
INTRODUCTION
Oral cancer, of which the vast majority of cases are oral squamous cell carcinoma (OSCC), is the major subtype of head and neck cancers. Epidemiologic studies have indicated that cigarette smoking might play a major role in the etiology of oral cancer [1–3]. Studies have estimated the etiological fraction of oral cancer in men attributable to smoking at 70% [4]. Using the mortality data from countries and regions such as Japan, Europe, Australasia, and North America, previous epidemiologic studies have indicated that the major etiological factor responsible for death from oral cancers is smoking followed by heavy alcohol consumption [5, 6]. Overall, these studies provide convincing evidence of an association between cigarette smoking and OSCC.
Promoter hypermethylation plays a crucial role in the inactivation of tumor suppressor genes during carcinogenesis. Accumulating evidence suggests that cigarette smoke might influence the development of various human diseases by inducing epigenetic changes. A genome-wide study revealed the loci of peripheral-blood DNA displaying differing methylation levels in current smokers, former smokers, and people who had never smoked [7]. By using pyrosequencing technology focused on DNA repetitive sequences and some tumor suppressor genes, Liu et al. demonstrated that cigarette smoke condensate (CSC) induces progressive global genomic hypomethylation and locoregional DNA hypermethylation in cultured human respiratory epithelia after incubation for up to 9 months [8]. Lin et al identified that the tobacco-specific carcinogen nicotine-derived nitrosamine ketone (NNK) induces the accumulation of DNA methyltransferase 1 (DNMT1) and the promoter hypermethylation of tumor suppressor genes, such as FHIT, p16INK4a, and RARB, in lung cancer patients [9]. Our previous study revealed global epigenetic aberrations in smoking-associated OSCC patients, and identified BEX1 and LDOC1 as 2 X-linked tumor suppressor genes with promoter methylated in 75% and 89% of OSCC tumor samples, respectively [10]. In this study, we investigated whether cigarette exposure induces profound epigenetic changes in oral cells, causing the silencing of tumor suppressor genes through promoter DNA methylation, which is involved in the development of oral cancer.
RESULTS
CSC exposure changes DNA methylation content of oral cells
To determine the effects of smoking on the global DNA methylation content of oral cells, we measured genomic 5-methyl-2-deoxycytidine (5mC) in CGHNK6 (an immortalized untransformed oral keratinocyte cell line) [11] and DOK (a dysplastic oral keratinocyte derived from a heavy smoker with OSCC) [12] cells after CSC exposure by using an enzyme-linked immunoassay (EIA)-based method. The genomic 5mC content of CGHNK6 cells changed markedly, with a significant (P < 0.01) increase at 4 and 6 weeks, followed by a decrease (P < 0.05) at 12 weeks in the CSC-treated cells compared with that in the DMSO-treated (vehicle control) cells (Figure 1). The CSC treatment resulted in a significant (P < 0.01) increase in the genomic 5mC content at 10 and 15 days in the DOK cells compared with that in the untreated and vehicle control cells. These results suggested that cigarette smoking modifies the DNA methylation content of oral untransformed CGHNK6 or partially transformed DOK cells.
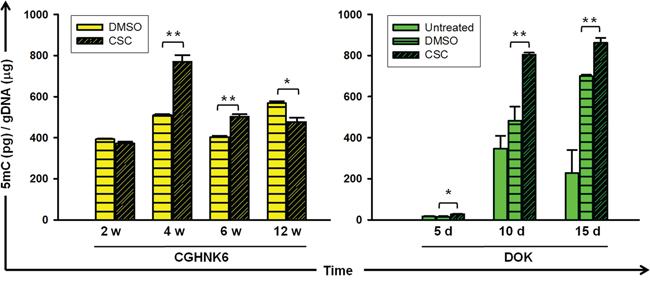
Figure 1: Genomic 5-methyl cytosine in oral cells with or without cigarette smoke exposure. The amount of genomic 5mC was quantified using a commercial EIA kit in CGHNK6, and DOK cells. The treatment conditions for CGHNK6 and DOK were with or without of CSC (0.1 μg/mL) exposure for indicated time. DMSO-treated cells were used as vehicle controls for the CSC-treated cells. Data are presented as mean ± SD (n = 3). *p < 0.05 and **p < 0.01.
CSC changes the nuclear accumulation of DNMT1 and DNMT3A in oral cells
S-adenosyl-methionine (SAM) is the major physiological methyl donor of DNMTs, including DNMT1, DNMT3A, and DNMT3B, which serve as the key enzymes in DNA methylation (Figure 2A). The ratio of intracellular SAM to its demethylated metabolite S-adenosyl-homocysteine (SAH) might provide an indirect indicator of DNMT activities, with an inverse correlation existing between the ratio of SAM/SAH and total DNMT activities. We established a liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) platform with which to measure intracellular SAM/SAH ratios to assess the effects of cigarette smoke on DNMT activities. To evaluate the usefulness of our platform, we measured the SAM/SAH ratios of CGHNK6 and DOK cells with or without treatment by the DNA methyltransferase inhibitor 5-aza-dC (5azaC). The SAM/SAH ratio was significantly (P < 0.001) higher in the 5azaC-treated CGHNK6 and DOK cells than in the vehicle control cells (Figure 2B), indicating that the intracellular SAM/SAH ratio measured using the LC-ESI-MS/MS system provides a sensitive indirect indicator of cellular DNA methyltransferase activities. The SAM/SAH ratio of the CGHNK6 cells decreased significantly (P < 0.01) after CSC (0.1 μg/ml) exposure for 3 weeks (Figure 2B). The SAM/SAH ratio of DOK cells decreased in a dose-dependent manner by the CSC treatment for 5 days (Figure 2B). These results suggested that the activities of DNMTs may change in response to CSC exposure. Subsequently, we evaluated the effects of CSC on the nuclear accumulation of DNMT1, DNMT3A and DNMT3B. We conducted Western blot analyses with the nuclear fractions of cell lysates isolated from CSC-treated CGHNK6 and DOK cells. Regarding short-term exposure (Figure 2C), we observed that CSC treatment rapidly increased the nuclear accumulation of DNMT1 in the CGHNK6 and DOK cells within 0.5 hours and reduced the accumulation after 2 hours. The nuclear accumulation of DNMT3A slightly increased at 2 and 4 hours in the CSC-treated CGHNK6 cells, but decreased in the CSC-treated DOK cells during the experimental period (Figure 2C). Regarding long-term exposure, we observed that nuclear DNMT1 substantially increased in the CGHNK6 cells after 14 and 28 days of CSC treatment (Figure 2D). The amount of nuclear DNMT1 increased in both DMSO- and CSC-treated DOK cells after 42 days of incubation, with slightly higher levels in the CSC-treated cells (Figure 2E). The nuclear accumulation of DNMT3A decreased markedly after 14 and 28 days in the CSC-treated CGHNK6 cells compared with that in the vehicle controls (Figure 2D). The amount of nuclear DNMT3A decreased substantially in both the DMSO- and CSC-treated DOK cells after incubation for 15 days (Figure 2E). None of the cell lines exhibited any significant changes in the amount of nuclear DNMT3B after CSC treatment under any of the analysis conditions. These results suggested that the changes in the nuclear accumulation of DNMT1 and DNMT3A after CSC exposure may contribute to the altered methylome of oral cells.
