
INTRODUCTION
Merkel cell carcinoma (MCC) is an aggressive skin cancer, and ~80% of MCC tumors have DNA from the Merkel cell polyomavirus (MCV) integrated into their genome [1, 2]. Up to 80% of patients with MCC develop metastases [3]. The relative 5-year survival has been reported to be 64% for patients in stages I or II, 39% in stage III, and 18% in stage IV [4]. Aside from tumor stage, there are no robust prognostic biomarkers for MCC.
Biomarkers for disease prognosis and early detection of recurrences improve the care of cancer patients. Although a variety of biomarkers exist for malignancies such as breast [5] or colorectal cancer [6], there is an essential need for MCC biomarkers. Based on reports of elevated serum levels in patients with non-cutaneous neuroendocrine tumors [7–12], some institutions follow neuron specific enolase (NSE) and chromogranin A (ChrA) blood levels in MCC patients. However, this practice is not part of consensus management guidelines [13, 14] and the utility of NSE and ChrA as MCC biomarkers has not been tested.
Circulating tumor cells (CTC) can be detected in the bloodstream and hold potential as cancer biomarkers [15]. Recent studies have highlighted the prognostic significance of CTC [16, 17]. Most CTC-identifying assays use antibodies against epithelial markers, e.g. EpCAM [18, 19], the epithelial cell adhesion molecule that is known to be expressed on many carcinomas [20] including MCC [21].
To assess their utility as biomarkers for MCC, we have conducted a retrospective analysis of clinical tests used at our institution. Despite their routine use in this patient population, we found NSE and ChrA ineffective as prognostic markers or for detection of MCC recurrence. In contrast, our recent experience with measuring CTC in MCC patients suggests they could be developed as a useful biomarker for this aggressive cancer.
RESULTS
Patient characteristics
A total of 60 MCC patients were included in the study. Patient characteristics are shown in Table 1. Median follow up time was 43 months (range 3–182 months). At the last date of contact, 37 patients were alive with no evidence of disease, 6 were alive with disease, 9 had died of disease, and 8 had died of other causes. The estimated 5-year progression free survival (PFS) was 58.6% and 5-year disease specific survival (DSS) was 81.3%. Survival varied significantly with tumor stage for both PFS (p<0.05) and DSS (p<0.005) (Figure S1).
Table 1: Characteristics of patients included in the study and their Merkel cell carcinoma tumors
Number |
% |
|
All patients |
60 |
100 |
Male |
37 |
62 |
Female |
23 |
38 |
Median age at diagnosis: 70 y (range 33–90) |
||
Median follow up: 43 months (range 3–182) |
||
Patients with progression (60 total events) |
24 |
40 |
Stage at diagnosis |
||
IA |
20 |
33 |
IB |
13 |
22 |
II |
11 |
18 |
III |
16 |
27 |
IV |
0 |
0 |
Cases with FFPE tumor for immunostaining |
46 |
77 |
NSE expression positive |
46 |
100 |
CK20 expression positive |
44 |
96 |
CD56 expression positive |
42 |
91 |
ChrA expression positive |
41 |
89 |
EpCAM expression positive |
33 |
72 |
Merkel cell polyomavirus positive |
30 |
65 |
Tumor characteristics
We used immunostaining to assess potential biomarker expression in MCC tumors. Tumor tissue was available for 46 patients (77%). Of the tested samples, 100% stained positive for NSE, 96% for CK20, 91% for CD56, 89% for ChrA, 72% for EpCAM, and 65% for MCV (Table 1), confirming frequent expression of these MCC tumor markers. Among the immunostained cases, 67% were positive for both EpCAM and CD56, and 72% for EpCAM and CK20 suggesting frequent co-expression of these marker combinations in MCC tumors. Staining intensities for individual markers were graded from 0–2. For each immunohistochemical marker, staining intensity failed to correlate with PFS or DSS (Figure S2), suggesting they are not useful as prognostic markers.
NSE and ChrA blood levels are not effective biomarkers
Among the 60 study patients there was a total of 342 NSE and 367 ChrA blood level assessments. We analyzed NSE and ChrA levels as categorical variables, scored as either within normal limits (WNL), above normal (Abv NL), or high. There was no significant difference in PFS or DSS detected based on the patients’ initial NSE or ChrA blood levels (first assessment after diagnosis of MCC, Figure 1), suggesting that initial NSE and ChrA levels are not effective as prognostic biomarkers. However, there was a trend (p = 0.0783) toward PFS correlating with first NSE levels. Similar analysis of maximum NSE and ChrA levels failed to detect any difference in PFS or DSS, suggesting that elevations in these markers fail to correlate with disease progression (Figure 1). To directly assess if NSE or ChrA levels varied with tumor burden, we used data visualization of all lab values for each patient plotted with all treatment completion and disease recurrence events. There was no obvious trend associating tumor treatment or recurrence events to changes in NSE or ChrA levels (Figure S3). Finally we assessed the distribution of NSE and ChrA values drawn when patients had no evidence of disease (NED) versus patients with active or recent tumor burden (28 days prior to 56 days after tumor being present). There was no increased likelihood of finding elevated NSE or ChrA levels in patients with tumor versus those with NED (Figure 2). Taken together these results suggest that NSE and ChrA blood levels are not effective at predicting outcomes, following treatment response, or detecting recurrences in patients with MCC.
