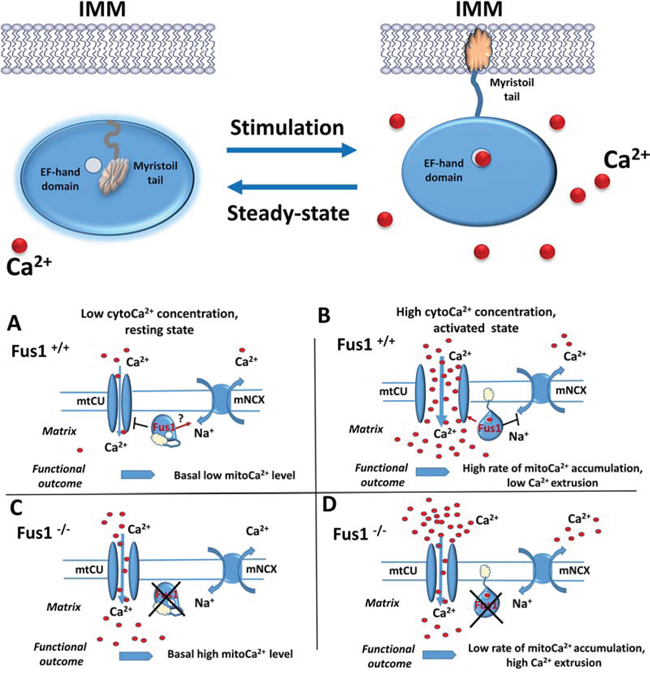
Figure 2: Hypothetic scheme of Fus1 functioning in mitochondria. Upper panel. Putative mechanism of molecular organization and activity of Fus1. At Ca2+- free or low Ca2+ conditions (steady-state), EF-hand domain (open circle) of Fus1 protein is not occupied by Ca2+ ions while its myristoyl tail is folded inside the hydrophobic pocket of the protein preventing Fus1 from anchoring the membranes (left). Upon stimulation, elevation of Ca2+ in cytosol (red circles) results in Ca2+ ion binding to the EF-hand domain, release of myristoyl tail and anchoring of Fus1 protein to IMM. Lower panel. A. At Ca2+-free or low Ca2+ conditions, Fus1 may inhibit mtCU while simultaneously maintain the mNCX active state thus preventing accumulating Ca2+ in mitochondria. B. Elevation of Ca2+ leads to Fus1 activation and anchoring in IMM. Membrane tethering allows Fus1 to activate mtCU and inhibit mNCX that leads to retention of Ca2+ in mitochondrial matrix. C. When Fus1-mediated gatekeeping function is lost at a steady state, the mtCU channel is not tightly closed allowing cytosolic Ca2+ uptake while mNCX does not return Ca2+ ions back to cytosol. Thus, Ca2+ concentration in mitochondria of Fus1-deficient cells is higher. D. When Fus1-mediated gatekeeping function is absent during activation, the mtCU channel does not open to a full extent leading to insufficient buffering of cytosolic Ca2+ signals. Additionally, mNCX is over-activated resulting in extrusion of Ca2+ from mitochondria and an overall slow rate of mitochondrial Ca2+ accumulation.
It is important to note that the levels of mitochondrial Ca2+, ROS, ΔΨm, and thiols define activities of mitochondrial channels such as inner membrane anion channel (IMAC) and mPTP, thereby defining cell fate [60, 69].
Overall, Ca2+ accumulation in mitochondria coordinates multiple coupled processes into cross-regulatory transduction loops and, thus, allows connecting metabolic demands with cell fate (apoptosis, proliferation, etc.).
Calcium-mediated cross-talk between mitochondria and ER
Currently, it is accepted that mitochondria shape intracellular Ca2+ signals, affect transcription of nuclear-encoded genes, and regulate multiple cellular functions [70, 71]. The mechanisms of mitochondrial involvement include the control of store-operated Ca2+ entry (SOCE) and cross-talk between mitochondria and endoplasmic reticulum (ER), a key calcium storage organelle [1, 2, 3]. SOCE represents a multi-step cascade of events that start from depletion of Ca2+ from ER induced by agonist stimulation that triggers Ca2+ entry across the plasma membrane. This process is tightly associated with the IP3 (inositol-3-phosphate) production that leads to the release of Ca2+ from the ER store. The decrease of ER Ca2+ levels is accompanied by dissociation of Ca2+ ions from EF-hands of STIM1 protein. This results in STIM1 conformational changes, its redistribution towards the plasma membrane, and interaction with an integral plasma membrane protein ORAI1, a pore subunit of Ca2+ Release-Activated Ca2+ (CRAC) channel. The interaction between ORAI1 and STIM1 maintains trans-membrane Ca2+ currents through CRAC resulting in the rise of free intracellular Ca2+. It was established that the magnitude of Ca2+ currents is strictly dependent on the strength of the ORAI1/STIM1 interaction, which is increased proportionally to the level of ER Ca2+ decrease. Upon Ca2+ elevations, CRAC and IP3 receptors are subjected to slow inactivation thus forming a negative regulatory loop [72, 73].
The involvement of mitochondria in SOCE includes buffering of Ca2+ released from IP3–induced ER store thereby promoting emptying ER stores and extended opening of CRAC channels. Moreover, due to the close proximity of mitochondria to the plasma membrane and ER stores, Ca2+ uptake by mitochondria reduces Ca2+-dependent slow inactivation of CRAC and IP3 receptors [2, 3].
Since mitochondria and ER form multiple contacts, the cross-talk between these two organelles plays an important role in the maintenance of overall Ca2+ homeostasis [74, 3] that occurs via several ways:
After initial uptake of Ca2+ mitochondria slowly release it through mNCX in the closest proximity to the ER Ca2+ ATPase pump (SERCA) thus refilling ER Ca2+ store.
Mitochondria-produced ROS oxidize proteins involved in Ca2+ transport and thus activate ryanodine (Ry) and inositol triphosphate (IP3) receptors involved in Ca2+ release and inhibit Ca2+ ATPases involved in Ca2+ uptake.
Mitochondria produce ATP that supplies SERCA and plasma membrane Ca2+ ATP pumps with energy.
Recently, the key role of MCU in SOCE-mediated Ca2+ transport has been confirmed. In particular, it was shown that MCU and UCP2 are required for decreasing inactivation of IP3 receptors and oligomerization and redistribution of STIM1, thus supporting store-operated Ca2+ entry [75, 76].
Role of mitochondria in cellular calcium oscillation
The first cytosolic Ca2+ spike determines generation of oscillatory Ca2+ signals in cytosol [77]. Being increased after cell activation, cytosolic Ca2+ is accumulated in mitochondria through Ca2+ uptake mechanisms. Following a drop in cytosolic Ca2+ concentration, mNCX releases Ca2+ ions from mitochondria that, due to the close proximity to ER, leads to sensitization of ryanodine and inositol triphosphate receptors and triggers subsequent (so-called regenerative) Ca2+ release from ER store. Cyclic repeats of Ca2+ shuttling between cytoplasmic, ER, and mitochondrial compartments form a pattern of Ca2+ oscillations [77]. The role of mitochondria in calcium oscillations has been shown for the number of cells including HeLa [77, 78], cardiomyocytes [79], neurons [80], astrocytes [81], etc. Significant role of mNCX was demonstrated for ER Ca2+ refilling and ER/mitochondria Ca2+ recycling in B-lymphocytes after B cell receptor activation [82]. The role of modulation of Ca2+ oscillations in the regulation of transcription factors’ activation is highlighted in the next chapter.