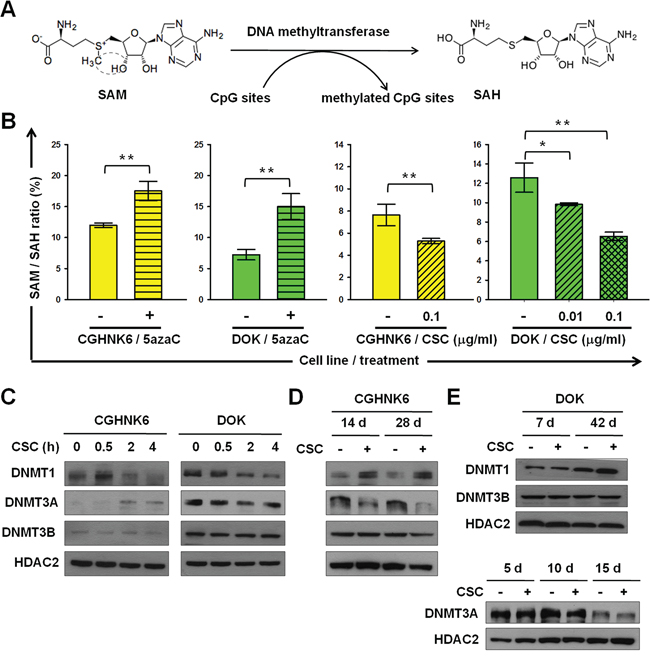
Figure 2: CSC changed the nuclear accumulation of DNMT1 and DNMT3A in CGHNK6 and DOK cells. A. DNA methylation reaction catalyzed by DNMTs. B. The SAM/SAH ratios in CGHNK6 and DOK cells with or without 5aza-C treatment for 5 days and with or without CSC treatment for 3 weeks (CGHNK6) or 5 days (DOK). Data are presented as mean ± SD (n = 3). *p < 0.05 and **p < 0.01. C, D. and E. the amounts of nuclear DNMT1, DNMT3A, and DNMT3B were determined using western blotting at the indicated CSC (0.1 μg/ml) treatment times. DMSO-treated cells were used as vehicle controls.
Identification of tumor suppressor genes epigenetically silenced by cigarette exposure in OSCC
According to the smoking history of DOK’s donor, the differentially expressed genes (DEGs) in DOK compared to normal human oral keratinocytes (HOK) may suggest a list of genes affected by long-term cigarette exposure and involved in the development of OSCC. The genes showing hypermethylation in tumors of smoking OSCC patients may include epigenetic targets important for smoking-related oral malignant transformation. To identify the potential epigenetically silencing tumor suppressor genes affected by cigarette smoke in OSCC, we conducted integrated analyses of the gene expression profiles obtained from the HOK and DOK, and the previously established methylation profiles of tumor-nontumor pairwise samples of smoking OSCC patients (Figure 3A). Using a Venn diagram analysis, we identified 5 genes (GPX3, LDOC1, LIPG, LXN, and UCHL1) with lower expression in DOK than in HOK, and increased methylation levels in OSCC tumors of smokers. Through a literature survey, we identified that GPX3, LDOC1, LXN, and UCHL1, but not LIPG, have been reported as tumor suppressor genes, and that their expression was mediated by promoter DNA methylation (Supplementary Table S1). We considered GPX3, LDOC1, LXN, and UCHL1 as candidate tumor suppressor genes involved in oral malignant transformation and potentially silenced by cigarette exposure. Figure 3B displays the clinical methylation profiles of the GPX3, LDOC1, LXN, and UCHL1 obtained from the 40 tumor-nontumor pairwise samples of smoking OSCC patients. We designed qPCR primers and promoter-located qMSP primers to validate our array data. Our results confirmed that GPX3, LDOC1, LXN, and UCHL1 were lost expression and promoter methylated in the DOK, but expressed and promoter hypomethylated in the HOK (Figure 3C). To identify which gene among GPX3, LDOC1, LXN, and UCHL1 was more susceptible to promoter methylation after CSC treatment, we conducted qMSP in the DOK cells after exposure to CSC for 5, 10, and 15 days. After CSC treatment for 5 days, LDOC1 exhibited significant upregulation of promoter methylation, whereas the remaining three genes, GPX3, LXN, and UCHL1, exhibited no significant changes in promoter methylation (Figure 3D). The promoter methylation of UCHL1 and GPX3 was markedly increased after exposure to CSC for 10 and 15 days, respectively. We did not observe any promoter methylation of LXN in this time course qMSP study. These results suggested that the promoter methylation of LDOC1 was more sensitive to CSC exposure than that of GPX3, LXN, and UCHL1. In a previous study, we identified LDOC1 as an X-linked tumor suppressor gene epigenetically silenced in smoking-associated OSCC cases [10].
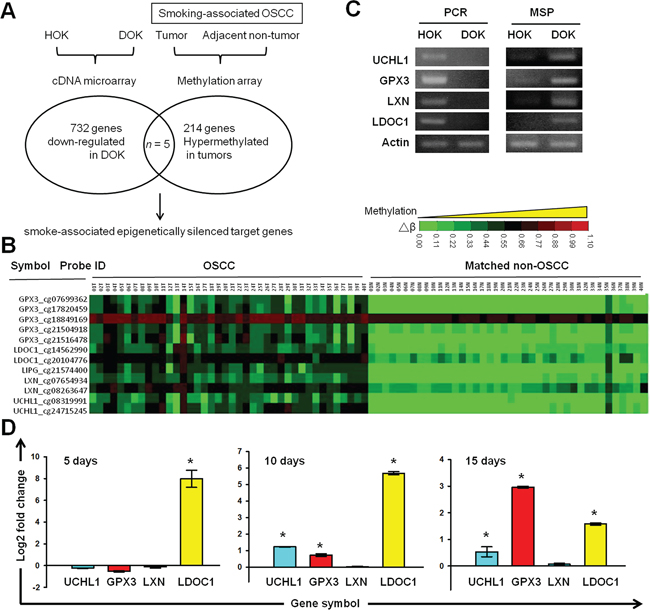
Figure 3: Identification of tumor suppressor genes silenced by cigarette exposure and DNA methylation in OSCC. A. Strategy used for integrated microarray analysis. The methylation database (GEO Accession No. GSE38532) was established from the smoking-associated OSCC patients evaluated in our previous study [10]. B. Clinical methylation profiles of GPX3, LDOC1, LIPG, LXN, and UCHL1 from tumor-nontumor pairwise smoking-associated OSCC. C. Gel images of qPCR and qMSP products from UCHL1, LXN, GPX33, and LDOC1 in DOK and HOK cells. D. Effects of CSC exposure on the promoter methylation of UCHL1, LXN, GPX33, and LDOC1. DOK cells were treated with 0.1 μg/mL of CSC for 5, 10, and 15 days and then analyzed using a qMSP.