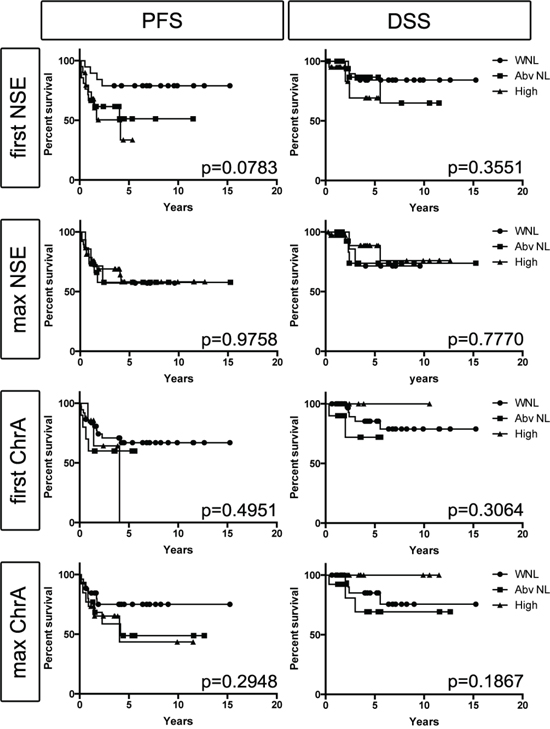
Figure 1: Blood levels of NSE or ChrA fail to correlate with PFS or DSS. Kaplan-Meier survival estimates for first (initial assessment after diagnosis of MCC) or max (maximal level measured during follow up) NSE and ChrA values categorized as: within normal limits (WNL; circles), above normal (Abv NL; squares), or high (triangles) (p-values as indicated).
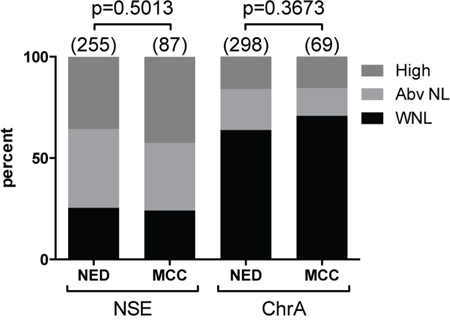
Figure 2: Blood levels of NSE or ChrA fail to correlate with tumor burden. Distribution of all analyzed NSE and ChrA blood levels as percentages (total numbers as indicated) categorized as within normal limit (WNL; black), above normal (Abv NL; light grey), or high (dark grey) in patients with no evidence of disease (NED) compared to patients with tumor (MCC; 28 days prior to 56 days after tumor being present) (p-values as indicated).
Circulating MCC cells are readily detected and reflect tumor burden
We used the maintrac system to quantify CTC in a total of 30 MCC patients (Table 2), as well as 10 healthy control individuals (5 male, 5 female). All 30 patients were analyzed at least once, and 13 patients had 2 or more serial tests for a total of 56 assays. Controls were tested once. EpCAM+ circulating cells were detected in 90% of all MCC patient samples (median 450 cells/ml, range 0–11,000) and 60% of controls (median 175 cells/ml, range 0–1,000). Among the 30 patients, 29 (97%) had detectable CTC in at least one blood sample. Counts of EpCAM+ circulating cells in MCC patients were significantly higher than in controls (p<0.05). The detection of circulating EpCAM+ cells in healthy volunteers using maintrac suggests that a portion of the CTC counted in MCC samples are non-malignant cells.