Global Ca2+ signals, mitochondrial Ca2+ accumulation, and regulation of transcription
Changes of cytosolic Ca2+ in response to cell activation are organized as oscillations with periodic drops and rises of Ca2+ [83, 84]. In 1997, it was established that frequency of Ca2+ oscillations determines the activities of NFAT and NF-κB, two major transcriptional factors important for T cell activation [85]. High-frequency Ca2+ oscillations with 6–7 min periodicity produce stimulatory effect on NFAT since its activation is linked with periodic cycles of phosphorylation-dephosphorylation by calcineurin [86]. On the contrary, the activity of NF-κB requires low-frequency Ca2+ signals since its activation is regulated by the degradation and re-synthesis of the protein inhibitor subunit IkB that takes a longer time span (about 30 min.) [87].
As described above, mitochondria regulate cellular Ca2+ oscillations in a cell type-specific manner. Thus, inhibition of mNCX with CGP37157 in HeLa cells leads to a switch of agonist-induced Ca2+ oscillations from high- to low-frequency pattern. At the same time, in fibroblasts with spontaneous Ca2+ oscillatory activity, CGP37157 enhances Ca2+ oscillation frequency [78]. This goes in line with the observation made on sensory neurons where impairment of trans-mitochondrial Ca2+ transport (this term refers to the combination of mitochondrial Ca2+ import and export) resulted in the elimination of intracellular Ca2+ plateau and diminished nuclear import of NFAT [80]. Altered Ca2+ accumulation in cells due to defects in trans-mitochondrial Ca2+ transport also leads to dephosphorylation of IkB and activation of NF-κB as well as ATF2 and the NF-κB-dependent HIF-1a axis, all of which have pro-tumorigenic activities [88]. Another example of decoding the frequency of Ca2+ oscillations is the modulation of Ca2+/calmodulin (CaM)-dependent kinase CaMKII activation. Higher-frequency Ca2+ oscillations or low-frequency spikes of long duration increase this enzyme activity through CaM trapping and, thus, maintain CaMKII autophosphorylation [89, 90]. Recently, Samanta et al. demonstrated that a pore-forming subunit of mtCU, MCU, is involved in the generation of Ca2+ oscillations evoked by physiological stimuli [91]. In particular, MCU maintains Ca2+ oscillatory pattern through sustained regenerative Ca2+ release and SOCE-mediated Ca2+ entry as well as via decrease in CRAC channel and IP3 receptor inactivation. As an outcome, knockdown of MCU leads to decrease in the activity of c-Fos and NFAT transcription factors [91]. Another mechanism of mitoCa2+ transport involvement in transcriptional regulation and metabolism has been described recently. Sustained mitochondrial Ca2+ loading regulates NAD+/NADH metabolism via TCA activation. Marcu et al. demonstrated that changes in the mitochondrial NAD+/NADH ratio can acutely influence protein acetylation by modulating both the activity and expression of sirtuins not only in mitochondria but also in the cytosolic and nuclear compartments [92].
Fus1 as a putative new mitochondrial Ca2+ sensor protein
Coordinated changes in ΔΨm and ROS production at the basal and activated states in immune cells with Fus1 KO prompted us to seek for common origin of these changes. Levels of ΔΨm and ROS are both dependent on the mitochondrial Ca2+ content [43, 93]. Using multiple protein alignment, we predicted that Fus1 has an EF-hand Ca2+ -binding domain and a hydrophobic pocket for interaction with the myristoyl tail [40], a saturated fatty acid covalently attached to the N-terminal glycine residue of the Fus1 protein [19]. This irreversible post-translational protein modification is called myristoylation. The similarity of the Fus1 protein structure to the one of the members of the Ca2+/myristoyl switch family allowed us to hypothesize that Fus1 is a potential Ca2+ sensor (Fig. 2, upper panel). The mechanism of action for Ca2+/myristoyl switch proteins was described about 20 years ago [94, 95, 41]. The proteins of Ca2+/myristoyl switch family have two main conformations (Fig. 2, upper panel). At a steady-state Ca2+ concentration, the myristoyl tail is clamped by aromatic residues inside the hydrophobic pocket. This conformation ensures inactive state of the proteins. When Ca2+ concentration raise (i.e., due to cell activation), Ca2+ ions bind to the EF-hand resulting in release of the myristoyl tail that anchors the protein to the membrane via intercalation into lipid bilayers (Fig. 2, upper panel). This mechanism helps to localize proteins to distinct signaling compartments, which is critical for coordinated regulation of signaling cascades [94, 41]. Recently, several myristoylated proteins residing in mitochondria have been described. Among them are proteins of the inner and outer mitochondrial membranes: GTPase ARL4D [96], ChChd3 [97], NADH-cytochrome b5 reductase [98, 99], truncated forms of actin [100], and Bid [101]. However, Fus1 is the first putative member of the Ca2+/myristoyl switch protein family localized to mitochondria. A distinct feature of the Fus1 protein structure is an overlap between the EF-hand (54–65 a.a.) and myristoyl-binding (45–110 a.a.) domains [40]. Proteins of the Ca2+/myristoyl switch family usually have these two regulatory elements separated from one another by a large amino acid stretch [41, 95]. However, in the ancient, evolutionary conserved Fus1 protein, Ca2+-binding motif (the EF-hand) resides inside the myristoyl-binding pocket [40].
The theoretically predicted Ca2+ sensor properties of Fus1 have been confirmed by measuring mitochondrial Ca2+ uptake in Fus1-deficient cells (Fig. 2). Depending on Ca2+ level in cytosol, Fus1 promoted a dual effect on the Ca2+ transport into mitochondria. At steady-state conditions, Fus1 maintained mitoCa2+ level by preventing its further uptake by mitochondria (Fig. 2, lower panel, A). However, during Ca2+ elevation in the cytosol induced by calcium agonist ionomycin or CD3/CD28 activation, Fus1 allowed accumulation of Ca2+ in mitochondria [34] (Fig. 2, lower panel, B). Thus, Fus1 belongs to a growing list of regulators of mitochondrial Ca2+ uptake identified in the last several years (MICU1–3, MCUR1 etc.) [102, 12].
We propose that being a Ca2+ sensor, Fus1 regulates a threshold for mitochondrial Ca2+ uptake. Indeed, basal Ca2+ levels in mitochondria of Fus1 KO cells were on average 2-fold higher than in WT cells (Fig. 2, lower panel, C). However, upon challenge with high-level Ca2+ pulses, mitochondria of Fus1 KO cells showed delayed and low-amplitude Ca2+ accumulations (Fig. 2, lower panel, D) [40]. Thus, Fus1 loss results in inefficient translation of cytosolic Ca2+ elevations into mitochondrial Ca2+ rises [40]. In this respect, Fus1 is similar to MICU1, a mitochondrial EF-hand Ca2+ sensor that interacts with the MCU pore subunits. MICU1 plays a gatekeeping role at low cytosolic Ca2+ concentrations while mediates an uptake of Ca2+ ions by mitochondria after stimulation with Ca2+-mobilizing agents [46, 103, 104, 105].
Cells possess a broad set of different Ca2+-binding EF-hand proteins for decoding Ca2+ signals. Each EF-hand protein has its own activation threshold that depends on the number and affinity of Ca2+-binding motifs. Thus, it is predicted that proteins containing only one such motif become activated earlier in response to Ca2+ changes than proteins with two or more Ca2+-binding domains, since occupation of more than one site with Ca2+ requires higher ion concentration [90]. Future comparative studies will determine if Fus1 that possesses a single Ca2+-binding domain [40] responds to calcium rises faster than proteins with multiple EF-hand domains.
Based on our experimental data [40], we suggest that one of the possible mechanisms of Fus1 involvement in Ca2+ regulation is inhibition of mNCX, a mitochondrial membrane antiporter protein that removes one Ca2+ ion from mitochondria in exchange for three Na+ ions and thus protects from excessive Ca2+ accumulation in mitochondria [54]. Noteworthy, similarly to Fus1 KO cells, MICU1 loss also results in the enhanced activity of mNCX [46]. In addition to mNCX regulation, Fus1 most likely employs other mechanisms of calcium control since mNCX inhibition by CGP37157 only partially restored accumulation of Ca2+ in mitochondria of Fus1-/- cells [40].