LDOC1 showed increased promoter methylation and silenced expression by CSC exposure in normal human oral cell line
Using Methyl Primer Expression software, we identified dense CpG sites in the LDOC1 promoter region and exon 1 at positions –1186 to +369. We designed 4 pairs of qMSP primers mapping at regions mLDOC1-1 (positions –567 to –460), mLDOC1-2 (positions –542 to –417), mLDOC1-3 (positions –354 to –227), and mLDOC1-4 (positions –6 to 124) within the CpG island proximal to the transcriptional start site (TSS) of LDOC1 (Figure 4A). The numbers of CpG sites in the 4 regions were 14, 15, 13, and 8, respectively (Table S2). Using these primers, we showed the CpG sites within the LDOC1 promoter region were nearly fully methylated in DOK cells (Supplementary Figure S1). To examine the effect of CSC on LDOC1 promoter methylation in untransformed oral cells, we conducted qMSP analysis with CSC-treated CGHNK6 cells. Results showed that the methylation of the regions covering the CpG sites proximal to the LDOC1 TSS, mLDOC1-1, mLDOC1-3, and mLDOC1-4, increased progressively following exposure to CSC for 3 weeks and 6 weeks (Figure 4B), and the CpG sites mapped to mLDOC1-2 were fully methylated within 3 weeks. As expected, increased promoter methylation was accompanied by the progressive downregulation of LDOC1 mRNA expression following exposure to CSC for 3 weeks and 6 weeks (Figure 4C). Western blotting showed the LDOC1 proteins were barely detectable after 3 and 6 weeks of CSC treatment (Figure 4C). We did not analyze the effect of CSC exposure on LDOC1 promoter methylation in other oral cell lines because LDOC1 has already silenced by hypermethylation in all OSCC cells available [10]. Since the LDOC1 protein expression markedly reduced to a very low level by CSC treatment within 3 weeks, we examined whether the CSC exposure for 3 weeks confers CGHNK6 cells tumorigenicity in vitro. As shown in Figure 4D, the CGHNK6 cells markedly increased proliferation after CSC exposure for 3 weeks. DMSO-treatment for 3 weeks increased cell proliferation to a less extent. Oncogenicity was determined by the abilities of sphere-forming and anchorage-independent growth. CSC treatment for 3 weeks resulted in the acquisition of cancer cell phenotypes by showing sphere-forming (Figure 4E) and anchorage-independent growth (Figure 4F), whereas the DMSO treatment for 3 weeks did not confer CGHNK6 cells these transformed properties.
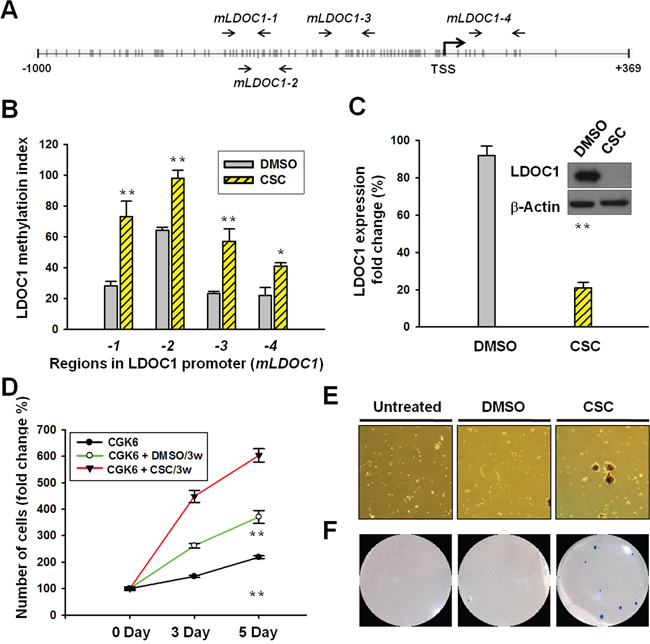
Figure 4: CSC treatment induced LDOC1 silencing and promoter methylation in untransformed CGHNK6 cells accompanied by acquiring oncogenic properties. A. Locations of qMSP primer pairs in the CpG island proximal to the LDOC1 TSS. B. The methylation of CpG sites proximal to the LDOC1 TSS was measured using qMSP. CGHNK6 cells treated with DMSO were used as vehicle controls. C. The expression of LDOC1 mRNA (bar chart) and protein (gel images) was quantified using qPCR and western blotting, respectively, in CGHNK6 cells after exposure to CSC for 3 weeks or 6 weeks. The proliferation D. sphere-forming E. and anchorage-independent growth F. of CGHNK6 cells with or without CSC treatment (0.1 μg/ml) for 3 weeks. In proliferation assay, data are presented as mean ± SD (n = 3) analysed using the Student t-test. **p < 0.01.