Table 2: Characteristics of Merkel cell carcinoma patients monitored for CTC
Number |
% |
|
All patients |
30 |
100 |
Male |
20 |
33 |
Female |
10 |
67 |
Median age at diagnosis: 68 y (range 54–90) |
||
Stage at time of CTC monitoring |
||
IA |
9 |
30 |
IB |
6 |
20 |
II |
2 |
7 |
III |
9 |
30 |
IV |
4 |
13 |
Individuals with circulating EpCAM+ cells/ml |
||
Patients (median: 450, range 0–11,000) |
29 |
97 |
Controls (n = 10) (median: 175, range 0–1,000)* |
6 |
60 |
Individuals with circulating EpCAM+ CD56+ cells/ml |
||
Patients (median: 150, range 0–8,230) |
23 |
77 |
Controls (n = 10) (median: 0, range 0–0)*** |
0 |
0 |
Individuals with circulating EpCAM+ CK20+ cells/ml |
||
Patients (median: 220, range 0–8,980) |
24 |
80 |
Controls (n = 10) (median: 0, range 0–630)* |
3 |
30 |
*p<0.05 ***p<0.0005
To more specifically detect CTC in MCC patients we added a second tumor marker to the assay (Figure 3A). In patient samples, the median percentage of CD56+ cells among EpCAM+ cells was 38% (range 0–100), and median percentage of CK20+ cells among EpCAM+ cells was 47% (range 0–100). EpCAM+, CD56+ CTC were detected in 23 of the 30 MCC patients (77%), whereas no (0%) control samples had EpCAM+, CD56+ double positive cells (p<0.0005). EpCAM+, CK20+ cells were detected in 24 MCC patients (80%) and 3 (30%) healthy controls (p<0.01). We also used immunoperoxidase staining for MCV large T-antigen on cell suspensions of two selected patients to confirm that MCV+ CTC are detectable in patient blood (Figure 3B). Unfortunately, we were unable to adapt fluorochrome labeled MCV large T-antigen antibody for routine use in our assay due to nonspecific staining. These results demonstrate that adding a second tumor marker can increase specificity of CTC analysis with EpCAM+, CD56+ double detection showing no false positives in healthy control patients.
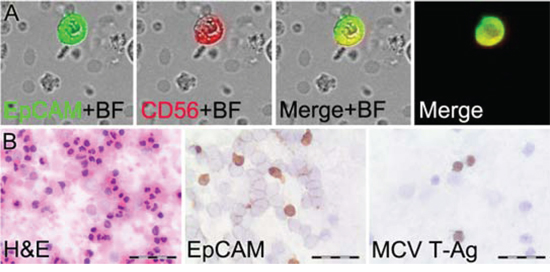
Figure 3: Detection of MCC circulating tumor cells (CTC). A. Laser Scanning Cytometry image of an EpCAM+, CD56+ CTC using immunofluorescence and brightfield (BF) detection. B. Light microscopy of peripheral blood cell suspensions stained with hematoxylin and eosin or immunoperoxidase staining of CTC with EpCAM and MCV large T-antigen (T-Ag). Scale bars=40 μm.
There was insufficient follow up time after measuring CTC counts to assess their prognostic value. We used disease stage at time of blood collection (Table 2, control samples as stage 0) as a proxy for progression risk. The mean counts of EpCAM+ CTC were significantly different by disease stage (one-way ANOVA p<0.005), however a linear trend test was not significant reflecting a lack of correlation between increasing CTC counts and higher disease stage (Figure 4B). Similar results were seen with EpCAM+, CD56+ and EpCAM+, CK20+ CTC (Figure S4). One confounding issue in this analysis is that not all patient samples were drawn at the same point relative to their diagnosis, recurrence events, and tumor treatments (median time after diagnosis=38 months, range=0–148 months). The prognostic implications of MCC stage after disease progression are not well defined, and thus stage in this context may not portend eventual outcomes. Further follow up is needed before we can assess if CTC counts will correlate with disease progression.
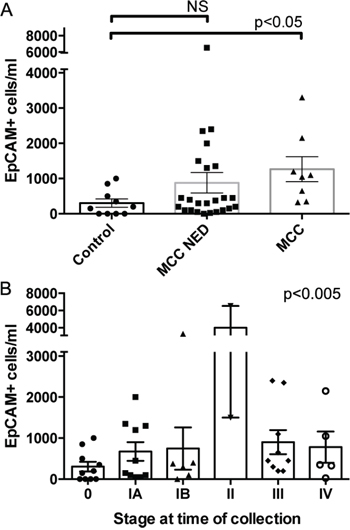
Figure 4: CTC counts correlate with MCC disease burden. Absolute numbers and mean values of EpCAM+ cells/ml blood detected in A. healthy controls, MCC patients with no evidence of disease (MCC NED), and MCC patients with active or recent tumor (MCC); or B. patients with different disease stages at time of CTC assessment compared by one-way ANOVA (error bars=SEM, p-values as indicated).