Taken together, these data point at the interaction of the described Ca2+-binding proteins with multiple targets aimed at the fine-tuning of mitochondrial Ca2+ transport through modulation of its uptake and extrusion. We hypothesize that this process might include several steps and involve mtCU activation and mNCX inhibition depending on the amplitude, frequency, and other parameters of Ca2+ response.
ROLES OF MITOCHONDRIA, CALCIUM, AND FUS1 IN TUMORIGENESIS AND IMMUNITY
Mitochondrial Ca2+ transport and tumorigenesis
Changes in intracellular Ca2+ concentration are used as a universal signaling mechanism to control diverse biological processes including proliferation and cell death, two hallmarks of tumorigenesis. Perturbation in the mechanisms of mitochondrial Ca2+ transport, e.g. via changes in the mtCU or mNCX activities, are translated into changes in signal transduction cascades as well as activities of transcription factors and their targets. Therefore, initial pro-tumorigenic events in cells as well as tumor cell fate are mediated via changes in intracellular calcium. Thus, it was shown that elevated cytosolic Ca2+ concentration increased activation of ERK1/2 and PKC that led to the activation of pro-tumorigenic transcription program [88]. Recently, LETM1, an EF-hand containing Ca2+/H+ transporter, was shown to control proliferation by shaping cellular bioenergetics via mitochondrial Ca2+ flux. Loss of LETM1 enhanced mitoROS production, AMPK (AMP activated kinase) activation, and promoted autophagy and cell cycle arrest [106].
Tumor cell survival was shown to be regulated by mitochondrial calcium fluxes [107]. Thus, aggressively growing malignant melanoma cells showed accelerated mitochondrial Ca2+ fluxes, which coincided with enhanced SOCE-mediated Ca2+ influx and high levels of constitutively active protein kinase B/Akt (PKB). Interruption of trans-mitochondrial Ca2+ transport abolished SOCE-mediated Ca2+ influx, inactivated PKB, slowed cell growth, and increased sensitivity to apoptosis [107].
The importance of NADPH oxidase (NOX) for tumor growth have been broadly demonstrated [108, 109]. The activity of NOX, which produces ROS and controls transcription of a number of genes, was shown to be dependent on mitochondrial calcium uniporter (mtCU). Suppression of mtCU leads to retention of Ca2+ in the cytosol and over-stimulation of PKC, which is necessary for phosphorylation of NADPH subunits and its activity [110].
The important role of mitochondria and Ca2+ in apoptosis makes mitochondrial Ca2+ transport an attractive target for designing novel therapeutic approaches for cancer treatment [111]. Indeed, some tumor suppressors and oncogenes promote their activities via modulation of mitochondrial Ca2+ uptake and, vice versa, established regulators of mitoCa2+ level demonstrated their involvement in carcinogenesis. Thus, pro-oncogenic constitutively active STAT3 protein inhibits expression of nuclear-encoded genes of the mitochondrial electron transport chain (ETC) thereby decreasing ΔΨm, a driving force for Ca2+ uptake, and, therefore, resulting in the decrease of mitoCa2+ levels [112]. Recently, it was shown that pro-tumorigenic, aberrantly high expression of miR-25 or its family members miR-92a and miR-363 selectively decreases calcium uniporter (MCU) levels and mitochondrial Ca2+ uptake that results ultimately in resistance to apoptosis and increased cancer cell survival [111, 113]. Furthermore, it was also shown that in the absence of MICU1, an essential regulator of mitochondrial Ca2+ threshold, mitochondria become constitutively loaded with Ca2+ triggering excessive ROS generation and increasing sensitivity to apoptotic stress [104]. Thus, MICU1 was characterized as a key molecule through which cancer cells acquire resistance to toxic effects of positively charged gold nanoparticles (+)AuNPs. Silencing of MICU1 allowed to overcome resistance to (+)AuNPs initiating the mitochondrial pathway for apoptosis [114]. Thus, modulation of mitochondrial Ca2+ levels by tumor suppressors and oncogenes is an important mechanism in cancer cell survival and apoptosis that could be targeted in anti-tumor therapeutic approaches.
Mitochondrial Ca2+ transport and T cell activation
Upon activation, T cells interact with professional antigen-presenting cells and form immune synapses (IS) with the latter. At the IS, the energy-consuming processes of ion transport take place. To meet energy demands, T cell activation is linked to mitochondrial translocation into the areas of the IS [115, 116] where mitochondria provide local ATP production and Ca2+ buffering. Local Ca2+ buffering at the IS facilitates larger and more sustained Ca2+ influx and concomitant activation of transcription factors NFAT, AP1, and NF-κB [115].
Ca2+ influx immobilizes mitochondria at the plasma membrane through interaction with the mitochondrial calcium-binding protein Miro, which is needed for kinesin-1-dependent mitochondrial transport mediated via an adaptor protein Milton [117, 118].
It was proposed that at the IS, increase in mitochondrial ATP synthesis occurs via mtCU-dependent activation of the TCA cycle [68]. Recently, a new mtCU-dependent mechanism of T cell activation was described by Ledderose et al. [119]. Immediately after activation, T lymphocytes rapidly increase ATP production in response to mtCU-mediated mitoCa2+ elevation. Consequently, ATP is translocated to the extracellular space by a hemi-channel Panx1 transporter where it binds to and activates membrane P2X purinergic receptors. These events help maintaining Ca2+ signaling in T cells [119].
Different signaling events during T cell activation are dependent on changes in mitochondrial Ca2+ levels. Thus, T cells activation requires production of mitochondrial ROS, which is induced by TCR-initiated calcium flux into mitochondria after mtCU opening that leads to increased complex III turnover [10, 111].
Major T helper cell subsets, Th1 and Th2, are distinct in their intracellular calcium handling. Thus, Th2 cells have lower activity of mtCU and mtCU-mediated CRAC currents and, therefore, slower Ca2+ influx. This explains a lower level of Th2 activation as compared to Th1 cells upon identical activating stimuli and production of different sets of cytokines [112].
Fus1-mediated calcium handling in the NFAT and NF-κB activation
NFAT and NF-κB are two major transcription factors (TFs) that drive the T cell activation program including expression of IL-2, IL-4, IL-6, IL-12, TNFa, IFNg, CD40L, FasL, etc [120, 121]. In our study, Fus1 KO CD4+ T cells showed elevated basal expression of NFAT and NF-κB target genes while the activation index for these genes (a fold difference in RNA expression before and after stimulation) was decreased as compared to WT CD4+ T cells [40]. These data point at aberrant activation of the NFAT and NF-κB pathways in Fus1–deficient T cells that may stem from alterations in mitochondrial Ca2+ accumulation, mitochondrial ROS production [9, 10], as well as mtCU and mNCX activities [80, 10, 91]. As mentioned above, in cancer cells increase in cytosolic Ca2+ leads to the activation of NFAT and NF-κB and, thus, to stimulation of a pro-tumorigenic gene network [88]. Prevention of the cytosolic Ca2+ uptake by mitochondria enhances resistance to apoptotic stimuli. On the contrary, excessive accumulation of mitoCa2+ via inhibition of mNCX results in cell death [62]. We showed that in normal epithelial cells, Fus1 loss alters accumulation of Ca2+ in mitochondria. Thus, in Fus1 KO cells mitoCa2+ level was elevated at basal conditions while at Ca2+-loading conditions it was decreased as compared to WT cells [40]. These changes in mitochondrial Ca2+ concentration correlated with chronic activation of NF-κB-dependent genes and unbalanced inflammatory response, thus making Fus1-deficient cells prone to malignant transformation and growth [40, 36].
How tumor suppressor proteins, mitochondrial calcium, anti-microbial response and autoimmunity are linked?