LDOC1 downregulation led to human untransformed oral cells acquiring clonogenicity and being associated with premalignant oral lesions and early stages of OSCC
To investigate whether LDOC1 functions as a tumor suppressor gene in the early event of OSCC, we analyzed the effect of the downregulation of LDOC1 on cell proliferation and oncogenicity of CGHNK6 and CGHNK2 (another untransformed oral keratinocyte cell line) [11] cells. We employed lentivirus-mediated shRNA method to reduce the expression of LDOC1 in CGHNK6 and CGHNK2 cells. Cells were infected with lentivirus carrying shRNA specifically targeting LDOC1. The infection efficiencies were approximately 90%, as determined by detecting the expression of green fluorescent protein (GFP) 48 h after infection (Supplementary Figure S2). Puromycin was used for selection of puromycin-resistant cell pools. The mRNA and protein expression of LDOC1 were analyzed by qPCR and Western blotting. The results showed that the LDOC1 mRNA and protein significantly reduced in the CGHNK6-shLDOC1 and CGHNK2-shLDOC1 cells as compared to the corresponding control groups (CGHNK6-shCtrl and CGHNK2-shCtrl) (Figure 5A). To analyze the effect to the downregulation of LDOC1 on cell proliferation, MTT assays were performed. As shown in Figure 5B, inhibition of LDOC1 expression slightly but significantly increased the proliferation of CGHNK6 and CGHNK2 cells. To examine the effect of LDOC1 on clonogenicity, soft agar colony formation assays were performed. The results showed that CGHNK6 cells with or without LDOC1 knockdown (CGHNK6-shLDOC1 and CGHNK6-shCtrl) failed to form colonies (Figure 5C). Interestingly, CGHNK2-shLDOC1 cells formed numerous colonies whereas CGHNK2-shCtrl cells remained unable to grow colony on soft agar (Figure 5C). Giving that anchorage-independent growth as an important trait of malignant transformation, these results suggested that LDOC1 downregulation not only induced cell proliferation but also drive the untransformed CGHNK2 cells toward carcinogenesis in vitro. These findings also implied that LDOC1 downregulation may play an important role in OSCC. To examine the expression of LDOC1 protein in oral malignant transformation, we carried out immunohistochemistry study with tissue microarrays containing oral biopsies covering normal, precancerous lesions, and all stages of cancer progression. The result demonstrated LDOC1 expression was mainly located in the cytoplasm and 76.5% (13/17) normal oral tissues showed LDOC1 high expression, compared to 53.3% (8/15) oral premalignant lesions (OPML, including hyperplasia squamous epithelium and benign tumors) showing no or low LDOC1 expression (Figure 5D). Among 35 of early stage (21 for stage I and 14 for II) OSCC samples, the percentage of low LDOC1 expression were increased with increasing clinical stage, 47.6% (10/21) and 64.3% (9/14) for stage I and stage II OSCC, respectively (Figure 5D). However, 7 of 10 (70%) late stages (stage III and IV) OSCC showed high LDOC1 expression (data not shown), implying LDOC1 may play different roles in early and late stages of OSCC. Representative immunohistochemical results for LDOC1 in normal oral tissues, OPML, and OSCC were shown in Figure 5D. Altogether, these results suggested that LDOC1 downregulation may promote tumorigenesis in early oral carcinogenesis.
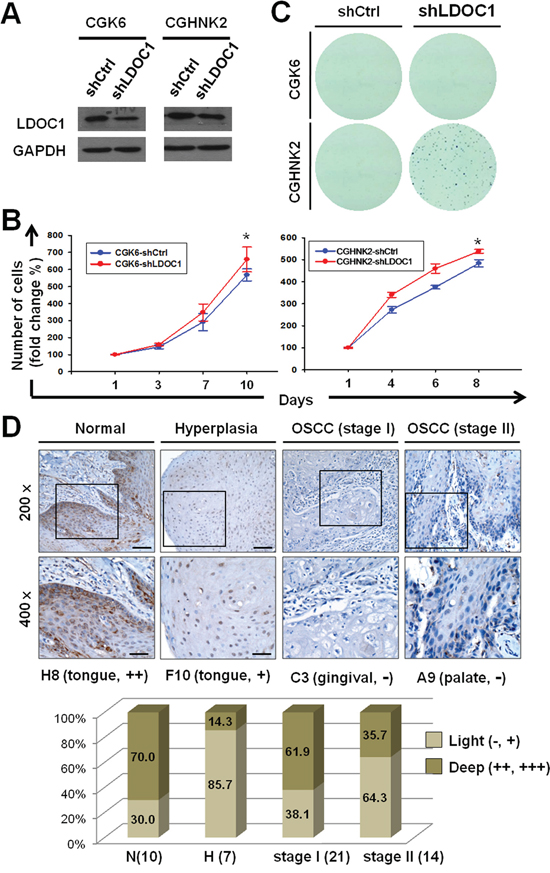
Figure 5: Downregulation of LDOC1 was involved in oral neoplastic transformation. A. Analysis of LDOC1 mRNA and protein expression in CGHNK2 and CGHNK6 cells infected with lentivirus carrying shLDOC1 or shCtrl (vector control) by quantitative real-time PCR (lower) and Western blotting analysis (upper), respectively. B. LDOC1 knockdown increased the cell proliferation of CGHNK6 (left) and CGHNK2 (right) cells. MTT assay was used to estimate the cell numbers at indicated time after seeding. Data are presented as mean ± SD (n = 3). *p < 0.05. C. Effect of LDOC1 knockdown on anchorage-independent colony forming in CGHNK6 and CGHNK2 cells. D. The expression of LDOC1 protein in normal, diagnosed tissues of oral premalignant lesion (OPML), and early stages OSCC (stage I and II). Representative images for immunohistochemical analysis of LDOC protein expression were shown. The bar chart plots percentage of high or low LDOC1 protein expression in each group of oral samples.
LDOC1 downregulation was associated with the smoking status of non-cancerous and cancerous lung tissues
Besides oral cavity, lung also frequently expose to cigarette smoke which has been well recognized as the major causative factor to lung cancer. Therefore, it is possible that LDOC1 might decrease expression by cigarette exposure in lung of smokers and smoking-associated lung cancer. To test this, we performed a search of the publicly available microarray studies in the Oncomine database (www.oncomine.org) to examine the expression of LDOC1 in normal and cancerous lung tissues of smokers and non-smokers. In Landi’s study [13], LDOC1 expression significantly decreased in lung tissues of 15 current smokers compared to those from 16 never smokers (Figure 6A). There were no differences in LDOC1 expression between the former smokers (n = 18) and never smokers. In Bhattacharjee’s study [14], LDOC1 expression markedly downregulated in all of the 6 small-cell lung carcinoma (SCLC), the lung cancer type most strongly associated with cigarette exposure, compared to the normal lung tissues (without information of smoke status, n = 17) (Figure 6B). The same dataset also showed that LDOC1 expression significantly decreased in tumors of non-small-cell lung carcinoma (NSCLC) patients with smoking habit (packs per year > 10, n = 95) as compared to normal lung (Figure 6C). These findings support the effect of cigarette smoke on LDOC1 expression as well as a role of LDOC1 in smoke-associated lung cancer.