To assess if CTC counts reflect MCC disease burden, we compared controls to NED patients and to patients with active disease (measurable tumor or treatment within the last 56 days). Of the 8 patients with active disease all 8 (100%) had detectable CTC regardless of the staining markers used. Moreover, despite the small sample size, patients with active disease had significantly higher CTC counts than controls for all staining combinations (Figure 4A and S4, p<0.05), suggesting that CTC counts may be useful in following the extent of disease or detecting recurrences in patients with MCC. Many MCC patients with NED had CTC counts comparable to those seen in healthy controls, but some had elevated CTC counts. It is unclear if the NED patients with elevated CTC counts had occult disease or are at higher risk for disease progression.
In patients with multiple CTC observations, we were able to examine CTC counts over time. Patients without any disease related events showed consistent CTC numbers over time with deviations of no more than 20%. Four patients were monitored while undergoing tumor treatment, and all displayed decreasing CTC numbers (Figure 5A–5C). Only 2 patients developed new metastases during serial monitoring, both associated with increasing CTC numbers (Figure 5C, 5D). Although anecdotal, these observations further suggest that CTC counts reflect MCC disease status and could be used to effectively monitor for treatment responses and detect recurrences.
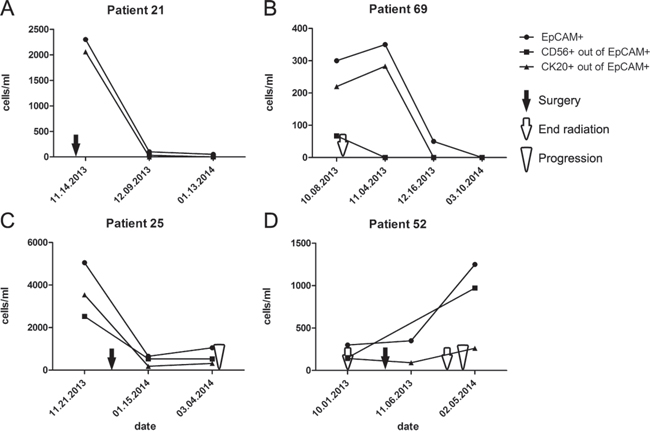
Figure 5: Longitudinal changes in CTC counts reflect MCC treatment response and tumor progression in individual patients. A. Patient 21 had surgery of a local recurrence on the 11th of November 2013 (black arrow), CTC numbers decreased within 4 weeks and then stayed stable during the next 4 weeks. B. Patient 69 had radiotherapy from the 4th of September 2013 until the 9th of October (white arrow), followed by a therapy with interferon α2b over the next 18 months, CTC numbers increased during the first weeks of interferon treatment, but then decreased dramatically. C. Patient 25 had surgery of regional metastases on the 27th of December 2013 (black arrow), CTC numbers decreased within 4 weeks followed by a CTC increase and the diagnosis of distant metastases on the 5th of March 2014 (white arrowhead). D. Patient 52 completed radiotherapy on the 1st of October 2013 (white arrow); on the 23rd of October a regional LN metastasis was resected (black arrow) and treated with radiotherapy. Shortly after completing radiation (white arrow), a distant metastasis was diagnosed on the 30th of January 2014 (white arrowhead).
DISCUSSION
We found that NSE and ChrA levels are not highly effective biomarkers to predict progression or detect recurrent MCC. In contrast, CTC counts demonstrate potential utility as a biomarker for MCC disease behavior.
Elevated blood levels of NSE and ChrA have been reported in MCC patients [22–27]. However, their diagnostic and prognostic value has only been investigated in single cases or small patient groups. A comparison between ChrA and NSE levels in neuroendocrine tumor (NET) patients demonstrated elevated ChrA in 25% and NSE in 50% of four MCC patients [28]. In contrast, Cimitan et al. found normal ChrA levels in six MCC patients [29]. Our observations from 60 patients over 259 person-years suggest changes in NSE and ChrA are not clinically meaningful in following patients with MCC. Most MCC tumors express NSE and ChrA, however both markers are also expressed in normal tissues [11, 30], requiring sufficient tumor burden to alter blood levels [7]. Thus, it is not surprising to find that NSE and ChrA might be more specific as biomarkers for lower grade NETs and less effective in aggressive MCC. However, as its retrospective design and inconsistent testing across the patient population limit our study of NSE and ChrA in MCC, it is possible that a weak correlation between these markers and MCC went undetected. A regimented prospective analysis would be required to investigate this possibility.
CTC enumeration has proven useful in establishing prognosis for patients with breast, colon, liver, and prostate cancer [16–19]. Breast CTC can reflect response to chemotherapy, predict relapse, and serve as markers for tumor aggressiveness [19]. Interestingly, breast cancer CTC can be found up to 22 years after mastectomy in NED patients [31]. Similarly, we detected elevated CTC counts (480 EpCAM+, CD56+ cells/ml) in a NED patient 11 years after MCC diagnosis. The source and nature of CTC detected in long-term cancer survivors remains unclear, and raises questions about their tumorigenic potential.