Fus1 is a typical tumor suppressor [18–20, 21, 22, 23] with a newly assigned function of a regulator of mitochondrial calcium handling [40]. For many years, roles of tumor suppressors and oncogenes in the immune system regulation have remained underappreciated. However, recent findings implicated many of these proteins in autoimmune disorders, while immuno-regulatory proteins (STAT3, STAT5, NF-κB, NFAT etc.) have been linked to tumorigenesis [122, 123, 124, 125, 126]. Mechanistically, many of these proteins execute their functions via modulation of Ca2+ signaling [127]. Thus, Bcl-2 protein, a member of the Bcl-2 family (Bcl-2, Bcl-xL, Bax, Mcl-1) that serves as a main barrier against autoimmune disorders [128], modulates calcium signaling via mtCU potentiation [129]. Anti-apoptotic Bcl-2 is also capable to suppress mtCU by raising the threshold for Ca2+ uptake in HEK293 cells [130, 131]. Another member of the Bcl-2 protein family, Mcl-1, was shown to play a key role in hematological and biliary malignancies as well as in fatal autoimmune disorders. Mcl-1 inhibits Ca2+ accumulation in mitochondria and prevents apoptosis, most likely through binding to p32, a positive regulator of mtCU [132, 133]. Mcl-1 plays a critical role in survival of Treg cells while other anti-apoptotic proteins, Bcl-2 and Bcl-xL, are dispensable for this process [134]. However, the importance of cooperation between Bcl-xL and transcription factor FoxP3 was demonstrated for development and persistence of Treg cells preventing arthritis [135]. Bcl-xL was found to be involved in the activation-induced cell death (AICD) initiated in T cells after TCR activation [136, 137]. Mechanistically, Bcl-xL interacts with VDAC promoting transport of Ca2+ from the cytosol to mitochondrial matrix and, thus, modulating calcium signaling [138]. In the absence of Bim, a tumor suppressor from the Bcl-2 family, T cells display impaired Ca2+ response and NFAT activation after TCR ligation due to prevalence of inhibitory interaction of Bcl-2 with IP3 receptor on the surface of ER. As a result, mice with targeted inactivation of Bim in hematopoietic system do not develop experimental autoimmune encephalomyelitis and induced diabetes [139].
Classical tumor suppressor p53 was shown to control Treg cell differentiation and, thus, protect against systemic autoimmune inflammation [140–142]. In cancer cells, p53 induces expression of a novel calcium channel TRPC6 and recruits it for Ca2+ release-dependent drug-induced apoptosis [143]. On the other hand, prevention of p53 transport to the nucleus and its re-localization to mitochondria lead to decreased accumulation of Ca2+ in mitochondria [144].
We showed that Fus1 KO mice spontaneously develop systemic lupus erythematosus, an autoimmune disease [36]. Ca2+ dynamics and associated signaling pathways are dramatically altered in autoimmune disorders [145–148]. Human peripheral blood lupus T cells are characterized by decreased NF-κB activation, elevated levels of ΔΨm, mitoCa2+ and cytoCa2+ after TCR activation [149]. It was reported that in inflammatory bowel disease, T cells from inflamed tissues have increased cytoCa2+ levels [150]. At the same time, importance of a proportional NF-κB activation was demonstrated in rheumatoid arthritis: enhanced TNFα-induced NF-κB activation led to diminished Ca2+ signaling but prolonged secretion of inflammatory cytokines [151]. Recently, the role of ORAI1 in autoimmunity has been established. Mouse T cells lacking SOCE Ca2+ current promoted via ORAI1 failed to induce colitis after adoptive transfer into lymphopenic mice [152]. Thus, organization of Ca2+ signaling in T cells meets demands of the complex systems controlled by different multi-level checkpoints to prevent pathological changes.
Regulatory T cells can suppress immune cell activities and, therefore, are important for evasion of tumor cells from immune surveillance [12]. Thus, a direct inhibitory effect on T lymphocytes or antigen-presenting cells was demonstrated for CD4+CD25+FoxP3+ Treg cells [153]. In our study, we found that in an asbestos-induced inflammatory environment favorable to tumor growth, Fus1 loss led to excessive amount of CD3+CD4-CD8- T cells called double-negative (DN) T cells that were accumulated locally in peritoneum [34]. Although the nature of DN T cells origin is not well defined, our observation of early down-regulation of surface CD4 molecule after activation of Fus1 KO CD4+ T cells [40] suggested that they could originate from CD4+ and CD8+ T cells after loss of the surface localization of CD4 or CD8 co-receptors, which is in-line with other studies [154]. Interestingly, patients with SLE, an autoimmune disease that Fus1 KO mice are prone to, have a significantly larger pool of circulated DN T cells [155]. Moreover, CD4+ T cells from SLE patients have decreased levels of surface CD4 and TCRzeta receptors due to increased activity of the HRES-1/Rab4 system involved in the endocytosis of cell surface molecules and regulated by mTOR [156, 157]. mTOR is regulated by multiple mechanisms and pathways including calcium signaling [158, 159, 160]. Reducing cytosolic calcium inhibits phosphorylation of S6K1, an mTOR effector [161]. In our study, bacteria-infected Fus1 KO lung tissues had higher levels of activated S6 protein, a downstream target of mTOR [42]. Mechanistically, lower mitochondrial Ca2+ and, as a result, higher cytoplasmic Ca2+ in Fus1 KO cells may activate mTOR and potentially promote differentiation of T cells towards the DN phenotype (Fig. 3). As a consequence, DN T cells could inhibit local immune response leading to the switch from acute to chronic disease course, which is conducive to cancer growth.
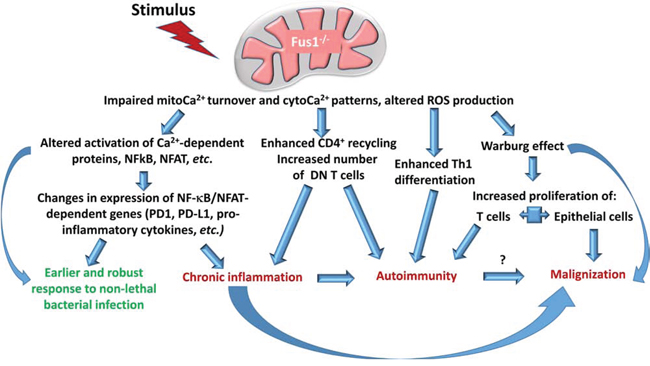
Figure 3: Role of Fus1 in the control of immune response and tumor growth. Stimulus (TCR triggering, cytokines, mediators, hormones, etc.) applied to Fus1-/- cells results in altered Ca2+ response due to impaired mitoCa2+ accumulation that, in turn, alters ROS response. These changes have several critical outcomes and include enhanced Th1 differentiation, changes in activity of NFkB and NFAT transcription factors, and potential predisposition to a profound metabolic switch (Warburg-like effect) providing these cells advantage toward proliferation. Taken together, it may lead to development of pathological systemic conditions (red font) such as chronic inflammation, autoimmune syndrome, or tumor growth. At the same time, these changes in immune cells may be advantageous (green font) allowing more effectively combat bacterial infections.
Th1 and CD8+ T cells represent hallmark subsets of autoimmunity since IL-2 and IFNy promote CD8+ T cell response directed towards parenchymal cells [162]. We showed that Fus1 deficiency leads to the shift in Th differentiation towards Th1 [40] that supports our early results on the autoimmune phenotype in Fus1 knockout mice [36]. The role of mitochondria in Th polarization is not well studied. However, it was shown that Th2 cells have more effective mechanisms of Ca2+ clearance from the cytosol while in Th1 cells cytosolic Ca2+ oscillate for the longer period of time after activation [163]. Furthermore, elevated ROS levels support Th2 polarization [164]. Thus, decreased mitochondrial Ca2+ accumulation, subsequent retention of Ca2+ in the cytosol, and decreased ROS production after stimulation in Fus1 KO T cells might promote Th1 stemming (Fig. 3). As a result, it will be accompanied by excessive production of IFN-gamma and tissue inflammation. We also cannot rule out that the autoimmune response induced by Fus1 loss may predispose to initiation of tumor growth since autoimmune inflammation has a chronic course.