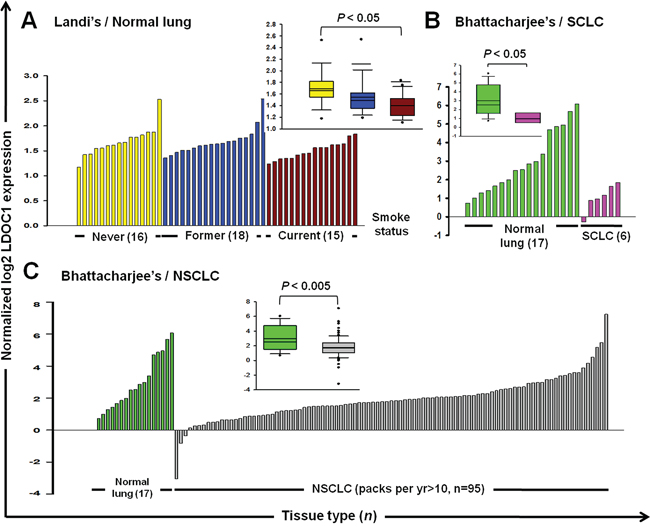
Figure 6: Downregulation of LDOC1 in non-cancerous and cancerous lung tissues of smokers. The LDOC1 expression profiles of A. normal lung with information about smoking status [36], B. small cell lung carcinoma and C. and non-small cell lung carcinoma with smoking history [37], were obtained from publicly available microarray data sets in Oncomine (https://www.oncomine.com). Each of the normal and tumour samples is plotted in order of increasing levels of LDOC1. Inset: box plots display the median values of the array data and the 25th and 75th percentiles. The minimum and maximum values are indicated as whiskers. Points indicate outliers. Statistical differences were determined by Student t test.
DISCUSSION
Cigarette contains numerous organic and inorganic carcinogens. Therefore, results from studies assessing the effects of single tobacco components, such as NNK or nicotine, might not reflect the comprehensive oncogenic effects of cigarette smoke. In this study, we prepared CSC according to a standard protocol and evaluated the epigenetic effects of CSC on oral cells. The effects of CSC on the methylome and DNMTs of oral untransformed CGHNK6 and partially transformed DOK cells were observed (Figures 1 and 2). Pickering’s study [15] supports our findings indirectly. By whole-exome sequencing and copy-number analysis, Pickering et al [15] failed to observe any genetic mutation signature associated with smoking in oral tongue tumors. They proposed that the epigenetic alterations are possibly involved in the oncogenesis of smoke-associated squamous cell carcinoma of the tongue. SAM is the methyl donor for numerous methyltransferases. In biological systems, SAM is generated from methionine and donates its methyl group to various acceptor molecules, such as nucleic acids, lipids, and proteins, to be converted to SAH [16]. SAH can be recycled through the production of homocysteine and methionine. Thus, methyltransferase activity catalyzed by DNMTs plays a role in the homeostasis between SAM and SAH. In previous studies, the plasma concentration of SAH, not the SAM/SAH ratio, provided an indicator of DNA methylation status. Yi et al identified a positive correlation between plasma SAH levels and lymphocyte DNA hypomethylation in healthy young women [17]. Castro et al observed that patients with vascular disease displayed significantly higher plasma SAH concentrations, decreased plasma SAM/SAH ratios, and lower global DNA methylation statuses compared with control [18]. In animal models and in humans, the SAM/SAH ratio does not correlate with DNA methylation levels because it can be affected by various factors, such as methionine or folate in diets, physiological status, medical treatment, and numbers of methyl transferring enzymes [19, 20]. However, in our cultivated cell model, we limited the factors modulating the methylation reaction; therefore, the SAM/SAH ratio provided a sensitive indirect indicator of DNMT activities. We showed that 5azaC treatment results in a substantial increase in the SAM/SAH ratio of CGHNK6 and DOK cells (Figure 2B), supporting the efficacy of the SAM/SAH ratio in the assessment of DNMT activities. Previous studies have observed DNMT1 overexpression in patients with lung and liver cancers who were smokers [21, 22]. Lin et al. demonstrated that the tobacco-specific carcinogen NNK increases the stability of DNMT1, leading to its nuclear accumulation [9]. Consistent with this result, we observed a remarkable increase in nuclear DNMT1 in the CGHNK6 cells after exposure to CSC for 14 or 28 days (Figure 2D). In the CGHNK6 cells, DNMT3A was slightly increased after CSC treatment for 2 or 4 hours; however, it was markedly reduced after CSC treatment for 14 or 28 days (Figure 2D). Overall, our data suggested that changes in the nuclear accumulation of DNMT1 and DNMT3A may contribute to the aberrant methylome of untransformed oral cells exposed to cigarette smoke.
The DOK cell line was derived from tongue tissue with epithelial dysplasia adjacent to the OSCC tumor of a heavy smoker [12]. Considering the smoking history of the OSCC patient from whom the DOK cells were derived, we speculated that the DOK cell line could provide invaluable information on OSCC pathogenesis mediated by cigarette smoke. Using integrated array analysis, we identified 5 DNA methylation targets, including GPX3, LDOC1, LIPG, LXN, and UCHL1 in the DOK and in a smoking OSCC patient cohort. Furthermore, we identified that the promoter methylation of GPX3, LDOC1, and UCHL1 is sensitive to CSC exposure because significantly increased promoter methylation levels of these three genes were observed in the DOK cells within 15 days after CSC treatment (Figure 3D). LDOC1, an epigenetically silenced tumor suppressor gene in smoking-associated OSCC, was most sensitive to CSC and showed a remarkable increase in promoter methylation after CSC treatment for only 5 days (Figure 3D). Using CGHNK6 cells, we demonstrated the silencing of LDOC1 expression and increased promoter methylation by CSC treatment within 3 weeks (Figure 4B and 4C). Additionally, we showed that the CpG islands within the promoter region of LDOC1 were intensively methylated in smoker-derived DOK cells (Supplementary Figure S1). Altogether, these results suggest the methylation levels of LDOC1 promoter are associated with cigarette smoke and may be used as a biomarker for tobacco exposure. Interestingly, the array data from lung tissues (Figure 6) provide supporting evidence for using LDOC1 as an indicator for tobacco exposure. Landi’s study [13] showed that, in normal lung, the expression of LDOC1 was significantly decreased in lung of current smokers than that of never smokers (Figure 6A). Bhattacharjee’s study [14] showed that the LDOC1 expression was relatively low in small cell lung carcinoma—a lung cancer type strongly associated with smoking (Figure 6B). Although the relevant methylation levels of LDOC1 for these studies were absent, the LDOC1 down-regulation was likely caused by promoter methylation because we have demonstrated that the expression of LDOC1 was majorly governed by promoter hypermethylation in our previous study [10]. In HOK and DOK cells, the methylation levels of LDOC1 inversely correlated with the LDOC1 mRNA expression (Figure 3C). We demonstrated that the methylation of LDOC1 promoter increased accompanied by a reduction in LDOC1 expression upon CSC treatment (Figure 4B and 4C). All these findings support that, in tissues of respiratory system, such as lung, which is frequently exposed to cigarette smoke, the expression and promoter methylation of LDOC1 will be altered. Further investigation regarding the feasibility of using LDOC1 as a molecular biomarker for cigarette smoke exposure by measuring the methylation of LDOC1 in sputum samples is warranted.