Numerous assays can detect CTC at single-cell resolution in peripheral blood [32], however some methods have shortcomings to consider. Flow cytometry cannot differentiate morphologically characteristic tumor cells from nonspecific events [33]. Density gradient purification leads to loss of relevant cells [34]. To date, CTC in the blood of MCC patients have been described in a handful of case reports and one case series [35–38]. Blom et al. [38] analyzed CTC from 34 MCC patients using the CellSearch system (Veridex LLC) that is based on magnetic bead enrichment. In contrast to maintrac, CellSearch is dependent on cells expressing a minimum level of surface antigen to be retained in the magnetic field, uses a large blood volume (7.5ml), and may result in cell destruction [39–42]. As a result, CellSearch shows lower sensitivity than other methods. Our study used the highly-sensitive maintrac assay that directly stains and quantifies cells from a single milliliter of patient blood.
Using the maintrac system we detected EpCAM+ CTC in 97% of MCC patients with a median of 450 cells/ml, and EpCAM+ CD56+ CTC in 70% of MCC patients with a median of 150 cells/ml. This contrasts with Blom et al. [38] who detected EpCAM+, CK8+ CTC in 41% of patients with a median of only 2 cells/7.5ml. Both studies demonstrated an association between CTC detection and extent of disease, as well as CTC reductions in response to therapy. In contrast to their findings, we found EpCAM+, CD56+ CTC in 13 of 22 (59%) NED patients, whereas Blom et al. did not detect CTC in NED patients. At the same time, their study failed to detect CTC in 15 of 27 cases (56%) that had active disease, whereas we found CTC in 8 of 8 samples (100%) from patients with active disease. Their study demonstrated a correlation between disease outcomes and CTC detection. Longer follow up is needed to determine the prognostic value of CTC levels in our study population.
Using maintrac we detected circulating EpCAM+ cells in some healthy controls. EpCAM is often expressed on epithelial tumors but also on some normal epithelia [20, 43], and normal epithelial cells can circulate in blood under certain circumstances [44, 45]. To increase the specificity of CTC detection, we used anti-EpCAM in combination with anti-CK20 [46, 47] or anti-CD56 [48–51]. Blood cells lack EpCAM expression, allowing for the exclusion of CK20 expression in granulocytes [52], and CD56 on NK T-cells [53]. However, EpCAM+, CK20+ cells in healthy volunteers suggest that some circulating cells may be normal intestinal epithelium that co-expresses both markers [54, 55]. In contrast, no circulating EpCAM+, CD56+ cells were found in healthy volunteers, making it the most specific marker combination tested.
CONCLUSIONS
We found that despite the reported utility of NSE and ChrA blood levels as biomarkers for low grade NETs, clinical use of NSE and ChrA levels in MCC patients failed to correlate with outcomes, disease progression, or tumor burden. In the same patient population we found that maintrac detection readily identified EpCAM+ CTC with high sensitivity. Moreover, adding CD56 as a second tumor marker increased the specificity of CTC detection. Although CTC counts reflected tumor burden, additional follow up is needed to determine how CTC correlate with disease outcomes. The use of CTC as biomarkers for MCC will require further development and validation, but our results and those of Blom et al. suggest CTC may be useful in the staging and longitudinal monitoring of MCC.
MATERIALS AND METHODS
Patients
Patients treated at the University of Heidelberg for MCC between 1998 and 2014 with at least one laboratory evaluation of blood NSE or ChrA levels were included in the study. MCC diagnosis was verified histopathologically. Clinical and biographical data were abstracted in a systematic chart review completed in October 2014. MCC staging was classified according to 7th edition AJCC guidelines [4].
All procedures have approved by the University of Heidelberg medical ethics committee (approval number S570/2013). Informed consent has been obtained.
Analysis of NSE and chrA blood levels
Blood samples were analyzed for NSE and ChrA in the Heidelberg University Hospital’s central laboratory. NSE (ug/ml) in serum was categorically interpreted as WNL <17, above normal ≥17–24.3, or high ≥24.3. Until December 2012 ChrA (U/ml) was measured in plasma, and after December 2012 ChrA (ng/ml) was measured in serum. A period of parallel analyses in plasma and serum assured good correlation between the two systems. A common categorical scale for ChrA levels was used as follows. ChrA plasma: WNL <25, above normal ≥25–53.75, high ≥53.75. ChrA serum: WNL <84.7, above normal ≥84.7–105.65, high ≥105.65.