Immune responses and autoimmunity are tightly controlled via expression of immunosuppressive/immunoregulatory molecules such as CTLA-4, PD-1/PD-L1, and BTLA that, in turn, are regulated via activation of transcription factors NFAT, NF-κB, STAT3 or STAT4 [165, 166, 167]. We demonstrated that Fus1 loss leads to a decreased PD-1 and PD-L1 expression in CD3/CD28-activated CD4+ T cells, which correlates with the impaired activation of NFAT/NF-κB-mediated transcription (Fig. 3) [40]. Thus, changes in the levels of immunoregulatory molecules may promote the autoimmune phenotype in Fus1 KO mice (Fig. 3). At the same time, we speculate that similar changes in the innate immune cells can enhance their protective potential. Indeed, PD-1-deficient dendritic cells demonstrated significantly improved protective response against Listeria [168].
In our study, we showed that Fus1 KO mice have increased resistance to A. baumannii pneumonia mediated by prompt and enhanced innate immune response to bacterial infection stemming from early activation of proinflammatory pathways (Fig. 3) [42]. The innate immune response is mediated by dendritic cells, neutrophils, macrophages, natural killers and some other cell types. Carrithers et al. demonstrated that in macrophages sodium channel NaV1.5 and sodium-calcium exchanger mNCX form a coupled system, which controls the generation of prolonged Ca2+ oscillations in the periphagosomal region important for uptake of mycobacteria [169]. Another molecular complex with important implication for anti-microbial defense is NADPH oxidase [170]. NADPH oxidase is sensitive to intracellular Ca2+ concentration since full activity of the enzyme requires stimulation of calcium-regulated PKC [171]. This signaling axis is controlled by mitochondrial Ca2+ uptake in a way that slower uptake results in retention of Ca2+ in cytosol and up-regulation of NADPH oxidase activity [110]. Loss of early effective Ca2+ sequestration of by mitochondria may result in increased activation of Ca2+-dependent enzymes such as NADPH oxidase, which is necessary in particular for activation of NLRP3, a component of inflammasome [172].
Fus1-mediated mitochondrial dysfunction may bring pro-tumorigenic changes not only to immune but also to epithelial cells (Fig. 3). Indeed, as we discussed above, Fus1 deficiency leads to a diminished mitochondrial Ca2+ uptake and, therefore, increased levels of Ca2+ in cytosol of epithelial cells, which could increase pro-survival potential of these cells as was shown for cancer cells [114, 104]. Moreover, since Ca2+ is involved in the activation of key TCA cycle enzymes, we assume that inefficient Ca2+ accumulation in mitochondria would reduce TCA cycle turnover, thus promoting switch to glycolytic metabolism (Warburg effect) (Fig. 3) [4–6]. Noteworthy, the same metabolic switch is crucial for activated T cells since glycolysis provides necessary level of NADPH equivalents, ATP and interplays with pentose-phosphate cycle fueling cells with monomers for nucleic acid, proteins, and lipid synthesis [4–6]. Thus, if Fus1-deficiency supports the same metabolic process in activated CD4+ T lymphocytes, it would result in chronic activation or permanent pre-activated T cells state as well as in triggering the avoiding mechanisms associated with immune tolerance (Fig. 3).
CONCLUDING REMARKS
Recent data suggest that many oncogenes, tumor suppressors, and immune regulators modulate mitochondrial homeostasis to perform their key activities. Calcium, a ubiquitous second messenger, is involved in a broad spectrum of physiological events. Remodeling of Ca2+ homeostasis that triggers changes in ROS and ATP production, calcium signaling and metabolic state is mediated through mitochondria and ER, two major intracellular calcium stores. Currently, there is as yet limited understanding of the role of specific modulators of Ca2+ metabolism/homeostasis/transport in controlling tumorigenesis and immune response. The mitochondrial protein Fus1, a tumor suppressor and immunoregulator highlighted in this review, is only “the tip of the iceberg” of a whole new concept on the key role of the calcium-regulating machinery in cancer and immunity [173, 174]. There is a long road ahead to determine the entire network maintaining and altering calcium homeostasis in a cell and assign a specific role to each of the Ca2+ regulators in cancer and immunity. Future pre-clinical studies should focus on designing/testing the potential agents (small molecules-, DNA-, or protein-based) that selectively target cancer- or immune cell-specific Ca2+ influx/efflux pathways and may serve as a basis for developing novel therapeutic interventions. Mitochondrial pharmacology nowadays is a fast developing field, which offers multiple tools to modulate mitochondrial processes (including Ca2+ transport, ROS production, etc.). These tools may be combined with classical schemes of chemo- or immunotherapy for maximal efficiency.
ACKNOWLEDGMENTS
We are grateful to Dr SV Ivanov for editorial help.
FUNDING
This work was supported by National Institutes of Health grants U54 CA163069, SC1 CA182843, U54 MD007593, R01 CA175370 and 1R21ES017496-01A1.
CONFLICTS OF INTEREST
No potential conflicts of interest.
REFERENCES
1. Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca(2)(+) and reactive oxygen species signaling. Journal of cell science. 2013; 126:2965–2978.
2. Parekh AB. Mitochondrial regulation of store-operated CRAC channels. Cell calcium. 2008; 44:6–13.
3. Watson R, Parekh AB. Mitochondrial regulation of CRAC channel-driven cellular responses. Cell calcium. 2012; 52:52–56.
4. Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Current opinion in immunology. 2014; 27:60–74.
5. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013; 342:1242454.
6. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunological reviews. 2012; 249:14–26.
7. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:8788–8793.
8. Gill T, Levine AD. Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction. The Journal of biological chemistry. 2013; 288:26246–26255.
9. Kaminski MM, Roth D, Sass S, Sauer SW, Krammer PH, Gulow K. Manganese superoxide dismutase: a regulator of T cell activation-induced oxidative signaling and cell death. Biochimica et biophysica acta. 2012; 1823:1041–1052.
10. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, Bryce PJ, Chandel NS. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013; 38:225–236.
11. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity. 2015; 42:406–417.
12. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 140:883–899.
13. Lerman MI, Minna JD. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer research. 2000; 60:6116–6133.
14. Prudkin L, Behrens C, Liu DD, Zhou X, Ozburn NC, Bekele BN, Minna JD, Moran C, Roth JA, Ji L, Wistuba II. Loss and reduction of FUS1 protein expression is a frequent phenomenon in the pathogenesis of lung cancer. Clin Cancer Res. 2008; 14:41–47.
15. Ivanov SV, Miller J, Lucito R, Tang C, Ivanova AV, Pei J, Carbone M, Cruz C, Beck A, Webb C, Nonaka D, Testa JR, Pass HI. Genomic events associated with progression of pleural malignant mesothelioma. Int J Cancer. 2009; 124:589–599.
16. Li G, Kawashima H, Ji L, Ogose A, Ariizumi T, Umezu H, Xu Y, Hotta T, Endo N. Frequent absence of tumor suppressor FUS1 protein expression in human bone and soft tissue sarcomas. Anticancer research. 31:11–21.
17. Demokan S, Chuang AY, Chang X, Khan T, Smith IM, Pattani KM, Dasgupta S, Begum S, Khan Z, Liegeois NJ, Westra WH, Sidransky D, Koch W, Califano JA. Identification of guanine nucleotide-binding protein gamma-7 as an epigenetically silenced gene in head and neck cancer by gene expression profiling. International journal of oncology. 2013; 42:1427–1436.
18. Kondo M, Ji L, Kamibayashi C, Tomizawa Y, Randle D, Sekido Y, Yokota J, Kashuba V, Zabarovsky E, Kuzmin I, Lerman M, Roth J, Minna JD. Overexpression of candidate tumor suppressor gene FUS1 isolated from the 3p21.3 homozygous deletion region leads to G1 arrest and growth inhibition of lung cancer cells. Oncogene. 2001; 20:6258–6262.