We carried out shRNA-mediated LDOC1 knockdown in the untransformed CGHNK6 and CGHNK2 cells to examine the tumor suppressor activity of LDOC1. Reduction in LDOC1 expression led to acquired anchorage-independent growth of CGHNK2 cells whereas no colonies grew by the knockdown of LDOC1 in CGHNK6 cells (Figure 5C). These differences in the tumorigenic potential driven by LDOC1 downregulation may be due to some unidentified genetic differences between CGHNK6 and CGHNK2 cells. However, these results still suggest that downregulation of LDOC1 may play a crucial role in the initiation stage of oral malignant transformation and contribute to the CSC-induced oncogenicity (Figure 4D–4F). In agreement with this hypothesis, the immunochemistry study revealed that LDOC1 downregulation was frequently observed in tissues of OPML and the early stage of OSCC (Figure 5D). These results echo our previous study findings [10], which identified LDOC1 as an X-linked tumor suppressor gene with promoter hypermethylation in 89% of OSCC patients who were habitual smokers. According to the tobacco-exposure sensitivity and in vitro anti-tumorigenic activity, we proposed that, in addition to oral cancer and lung cancer, LDOC1 may function as a tumor suppressor gene and is downregulated in several human cancers associated with smoking. Publically available array dataset support our hypothesis. As shown in Supplementary Figure S3, the expression of LDOC1 is also decreased in several smoking-associated cancers including cervical cancer [23], esophageal adenocarcinoma [24], pancreatic ductal adenocarcinoma [25], and head and neck cancers [26, 27]. Consistent with these array data, Nagasaki et al reported that LDOC1 is a proapoptotic tumor suppressor gene in pancreatic cancer [28]. Using comparative proteomic analyses, Lui et al demonstrated the significant downregulation of LDOC1 in serpin B5 (a tumor suppressor)-knockdown lung cancer cells [29]. Buchholtz et al further identified that LDOC1 is frequently silenced by promoter hypermethylation in cervical cancer [30]. These findings support a potential etiological role of LDOC1 in smoking-associated human cancers. The methylation status of LDOC1 could potentially provide a molecular marker for the screening of smokers at high risk of cancer.
In summary, our findings not only strengthen the association between cigarette smoke and altered epigenome in OSCC, they also suggest a critical role of LDOC1 in other tobacco-related cancers.
MATERIALS AND METHODS
Chemicals
S-(5′-Adenosyl)-L-methionine chloride dihydrochloride (SAM chloride dihydrochloride), and S-(5′-Adenosyl)-L-homocysteine crystalline (SAH crystalline), 5-aza-2-deoxycytidine (5-aza-dC) were purchased from Sigma-Aldrich. S-Adenosyl-L-methionine-d3 (S-methyl-d3) tetra (p-toluenesulfonate) (d3-SAM) salt was purchased from C/D/N Isotopes Inc, acetic acid from J.T.Baker, acetonitrile and methanol were HPLC grade from Merck, water from Millipore.
Cell lines and cell culture
Human oral keratinocytes (HOK) were obtained from the ScienCell and grown in Oral Keratinocyte Medium (OKM) with supplements (ScienCell). Immortalized untransformed human oral cell lines CGHNK6 and CGHNK2 was gift from Dr. Cheng (Chang Gung University, Taiwan) and grown in keratinocyte serum-free medium (KSFM) with supplements (Gibco) as previously described [11]. Briefly, CGHNK2, and CGHNK6 were primary culture cells from tissue biopsies of grossly normal oral mucosa with human papilloma virus (HPV) immortalization. Both cell lines were derived from patients with OSCC with the habits of smoking habit [11]. Human dysplastic oral keratinocyte DOK cell line were derived from dysplastic tissues neighboring to a tumor of an OSCC patient who was a heavy smoker before diagnosed as OSCC [12]. DOK cells were maintained in DMEM supplemented with 10% fetal calf serum, 100 g/ml penicillin, and 100 g/ml streptomycin.
Preparation and treatment of cigarette smoke condensates (CSC)
CSCs were generated from Kentucky Reference Cigarettes 3R4F (University of Kentucky, Tobacco and Health Research Institute, Lexington, KY, USA) using a home-made smoking machine resembling that used by Pieraccini et al. [31]. Smoking was performed following a standard procedure agreed internationally by organizations such as the International Standards Organization and USA Federal Trade Commission [32]. “The modified Cambridge Filter method ultimately adopted by the Commission” is often referred to as the “FTC method”. The smoke condensates were trapped on glass fiber filters, weighed, and dissolved in DMSO according to the required concentrations. DOK and CGHNK6 cells were constantly treated CSC at indicated dose; and fresh medium containing CSC was replaced every 3 days. The DMSO-treated cells were used as controls. At appropriate times, cells were harvested, and processed for further analysis.
Quantification of global DNA methylation
The global methylation of DNA was determined using competitive Enzyme Immunoassay (EIA) based commercial kit (Cayman’s DNA Methylation EIA Kit), as the manufacturer’s protocol. This assay is based on the competition between 5-methyl-2-deoxycytidine (5mC) in the sample and a 5mC-acetylcholinesterase (AChE) conjugate (5mC tracer) for a limited amount of 5mC monoclonal antibody (mAb). The amount of 5 mC tracer that is able to bind to the 5mC EIA mAb will be inversely proportional to the concentration of 5mC in the sample. The amount of mAb-5mC tracer complex binding to goat polyclonal anti-mouse IgG-coated ELISA plate was quantified by Ellman’s reagent with a distinct yellow color and absorbs strongly at 412 nm. The relative global DNA methylation content of sample was obtained from calculating the amount of 5mC in the sample relative to those of CpGenome Universal Methylated DNA (Chemicon), a commercial fully CpG methylated DNA. All samples were analyzed in biological and technical duplicates.