Detection of CTC
Patients treated for MCC between September 2013 and May 2014 who consented to participate were screened for CTC. Detection of CTC (cells/ml blood) using the maintrac system (SIMFO, Bayreuth, Germany) was performed as previously described [56]. EpCAM staining was used to identify all putative tumor cells, and CD56 or CK20 staining was added to confirm MCC tumor cells. Only appropriately stained cells that met the morphological criteria of a tumor cell were counted as CTC.
Immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) tissue was cut in 2 μm sections, stained using standard immunohistochemistry protocols, and visualized using the Envision System (Dako) as described by the manufacturer. For immunohistochemical staining of CTC, cell suspensions were dropped on slides, air-dried and stained with the primary antibodies.
Statistics
Progression free survival (PFS) and disease specific survival (DSS) were calculated from the date of first treatment until the date of first progression, death, or last follow-up. Two-tailed p-values were used for all comparisons.
Detailed methods are available in the Supplementary Materials.
ACKNOWLEDGMENTS
We thank all participating patients as well as Katrin Wolk for excellent technical work and Helga Schüren-Münch for admirably coordinating the clinic.
GRANT SUPPORT
This work was supported by the Olympia-Morata-Program of the University of Heidelberg to MRG, and the National Institutes of Health Intramural Research Program, Center of Cancer Research, National Cancer Institute to IB. The opinions expressed are those of the authors and do not necessarily represent those of the National Institutes of Health or the National Cancer Institute.
CONFLICTS OF INTEREST
The authors declare that they do not have financial or other conflicts of interest to declare.
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the University of Heidelberg institutional review board.
REFERENCES
1. Schrama D, Becker JC. Merkel cell carcinoma—pathogenesis, clinical aspects and treatment. J Eur Acad Dermatol Venereol. 2011; 25:1121–1129.
2. Nicolaidou E, Mikrova A, Antoniou C, Katsambas AD. Advances in Merkel cell carcinoma pathogenesis and management: a recently discovered virus, a new international consensus staging system and new diagnostic codes. Br J Dermatol. 2012; 166:16–21.
3. Tai P. Merkel cell cancer: update on biology and treatment. Curr Opin Oncol. 2008; 20:196–200.
4. Lemos BD, Storer BE, Iyer JG, Phillips JL, Bichakjian CK, Fang LC, Johnson TM, Liegeois-Kwon NJ, Otley CC, Paulson KG, Ross MI, Yu SS, Zeitouni NC, et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. 2010; 63:751–761.
5. Kittaneh M, Montero AJ, Gluck S. Molecular profiling for breast cancer: a comprehensive review. Biomarkers in cancer. 2013; 5:61–70.
6. Zeestraten EC, Benard A, Reimers MS, Schouten PC, Liefers GJ, van de Velde CJ, Kuppen PJ. The prognostic value of the apoptosis pathway in colorectal cancer: a review of the literature on biomarkers identified by immunohistochemistry. Biomarkers in cancer. 2013; 5:13–29.
7. Granberg D, Stridsberg M, Seensalu R, Eriksson B, Lundqvist G, Oberg K, Skogseid B. Plasma chromogranin A in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1999; 84:2712–2717.
8. O’Connor DT, Deftos LJ. Secretion of chromogranin A by peptide-producing endocrine neoplasms. N Engl J Med. 1986; 314:1145–1151.
9. Hsiao RJ, Seeger RC, Yu AL, O’Connor DT. Chromogranin A in children with neuroblastoma. Serum concentration parallels disease stage and predicts survival. J Clin Invest. 1990; 85:1555–1559.
10. Riley RD, Heney D, Jones DR, Sutton AJ, Lambert PC, Abrams KR, Young B, Wailoo AJ, Burchill SA. A systematic review of molecular and biological tumor markers in neuroblastoma. Clin Cancer Res. 2004; 10:4–12.
11. Burghuber OC, Worofka B, Schernthaner G, Vetter N, Neumann M, Dudczak R, Kuzmits R. Serum neuron-specific enolase is a useful tumor marker for small cell lung cancer. Cancer. 1990; 65:1386–1390.
12. Portela-Gomes GM, Hacker GW, Weitgasser R. Neuroendocrine cell markers for pancreatic islets and tumors. Appl Immunohistochem Mol Morphol. 2004; 12:183–192.
13. Becker J, Mauch C, Kortmann RD, Keilholz U, Bootz F, Garbe C, Hauschild A, Moll I. Short German guidelines: Merkel cell carcinoma. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology: JDDG. 2008; 1:S15–16.