19. Uno F, Sasaki J, Nishizaki M, Carboni G, Xu K, Atkinson EN, Kondo M, Minna JD, Roth JA, Ji L. Myristoylation of the fus1 protein is required for tumor suppression in human lung cancer cells. Cancer research. 2004; 64:2969–2976.
20. Ito I, Ji L, Tanaka F, Saito Y, Gopalan B, Branch CD, Xu K, Atkinson EN, Bekele BN, Stephens LC, Minna JD, Roth JA, Ramesh R. Liposomal vector mediated delivery of the 3p FUS1 gene demonstrates potent antitumor activity against human lung cancer in vivo. Cancer Gene Ther. 2004; 11:733–739.
21. Lin J, Sun T, Ji L, Deng W, Roth J, Minna J, Arlinghaus R. Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene. 2007; 26:6989–6996.
22. Deng WG, Kawashima H, Wu G, Jayachandran G, Xu K, Minna JD, Roth JA, Ji L. Synergistic tumor suppression by coexpression of FUS1 and p53 is associated with down-regulation of murine double minute-2 and activation of the apoptotic protease-activating factor 1-dependent apoptotic pathway in human non-small cell lung cancer cells. Cancer research. 2007; 67:709–717.
23. Deng WG, Wu G, Ueda K, Xu K, Roth JA, Ji L. Enhancement of antitumor activity of cisplatin in human lung cancer cells by tumor suppressor FUS1. Cancer Gene Ther. 2008; 15:29–39.
24. Du L, Schageman JJ, Subauste MC, Saber B, Hammond SM, Prudkin L, Wistuba II, Ji L, Roth JA, Minna JD, Pertsemlidis A. miR-93, miR-98, and miR-19 regulate expression of tumor suppressor gene FUS1. Mol Cancer Res. 2009; 7:1234–1243.
25. Lin J, Xu K, Gitanjali J, Roth JA, Ji L. Regulation of tumor suppressor gene FUS1 expression by the untranslated regions of mRNA in human lung cancer cells. Biochemical and biophysical research communications. 2011; 410:235–241.
26. Rutnam ZJ, Du WW, Yang W, Yang X, Yang BB. The pseudogene TUSC2P promotes TUSC2 function by binding multiple microRNAs. Nature communications. 2014; 5:2914.
27. Ji L, Roth JA. Tumor suppressor FUS1 signaling pathway. J Thorac Oncol. 2008; 3:327–330.
28. Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME, Grimm EA, Reuben JM, Baladandayuthapani V, Templeton NS, McMannis JD, Roth JA. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PloS one. 2012; 7:e34833.
29. Ren J, Yu C, Wu S, Peng F, Jiang Q, Zhang X, Zhong G, Shi H, Chen X, Su X, Luo X, Zhu W, Wei Y. Cationic liposome mediated delivery of FUS1 and hIL-12 coexpression plasmid demonstrates enhanced activity against human lung cancer. Current cancer drug targets. 2014; 14:167–180.
30. Li L, Yu C, Ren J, Ye S, Ou W, Wang Y, Yang W, Zhong G, Chen X, Shi H, Su X, Chen L, Zhu W. Synergistic effects of eukaryotic coexpression plasmid carrying LKB1 and FUS1 genes on lung cancer in vitro and in vivo. Journal of cancer research and clinical oncology. 2014; 140:895–907.
31. Meng J, Majidi M, Fang B, Ji L, Bekele BN, Minna JD, Roth JA. The tumor suppressor gene TUSC2 (FUS1) sensitizes NSCLC to the AKT inhibitor MK2206 in LKB1-dependent manner. PloS one. 2013; 8:e77067.
32. Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011; 35:467–477.
33. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nature reviews Cancer. 2013; 13:759–771.
34. Uzhachenko R, Issaeva N, Boyd K, Ivanov SV, Carbone DP, Ivanova AV. Tumour suppressor Fus1 provides a molecular link between inflammatory response and mitochondrial homeostasis. J Pathol. 2012; 227:456–469.
35. Ivanova AV, Ivanov SV, Prudkin L, Nonaka D, Liu Z, Tsao A, Wistuba I, Roth J, Pass HI. Mechanisms of FUS1/TUSC2 deficiency in mesothelioma and its tumorigenic transcriptional effects. Molecular cancer. 2009; 8:91.
36. Ivanova AV, Ivanov SV, Pascal V, Lumsden JM, Ward JM, Morris N, Tessarolo L, Anderson SK, Lerman MI. Autoimmunity, spontaneous tumourigenesis, and IL-15 insufficiency in mice with a targeted disruption of the tumour suppressor gene Fus1. J Pathol. 2007; 211:591–601.
37. Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene. 2007; 26:7283–7301.
38. Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008; 27:4385–4391.
39. Jayachandran G, Roth JA, Ji L. Analysis of protein-protein interaction using proteinchip array-based SELDI-TOF mass spectrometry. Methods in molecular biology. 2012; 818:217–226.
40. Uzhachenko R, Ivanov SV, Yarbrough WG, Shanker A, Medzhitov R, Ivanova AV. Fus1/Tusc2 is a novel regulator of mitochondrial calcium handling, Ca2+-coupled mitochondrial processes, and Ca2+-dependent NFAT and NF-kappaB pathways in CD4+ T cells. Antioxidants & redox signaling. 2014; 20:1533–1547.
41. Zozulya S, Stryer L. Calcium-myristoyl protein switch. Proceedings of the National Academy of Sciences of the United States of America. 1992; 89:11569–11573.
42. Hood MI, Uzhachenko R, Boyd K, Skaar EP, Ivanova AV. Loss of mitochondrial protein Fus1 augments host resistance to Acinetobacter baumannii infection. Infection and immunity. 2013; 81:4461–4469.
43. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004; 287:C817–833.
44. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011; 476:341–345.
45. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011; 476:336–340.
46. Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell metabolism. 2013; 17:976–987.
47. Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010; 467:291–296.
48. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PloS one. 2013; 8:e55785.
49. Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, et al. MCUR1 is an essential component of mitochondrial Ca(2+) uptake that regulates cellular metabolism. Nature cell biology. 2012; 14:1336–1343.
50. Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial Ca(2+) uniporter. Cell calcium. 2014; 55:139–145.
51. Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochimica et biophysica acta. 2005; 1717:1–10.
52. Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nature cell biology. 2007; 9:445–452.
53. Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009; 326:144–147.
54. Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. The Journal of biological chemistry. 2012; 287:31650–31657.
55. Palty R, Sekler I. The mitochondrial Na(+)/Ca(2+) exchanger. Cell calcium. 2012; 52:9–15.
56. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012; 51:2959–2973.
57. Brookes PS. Mitochondrial H(+) leak and ROS generation: an odd couple. Free radical biology & medicine. 2005; 38:12–23.
58. Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of signal transduction. 2012; 2012:329635.
59. Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009; 417:1–13.
60. Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. The Journal of biological chemistry. 2007; 282:21889–21900.
61. Ben-Hail D, Palty R, Shoshan-Barmatz V. Measurement of mitochondrial Ca2+ transport mediated by three transport proteins: VDAC1, the Na+/Ca2+ exchanger, and the Ca2+ uniporter. Cold Spring Harbor protocols. 2014; 2014:161–166.
62. Kaddour-Djebbar I, Choudhary V, Brooks C, Ghazaly T, Lakshmikanthan V, Dong Z, Kumar MV. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. International journal of oncology. 2010; 36:1437–1444.
63. Takeuchi A, Kim B, Matsuoka S. The destiny of Ca(2+) released by mitochondria. The journal of physiological sciences : JPS. 2015; 65:11–24.
64. De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. The Journal of biological chemistry. 2014; 289:20377–20385.
65. Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circulation research. 2008; 103:279–288.
66. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circulation research. 2006; 99:172–182.
67. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010; 121:1606–1613.
68. Liu T, O’Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. Journal of bioenergetics and biomembranes. 2009; 41:127–132.
69. Halestrap AP. What is the mitochondrial permeability transition pore? Journal of molecular and cellular cardiology. 2009; 46:821–831.
70. Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013; 13:577–591.
71. Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends in cell biology. 2014; 24:761–770.
72. Bergmeier W, Weidinger C, Zee I, Feske S. Emerging roles of store-operated Ca(2)(+) entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels. 2013; 7:379–391.
73. Shaw PJ, Qu B, Hoth M, Feske S. Molecular regulation of CRAC channels and their role in lymphocyte function. Cellular and molecular life sciences: CMLS. 2013; 70:2637–2656.
74. Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. Landmark Ed2009; 14:1197–1218.
75. Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, Graier WF, Malli R. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. Journal of cell science. 2014; 127:2944–2955.
76. Samanta K, Bakowski D, Parekh AB. Key role for store-operated Ca2+ channels in activating gene expression in human airway bronchial epithelial cells. PloS one. 2014; 9:e105586.
77. Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO reports. 2006; 7:390–396.
78. Hernandez-SanMiguel E, Vay L, Santo-Domingo J, Lobaton CD, Moreno A, Montero M, Alvarez J. The mitochondrial Na+/Ca2+ exchanger plays a key role in the control of cytosolic Ca2+ oscillations. Cell calcium. 2006; 40:53–61.
79. Takeuchi A, Kim B, Matsuoka S. The mitochondrial Na+-Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Scientific reports. 2013; 3:2766.
80. Kim MS, Usachev YM. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009; 29:12101–12114.
81. Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, Sekler I, Nolte C. Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013; 33:7206–7219.
82. Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. Pivotal role of mitochondrial Na(+)(-)Ca(2)(+) exchange in antigen receptor mediated Ca(2)(+) signalling in DT40 and A20 B lymphocytes. The Journal of physiology. 2012; 590:459–474.
83. Dupont G, Combettes L, Bird GS, Putney JW. Calcium oscillations. Cold Spring Harbor perspectives in biology. 2011; 3.
84. Smedler E, Uhlen P. Frequency decoding of calcium oscillations. Biochimica et biophysica acta. 2014; 1840:964–969.
85. Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997; 386:855–858.
86. Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998; 392:933–936.
87. Lewis RS. Calcium oscillations in T-cells: mechanisms and consequences for gene expression. Biochemical Society transactions. 2003; 31:925–929.
88. Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Frontiers in oncology. 2013; 3:292.
89. De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998; 279:227–230.
90. Parekh AB. Decoding cytosolic Ca2+ oscillations. Trends in biochemical sciences. 2011; 36:78–87.
91. Samanta K, Douglas S, Parekh AB. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PloS one. 2014; 9:e101188.
92. Marcu R, Wiczer BM, Neeley CK, Hawkins BJ. Mitochondrial matrix Ca(2)(+) accumulation regulates cytosolic NAD(+)/NADH metabolism, protein acetylation, and sirtuin expression. Molecular and cellular biology. 2014; 34:2890–2902.
93. Cortassa S, Aon MA, Marban E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003; 84:2734–2755.
94. Tanaka T, Ames JB, Harvey TS, Stryer L, Ikura M. Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature. 1995; 376:444–447.
95. Ames JB, Ishima R, Tanaka T, Gordon JI, Stryer L, Ikura M. Molecular mechanics of calcium-myristoyl switches. Nature. 1997; 389:198–202.
96. Li CC, Wu TS, Huang CF, Jang LT, Liu YT, You ST, Liou GG, Lee FJ. GTP-binding-defective ARL4D alters mitochondrial morphology and membrane potential. PloS one. 2012; 7:e43552.
97. Darshi M, Trinh KN, Murphy AN, Taylor SS. Targeting and import mechanism of coiled-coil helix coiled-coil helix domain-containing protein 3 (ChChd3) into the mitochondrial intermembrane space. The Journal of biological chemistry. 2012; 287:39480–39491.
98. Borgese N, Aggujaro D, Carrera P, Pietrini G, Bassetti M. A role for N-myristoylation in protein targeting: NADH-cytochrome b5 reductase requires myristic acid for association with outer mitochondrial but not ER membranes. The Journal of cell biology. 1996; 135:1501–1513.
99. Colombo S, Longhi R, Alcaro S, Ortuso F, Sprocati T, Flora A, Borgese N. N-myristoylation determines dual targeting of mammalian NADH-cytochrome b5 reductase to ER and mitochondrial outer membranes by a mechanism of kinetic partitioning. The Journal of cell biology. 2005; 168:735–745.
100. Utsumi T, Sakurai N, Nakano K, Ishisaka R. C-terminal 15 kDa fragment of cytoskeletal actin is posttranslationally N-myristoylated upon caspase-mediated cleavage and targeted to mitochondria. FEBS letters. 2003; 539:37–44.
101. Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science. 2000; 290:1761–1765.
102. Hajnoczky G, Booth D, Csordas G, Debattisti V, Golenar T, Naghdi S, Niknejad N, Paillard M, Seifert EL, Weaver D. Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Current opinion in cell biology. 2014; 29:133–141.
103. de la Fuente S, Matesanz-Isabel J, Fonteriz RI, Montero M, Alvarez J. Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. The Biochemical journal. 2014; 458:33–40.
104. Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012; 151:630–644.
105. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Molecular cell. 2014; 53:726–737.
106. Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK, Cheung JY, Houser SR, Madesh M. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014; 28:4936–49.
107. Feldman B, Fedida-Metula S, Nita J, Sekler I, Fishman D. Coupling of mitochondria to store-operated Ca(2+)-signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell calcium. 2010; 47:525–537.
108. Bauer G. Targeting extracellular ROS signaling of tumor cells. Anticancer research. 2014; 34:1467–1482.
109. Bonner MY, Arbiser JL. Targeting NADPH oxidases for the treatment of cancer and inflammation. Cellular and molecular life sciences : CMLS. 2012; 69:2435–2442.
110. Dikalov SI, Li W, Doughan AK, Blanco RR, Zafari AM. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. American journal of physiology Regulatory, integrative and comparative physiology. 2012; 302:R1134–1142.
111. Marchi S, Pinton P. Mitochondrial calcium uniporter, MiRNA and cancer: Live and let die. Communicative & integrative biology. 2013; 6:e23818.
112. Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, Gough DJ, Turkson J, Levy DE, Watson CJ, Wieckowski MR, Provero P, Pinton P, Poli V. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging. 2010; 2:823–842.
113. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Current biology: CB. 2013; 23:58–63.
114. Arvizo RR, Moyano DF, Saha S, Thompson MA, Bhattacharya R, Rotello VM, Prakash YS, Mukherjee P. Probing novel roles of the mitochondrial uniporter in ovarian cancer cells using nanoparticles. The Journal of biological chemistry. 2013; 288:17610–17618.
115. Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, Hoth M. T cell activation requires mitochondrial translocation to the immunological synapse. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:14418–14423.
116. Baixauli F, Martin-Cofreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E, Serrador JM, Sanchez-Madrid F. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. The EMBO journal. 30:1238–1250.
117. Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009; 136:163–174.
118. Junker C, Hoth M. Immune synapses: mitochondrial morphology matters. The EMBO journal. 30: 1187–1189.
119. Ledderose C, Bao Y, Lidicky M, Zipperle J, Li L, Strasser K, Shapiro NI, Junger WG. Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling. The Journal of biological chemistry. 2014; 289:25936–25945.
120. Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 109 Suppl. 2002; 109:S67–79.
121. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor perspectives in biology. 2009; 1:a000034.
122. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer. 2013; 12:86.
123. Bourgeais J, Gouilleux-Gruart V, Gouilleux F. Oxidative metabolism in cancer: A STAT affair? Jak-Stat. 2013; 2:e25764.
124. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harbor perspectives in biology. 2009; 1:a000141.
125. Qin JJ, Nag S, Wang W, Zhou J, Zhang WD, Wang H, Zhang R. NFAT as cancer target: mission possible? Biochimica et biophysica acta. 2014; 1846:297–311.
126. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nature reviews Cancer. 2014; 14:736–746.
127. Akl H, Bultynck G. Altered Ca(2+) signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochimica et biophysica acta. 2013; 1835:180–193.
128. Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell death & disease. 2010; 1:e48.
129. Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:9893–9898.
130. Hanson CJ, Bootman MD, Distelhorst CW, Maraldi T, Roderick HL. The cellular concentration of Bcl-2 determines its pro- or anti-apoptotic effect. Cell calcium. 2008; 44:243–258.
131. Hanson CJ, Bootman MD, Distelhorst CW, Wojcikiewicz RJ, Roderick HL. Bcl-2 suppresses Ca2+ release through inositol 1, 5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell calcium. 2008; 44:324–338.
132. Minagawa N, Kruglov EA, Dranoff JA, Robert ME, Gores GJ, Nathanson MH. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. The Journal of biological chemistry. 2005; 280:33637–33644.
133. Xiao K, Wang Y, Chang Z, Lao Y, Chang DC. p32, a novel binding partner of Mcl-1, positively regulates mitochondrial Ca(2+) uptake and apoptosis. Biochemical and biophysical research communications. 2014; 451:322–328.
134. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, Humblet-Baron S, Schonefeldt S, Herold MJ, Hildeman D, Strasser A, Bouillet P, Lu LF, Matthys P, Freitas AA, Luther RJ, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nature immunology. 2013; 14:959–965.
135. Haque R, Lei F, Xiong X, Wu Y, Song J. FoxP3 and Bcl-xL cooperatively promote regulatory T cell persistence and prevention of arthritis development. Arthritis research & therapy. 2010; 12:R66.
136. Koenen P, Heinzel S, Carrington EM, Happo L, Alexander WS, Zhang JG, Herold MJ, Scott CL, Lew AM, Strasser A, Hodgkin PD. Mutually exclusive regulation of T cell survival by IL-7R and antigen receptor-induced signals. Nature communications. 2013; 4:1735.
137. Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, Ho B, Chen J, Ding JL. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell death and differentiation. 2013; 20:478–489.
138. Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. The Journal of biological chemistry. 2013; 288:19870–19881.
139. Ludwinski MW, Sun J, Hilliard B, Gong S, Xue F, Carmody RJ, DeVirgiliis J, Chen YH. Critical roles of Bim in T cell activation and T cell-mediated autoimmune inflammation in mice. The Journal of clinical investigation. 2009; 119:1706–1713.
140. Campbell HG, Slatter TL, Jeffs A, Mehta R, Rubio C, Baird M, Braithwaite AW. Does Delta133p53 isoform trigger inflammation and autoimmunity? Cell cycle. 2012; 11:446–450.
141. Kawashima H, Takatori H, Suzuki K, Iwata A, Yokota M, Suto A, Minamino T, Hirose K, Nakajima H. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. Journal of immunology. 2013; 191:3614–3623.
142. Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, Chen YH. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005; 54:1423–1428.
143. Madan E, Gogna R, Keppler B, Pati U. p53 increases intra-cellular calcium release by transcriptional regulation of calcium channel TRPC6 in GaQ3-treated cancer cells. PloS one. 2013; 8:e71016.
144. Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, Mantovani F, Del Sal G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell death and differentiation. 2013; 20:198–208.
145. Tsokos GC. Calcium signaling in systemic lupus erythematosus lymphocytes and its therapeutic exploitation. Arthritis and rheumatism. 2008; 58:1216–1219.
146. Feske S. Calcium signalling in lymphocyte activation and disease. Nature reviews Immunology. 2007; 7:690–702.
147. Izquierdo JH, Bonilla-Abadia F, Canas CA, Tobon GJ. Calcium, channels, intracellular signaling and autoimmunity. Reumatologia clinica. 2014; 10:43–47.
148. Zhang W, Zhou Q, Xu W, Cai Y, Yin Z, Gao X, Xiong S. DNA-dependent activator of interferon-regulatory factors (DAI) promotes lupus nephritis by activating the calcium pathway. The Journal of biological chemistry. 2013; 288:13534–13550.
149. Perl A, Fernandez DR, Telarico T, Doherty E, Francis L, Phillips PE. T-cell and B-cell signaling biomarkers and treatment targets in lupus. Curr Opin Rheumatol. 2009; 21:454–464.
150. Schwarz A, Tutsch E, Ludwig B, Schwarz EC, Stallmach A, Hoth M. Ca2+ signaling in identified T-lymphocytes from human intestinal mucosa. Relation to hyporeactivity, proliferation, and inflammatory bowel disease. The Journal of biological chemistry. 2004; 279:5641–5647.
151. Cope AP. Studies of T-cell activation in chronic inflammation. Arthritis research. 2002; 4:S197–211.
152. McCarl CA, Khalil S, Ma J, Oh-hora M, Yamashita M, Roether J, Kawasaki T, Jairaman A, Sasaki Y, Prakriya M, Feske S. Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. Journal of immunology. 2010; 185:5845–5858.
153. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunological reviews. 2014; 259:88–102.
154. Hillhouse EE, Lesage S. A comprehensive review of the phenotype and function of antigen-specific immunoregulatory double negative T cells. Journal of autoimmunity. 2013; 40:58–65.
155. Crispin JC, Tsokos GC. Human TCR-alpha beta+ CD4- CD8- T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. Journal of immunology. 2009; 183:4675–4681.
156. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, Phillips PE, Crow MK, Oess S, Muller-Esterl W, Perl A. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. Journal of immunology. 2009; 182:2063–2073.
157. Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discovery medicine. 2010; 9:173–178.
158. Wing LY, Chen HM, Chuang PC, Wu MH, Tsai SJ. The mammalian target of rapamycin-p70 ribosomal S6 kinase but not phosphatidylinositol 3-kinase-Akt signaling is responsible for fibroblast growth factor-9-induced cell proliferation. The Journal of biological chemistry. 2005; 280:19937–19947.
159. Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nature medicine. 2013; 19:101–106.
160. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nature reviews Immunology. 2012; 12:325–338.
161. Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes. 2004; 53:S225–232.
162. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. Journal of autoimmunity. 2008; 31:252–256.
163. Fanger CM, Neben AL, Cahalan MD. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. Journal of immunology. 2000; 164:1153–1160.
164. Frossi B, De Carli M, Piemonte M, Pucillo C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Molecular immunology. 2008; 45:58–64.
165. Murakami N, Riella LV. Co-inhibitory pathways and their importance in immune regulation. Transplantation. 2014; 98:3–14.
166. Pasero C, Olive D. Interfering with coinhibitory molecules: BTLA/HVEM as new targets to enhance anti-tumor immunity. Immunology letters. 2013; 151:71–75.
167. Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. Journal of immunology. 2014; 192:4876–4886.
168. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi IH, Tamada K, Chen L. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009; 113:5811–5818.
169. Carrithers LM, Hulseberg P, Sandor M, Carrithers MD. The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS immunology and medical microbiology. 2011; 63:319–327.
170. Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS. Regulation of innate immunity by NADPH oxidase. Free radical biology & medicine. 2012; 53:72–80.
171. Nunes P, Demaurex N, Dinauer MC. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic. 2013; 14:1118–1131.
172. Sun B, Wang X, Ji Z, Wang M, Liao YP, Chang CH, Li R, Zhang H, Nel AE, Xia T. NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Lung Fibrosis by Multiwalled Carbon Nanotubes. Small. 2015; 11:2087–97.
173. Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochimica et biophysica acta. 2009; 1787:1342–1351.
174. Csordas G, Hajnoczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochimica et biophysica acta. 2009; 1787:1352–1362.