Intracellular SAM and SAH analysis
Intracellular metabolites extraction as described previously [33]. Liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) consist of a PE series 200 binary pump and autosampler (PE series 200, Perkin-Elmer) system coupled with an API 3000 triple quadruple tandem mass spectrometer (Applied Biosystem). Liquid Chromatography separation against SAM, SAH, and d3-SAM were performed on an Atiantis T3 3um (2.1 mm i.d. × 150 mm) reversed phase column (Waters) at a flow rate of 250 uL/min. The compositions of mobile phase A and B are 0.1% acetic acid and 0.1% acetic acid in acetonitrile, respectively. The gradient profile were 0–3 min linear increase from 0 to 20% B, 3–6 min linear increase from 20 to 100% B, hold at 100% B for 3 min, 9–9.1 min from 100 to 0% B and hold at 0% B for 2.9 min. The detection of SAM, SAH and d3-SAM was carried out using positive electro-spray ionization (ESI) interface mode in a multiple reactions monitoring (MRM) scan mode. The MRM parameters of SAM, SAH and d3-SAM, were optimized: declustering potential (DP): 26, 31 and 61 V for SAM, SAH and d3-SAM, respectively, focusing Potential (FP): 190, 270 and 340 V for SAM, SAH and d3-SAM, respectively, collision energy (CE): 23, 29 and 23 V for SAM, SAH and d3-SAM, respectively, Collision Cell Exit Potential (CXP): 20, 8 and 16 V for SAM, SAH and d3-SAM, respectively.
Extraction of DNA and RNA and bisulfite conversion of genomic DNA
Genomic DNA and total RNA from each sample were extracted using a QIAamp DNA mini Kit (Qiagen) and an RNeasy mini kit (Qiagen), respectively, according to the manufacturer’s instructions. The extracted genomic DNA and total RNA were quantified and confirmed for OD 260/280 values between 1.8 and 2.2 and OD 260/230 values greater than 1. For methylation experiments, genomic DNA was bisulfite converted using BisulFlash DNA modification kit (Epigentek) according to the manufacturer’s protocols. The converted DNA was stored at –80°C until ready to use.
Expression microarray analysis
The Illumina HumanHT-12_V4 Expression BeadChip was used for genome-wide expression studies with HOK and DOK cells. Data analysis was performed with GeneSpring GX software (Agilent Technologies). Quantile normalization was performed. The microarray data used in this study have been deposited in NCBIs Gene Expression Omnibus (GEO) http://www.ncbi.nlm.nih.gov/geo/ and can be accessed through GEO Series accession number GSE54861 and GSE38532 [10].
Quantitative real-time qPCR (qPCR)
The synthesis of cDNA from total RNA was performed with M-MLV reverse transcriptase (Promega). The primers required were designed by the online tool at the Universal ProbeLibrary Assay Design Center (Roche Applied Science, https://www.roche-applied-science.com) and shown in Supplementary Table S3. QPCR was performed with FastStart Universal Probe Master Kit (Roche Applied Science). The cycling parameters began with 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec, followed by a melting curve analysis.
Real-time quantitative methylation-specific PCR (qMSP)
The bisultife converted DNAs were subjected to qMSP using the primers shown in the Supplementary Table S2 and S4. A primer pair specific for the CpG-free genomic region of the ACTB gene was used for normalization: 5′-TGGTGATGGAGGAGGTTTAGTAAGT-3′ (F) and 5′-AACCAATAAAACCTACTCCTCCCTTAA-3′. CpGenome Universal CpG Methylated and Unmethylated DNA (Chemicon) was used as positive and negative control, respectively. QMSP was performed using SYBR Advantage qPCR Premix (Clontech) as described previously [10].
Knockdown of LDOC1 in CGHNK6 and CGHNK2 cells
For knockdown of LDOC1 expression, we purchased GIPZ lentivirus particles carrying shRNA specifically targeting LDOC1 from Dharmacon. To cell infection, 50% confluent of CGHNK6 and CGHNK2 cells were incubated with lentivirus for 24 h, and the medium containing puromycin (5 μg/ml; Sigma-Aldrich) was replaced to select stable cell pools for at least 2 weeks before usage. Cells infected with lentivirus with empty vector (shCtrl) were used as controls.
Proliferation assay
Cell proliferation was measured by the MMT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide] assay. Cells were seeded at 5 × 103 per well on 96-well plates. 10 μl of MMT (5 mg/ml) was added to each well at indicated time; the cells were further incubated for 4 h at 37°C and then subcultured in the medium with 100 μl of DMSO. The absorbance at 570 nm was measured on a micro ELISA reader (Bio-Rad).
Soft agar assay
Soft agar assay was performed as described previously [34]. Briefly, cells were resuspended in culture medium containing 0.3% agarose at a density of 5 × 103 cells in 6-well plates, then overlaid with 0.6% agarose and fed with fresh medium weekly for 3 weeks. After fixation and staining with violet blue, colonies were photographed and quantified using MetaMorph software (Molecular Devices). The experiment was performed three times, each time with four replicates.
Immunohistochemistry
Tissue microarray covering tissue sections of oral cavity carcinoma progression, OR802 (10 cancer adjacent tissues or normal tissues, 7 hyperplasia epithelia, 8 benign tumors, and 45 oral carcinoma) were purchased from US BIOMAX. Immunohistochemistry was performed as previously described [35]. Briefly, all paraffin sections were dewaxed and rehydration followed by heating in a 0.01 M citrate buffer for 20 min. Subsequently, anti-LDOC1 (1:2000 dilution, LifeSpan, Newton, MA, USA) was incubated overnight at room temperature. The secondary biotinylated antibody and the streptavidin–peroxidase conjugate (BioGenex, Netherlands) were then incubated on the sections for each 20 min. The sections were stained with diaminobenzidine (DakoCytomation) for detection and stained with hematoxylin QS for counterstaining (Vector, Burlingame). Normal serum and phosphate buffer instead of specific antibodies was used as negative controls. Immunoreactivity was scored by the staining intensity using MetaMorph software (Molecular Devices). Staining intensities < 0.15 was described to be low LDOC1 protein expression, staining intensities > or = 0.15 was described to be high LDOC1 protein expression.
Statistical analysis
Comparison of the results between various experimentally treated groups and their corresponding controls was carried out by Student’s t-test. All comparisons were considered significant when p < 0.05.
ACKNOWLEDGMENTS
We would like to thank Dr. Tsang-Wu Liu from National Institute of Cancer Research, National Health Research Institutes, Taiwan, for providing the human normal oral keratinocyte HOK.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Graham S, Dayal H, Rohrer T, Swanson M, Sultz H, Shedd D, Fischman S. Dentition, diet, tobacco, and alcohol in the epidemiology of oral cancer. Journal of the National Cancer Institute. 1977; 59:1611–1618.
2. Kademani D. Oral cancer. Mayo Clinic proceedings Mayo Clinic. 2007; 82:878–887.
3. Ide R, Mizoue T, Fujino Y, Hoshiyama Y, Sakata K, Tamakoshi A, Yoshimura T, Group JS. Cigarette smoking, alcohol drinking, and oral and pharyngeal cancer mortality in Japan. Oral diseases. 2008; 14:314–319.
4. Gandini S, Botteri E, Iodice S, Boniol M, Lowenfels AB, Maisonneuve P, Boyle P. Tobacco smoking and cancer: a meta-analysis. International journal of cancer Journal international du cancer. 2008; 122:155–164.
5. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011; 61:69–90.
6. Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M. Comparative Risk Assessment collaborating g. Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005; 366:1784–1793.
7. Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. American journal of human genetics. 2011; 88:450–457.
8. Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, Rao M, Meltzer PA, Schrump DS. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010; 29:3650–3664.
9. Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. The Journal of clinical investigation. 2010; 120:521–532.
10. Lee CH, Wong TS, Chan JY, Lu SC, Lin P, Cheng AJ, Chen YJ, Chang JS, Hsiao SH, Leu YW, Li CI, Hsiao JR, Chang JY. Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. The Journal of pathology. 2013; 230:298–309.
11. Lu YC, Chen YJ, Wang HM, Tsai CY, Chen WH, Huang YC, Fan KH, Tsai CN, Huang SF, Kang CJ, Chang JT, Cheng AJ. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer prevention research. 2012; 5:665–674.
12. Chang SE, Foster S, Betts D, Marnock WE. DOK, a cell line established from human dysplastic oral mucosa, shows a partially transformed non-malignant phenotype. International journal of cancer Journal international du cancer. 1992; 52:896–902.
13. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS one. 2008; 3:e1651.
14. Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:13790–13795.
15. Pickering CR, Zhang J, Neskey DM, Zhao M, Jasser SA, Wang J, Ward A, Tsai CJ, Ortega Alves MV, Zhou JH, Drummond J, El-Naggar AK, Gibbs R, et al. Squamous Cell Carcinoma of the Oral Tongue in Young Non-Smokers Is Genomically Similar to Tumors in Older Smokers. Clin Cancer Res. 2014; 20:3842–3808.
16. Loenen WA. S-adenosylmethionine: jack of all trades and master of everything? Biochemical Society transactions. 2006; 34:330–333.
17. Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. The Journal of biological chemistry. 2000; 275:29318–29323.
18. Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, Blom HJ, Jakobs C, Tavares de Almeida I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clinical chemistry. 2003; 49:1292–1296.
19. Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, Huang Y, Young D, Jacob ST. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. The Journal of nutrition. 2006; 136:1522–1527.
20. Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007; 45:1306–1312.
21. Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT, Wang YC. Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung cancer. 2007; 55:205–213.
22. Hammons GJ, Yan Y, Lopatina NG, Jin B, Wise C, Blann EB, Poirier LA, Kadlubar FF, Lyn-Cook BD. Increased expression of hepatic DNA methyltransferase in smokers. Cell biology and toxicology. 1999; 15:389–394.
23. Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer research. 2007; 67:10163–10172.
24. Kim SM, Park YY, Park ES, Cho JY, Izzo JG, Zhang D, Kim SB, Lee JH, Bhutani MS, Swisher SG, Wu X, Coombes KR, Maru D, et al. Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PloS one. 2010; 5:e15074.
25. Ishikawa M, Yoshida K, Yamashita Y, Ota J, Takada S, Kisanuki H, Koinuma K, Choi YL, Kaneda R, Iwao T, Tamada K, Sugano K, Mano H. Experimental trial for diagnosis of pancreatic ductal carcinoma based on gene expression profiles of pancreatic ductal cells. Cancer science. 2005; 96:387–393.
26. Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer research. 2004; 64:55–63.
27. Peng B, Gu Y, Xiong Y, Zheng G, He Z. Microarray-assisted pathway analysis identifies MT1X & NFkappaB as mediators of TCRP1-associated resistance to cisplatin in oral squamous cell carcinoma. PloS one. 2012; 7:e51413.
28. Nagasaki K, Schem C, von Kaisenberg C, Biallek M, Rosel F, Jonat W, Maass N. Leucine-zipper protein, LDOC1, inhibits NF-kappaB activation and sensitizes pancreatic cancer cells to apoptosis. International journal of cancer Journal international du cancer. 2003; 105:454–458.
29. Liu Y, Geng Y, Li K, Wang F, Zhou H, Wang W, Hou J, Liu W. Comparative proteomic analysis of the function and network mechanisms of MASPIN in human lung cells. Experimental and therapeutic medicine. 2012; 3:470–474.
30. Buchholtz ML, Juckstock J, Weber E, Mylonas I, Dian D, Bruning A. Loss of LDOC1 expression by promoter methylation in cervical cancer cells. Cancer investigation. 2013; 31:571–577.
31. Pieraccini G, Luceri F, Moneti G. New gas-chromatographic/mass spectrometric method for the quantitative analysis of primary aromatic amines in main- and side-stream cigarette smoke. I. Rapid Communications in Mass Spectrometry. 1992; 6:406–409.
32. Peeler CL. (1996). Cigarette testing and federal trade commission: a historical overview. In: NCI Expert Committee, U.D.o.H.a.H.S., Public Health Service, eds. Smoking and Tobacco Control, Monograph 7. (Bethesda, MD, USA: National Cancer Institute, National Institutes of Health), pp. 1–8.
33. Stevens AP, Dettmer K, Kirovski G, Samejima K, Hellerbrand C, Bosserhoff AK, Oefner PJ. Quantification of intermediates of the methionine and polyamine metabolism by liquid chromatography-tandem mass spectrometry in cultured tumor cells and liver biopsies. Journal of chromatography A. 2010; 1217:3282–3288.
34. Sheu JJ, Lee CC, Hua CH, Li CI, Lai MT, Lee SC, Cheng J, Chen CM, Chan C, Chao SC, Chen JY, Chang JY, Lee CH. LRIG1 modulates aggressiveness of head and neck cancers by regulating EGFR-MAPK-SPHK1 signaling and extracellular matrix remodeling. Oncogene. 2014; 33:1375–1384.
35. Wang CK, Chang LW, Chang H, Yang CH, Tsai MH, Tsai HT, Lin P. Pulmonary changes induced by trans, trans-2, 4-decadienal, a component of cooking oil fumes. The European respiratory journal. 2010; 35:667–675.
36. Peng CH, Liao CT, Peng SC, Chen YJ, Cheng AJ, Juang JL, Tsai CY, Chen TC, Chuang YJ, Tang CY, Hsieh WP, Yen TC. A novel molecular signature identified by systems genetics approach predicts prognosis in oral squamous cell carcinoma. PloS one. 2011; 6:e23452.
37. Estilo CL, P Oc, Talbot S, Socci ND, Carlson DL, Ghossein R, Williams T, Yonekawa Y, Ramanathan Y, Boyle JO, Kraus DH, Patel S, Shaha AR, et al. Oral tongue cancer gene expression profiling: Identification of novel potential prognosticators by oligonucleotide microarray analysis. BMC cancer. 2009; 9:11.