14. National Comprehensive Cancer Network - Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel cell carcinoma. 2015. http://www.merkelcell.org/usefulInfo/index.php. Accessed 13 May 2015.
15. Zhe X, Cher ML, Bonfil RD. Circulating tumor cells: finding the needle in the haystack. Am J Cancer Res. 2011; 1:740–751.
16. Friedlander TW, Premasekharan G, Paris PL. Looking back, to the future of circulating tumor cells. Pharmacol Ther. 2013; 142:271–80.
17. Pantel K and Alix-Panaberes C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013; 73:6384–6388.
18. Schulze K, Gasch C, Staufer K, Nashan B, Lohse AW, Pantel K, Riethdorf S, Wege H. Presence of EpCAM-positive circulating tumor cells as biomarker for systemic disease strongly correlates to survival in patients with hepatocellular carcinoma. Int J Cancer. 2013; 133:2165–2171.
19. Pachmann K, Camara O, Kavallaris A, Krauspe S, Malarski N, Gajda M, Kroll T, Jorke C, Hammer U, Altendorf-Hofmann A, Rabenstein C, Pachmann U, Runnebaum I, et al. Monitoring the response of circulating epithelial tumor cells to adjuvant chemotherapy in breast cancer allows detection of patients at risk of early relapse. J Clin Oncol. 2008; 26:1208–1215.
20. Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007; 171:386–395.
21. Kurzen H, Kaul S, Egner U, Deichmann M, Hartschuh W. Expression of MUC 1 and Ep-CAM in Merkel cell carcinomas: implications for immunotherapy. Archives of dermatological research. 2003; 295:146–154.
22. Giannone L, Johnson DH, Grosh WW, Davis BW, Marangos PJ, Greco FA. Serum neuron-specific enolase in metastatic Merkel cell tumors. Med Pediatr Oncol. 1985; 13:357–362.
23. Gu J, Polak JM, Van Noorden S, Pearse AG, Marangos PJ, Azzopardi JG. Immunostaining of neuron-specific enolase as a diagnostic tool for Merkel cell tumors. Cancer. 1983; 52:1039–1043.
24. Plowman PN. Serum marker for Merkel cell tumour. Clin Radiol. 1989; 40:542.
25. Hall PA, d’Ardenne AJ, Butler MG, Stearn PM, Subbuswamy SG, Blackshaw AJ, Levison DA. Cytokeratin and laminin immunostaining in the diagnosis of cutaneous neuro-endocrine (Merkel cell) tumours. Histopathology. 1986; 10:1179–1190.
26. Wilson BS, Lloyd RV. Detection of chromogranin in neuroendocrine cells with a monoclonal antibody. Am J Pathol. 1984; 115:458–468.
27. Battifora H, Silva EG. The use of antikeratin antibodies in the immunohistochemical distinction between neuroendocrine (Merkel cell) carcinoma of the skin, lymphoma, and oat cell carcinoma. Cancer. 1986; 58:1040–1046.
28. Nobels FR, Kwekkeboom DJ, Coopmans W, Schoenmakers CH, Lindemans J, De Herder WW, Krenning EP, Bouillon R, Lamberts SW. Chromogranin A as serum marker for neuroendocrine neoplasia: comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J Clin Endocrinol Metab. 1997; 82:2622–2628.
29. Cimitan M, Buonadonna A, Cannizzaro R, Canzonieri V, Borsatti E, Ruffo R, De Apollonia L. Somatostatin receptor scintigraphy versus chromogranin A assay in the management of patients with neuroendocrine tumors of different types: clinical role. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2003; 14:1135–1141.
30. Haimoto H, Takahashi Y, Koshikawa T, Nagura H, Kato K. Immunohistochemical localization of gamma-enolase in normal human tissues other than nervous and neuroendocrine tissues. Lab Invest. 1985; 52:257–263.
31. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004; 10:8152–8162.
32. Muller V, Alix-Panabieres C, Pantel K. Insights into minimal residual disease in cancer patients: implications for anti-cancer therapies. Eur J Cancer. 2010; 46:1189–1197.
33. Pachmann K, Heiss P, Demel U, Tilz G. Detection and quantification of small numbers of circulating tumour cells in peripheral blood using laser scanning cytometer (LSC). Clin Chem Lab Med. 2001; 39:811–817.
34. Fleisher TA, Marti GE. Detection of unseparated human lymphocytes by flow cytometry. Current protocols in immunology/edited by John E Coligan, [et al]. 2001; Chapter 7:Unit 7 9.
35. Tam CS, Turner P, McLean C, Whitehead S, Cole-Sinclair M. ‘Leukaemic’ presentation of metastatic Merkel cell carcinoma. Br J Haematol. 2005; 129:446.
36. Nemoto I, Sato-Matsumura KC, Fujita Y, Natsuga K, Ujiie H, Tomita Y, Kato N, Kondo M, Ohnishi K. Leukaemic dissemination of Merkel cell carcinoma in a patient with systemic lupus erythematosus. Clin Exp Dermatol. 2008; 33:270–272.
37. Hartley MA, Tao J, Baz R. Merkel cell carcinoma in the peripheral blood of a patient with concomitant chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol. 2010; 28:e113–114.
38. Blom A, Bhatia S, Pietromonaco S, Koehler K, Iyer JG, Nagase K, Paulson K, Sabath DE, Nghiem P. Clinical utility of a circulating tumor cell assay in Merkel cell carcinoma. J Am Acad Dermatol. 2013.
39. Scriba TJ, Purbhoo M, Day CL, Robinson N, Fidler S, Fox J, Weber JN, Klenerman P, Sewell AK, Phillips RE. Ultrasensitive detection and phenotyping of CD4+ T cells with optimized HLA class II tetramer staining. J Immunol. 2005; 175:6334–6343.
40. Pizon M, Zimon D, Carl S, Pachmann U, Pachmann K, Camara O. Heterogeneity of circulating epithelial tumour cells from individual patients with respect to expression profiles and clonal growth (sphere formation) in breast cancer. Ecancermedicalscience. 2013; 7:343.
41. Coumans FA, Doggen CJ, Attard G, de Bono JS, Terstappen LW. All circulating EpCAM+CK+CD45-objects predict overall survival in castration-resistant prostate cancer. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2010; 21:1851–1857.
42. Panteleakou Z, Lembessis P, Sourla A, Pissimissis N, Polyzos A, Deliveliotis C, Koutsilieris M. Detection of circulating tumor cells in prostate cancer patients: methodological pitfalls and clinical relevance. Molecular medicine. 2009; 15:101–114.
43. Gaiser MR, Lammermann T, Feng X, Igyarto BZ, Kaplan DH, Tessarollo L, Germain RN, Udey MC. Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc Natl Acad Sci U S A. 2012; 109:E889–897.
44. Hardingham JE, Hewett PJ, Sage RE, Finch JL, Nuttall JD, Kotasek D, Dobrovic A. Molecular detection of blood-borne epithelial cells in colorectal cancer patients and in patients with benign bowel disease. Int J Cancer. 2000; 89:8–13.
45. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420:860–867.
46. Moll R, Franke WW. Cytoskeletal differences between human neuroendocrine tumors: a cytoskeletal protein of molecular weight 46, 000 distinguishes cutaneous from pulmonary neuroendocrine neoplasms. Differentiation. 1985; 30:165–175.
47. Bellizzi AM. Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anat Pathol. 2013; 20:285–314.
48. Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, Amano M, Ogata K, Setoyama M. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci. 2003; 31:219–224.
49. Gallego R, Garcia-Caballero T, Fraga M, Beiras A, Forteza J. Neural cell adhesion molecule immunoreactivity in Merkel cells and Merkel cell tumours. Virchows Arch. 1995; 426:317–321.
50. LeBoit PE. “Magic bullets” in immunohistochemistry. Am J Dermatopathol. 2002; 24:518–520.
51. McNiff JM, Cowper SE, Lazova R, Subtil A, Glusac EJ. CD56 staining in Merkel cell carcinoma and natural killer-cell lymphoma: magic bullet, diagnostic pitfall, or both? J Cutan Pathol. 2005; 32:541–545.
52. Jung R, Petersen K, Kruger W, Wolf M, Wagener C, Zander A, Neumaier M. Detection of micrometastasis by cytokeratin 20 RT-PCR is limited due to stable background transcription in granulocytes. Br J Cancer. 1999; 81:870–873.
53. Imamura N, Kusunoki Y, Kajihara H, Okada K, Kuramoto A. Aggressive natural killer cell leukemia/lymphoma with N901-positive surface phenotype: evidence for the existence of a third lineage in lymphoid cells. Acta haematologica. 1988; 80:121–128.
54. Bustin SA, Gyselman VG, Siddiqi S, Dorudi S. Cytokeratin 20 is not a tissue-specific marker for the detection of malignant epithelial cells in the blood of colorectal cancer patients. International journal of surgical investigation. 2000; 2:49–57.
55. Pantel K, Alix-Panabieres C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010; 16:398–406.
56. Pachmann K, Clement JH, Schneider CP, Willen B, Camara O, Pachmann U, Hoffken K. Standardized quantification of circulating peripheral tumor cells from lung and breast cancer. Clin Chem Lab Med. 2005; 43:617–627.