
INTRODUCTION
Sorafenib, a multikinase inhibitor, is the current standard systemic therapy for patients with advanced hepatocellular carcinoma (HCC) [1]. The major mechanism of action includes inhibition of the Raf/mitogen-activated protein kinase-extracellular signal-regulated kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling and inhibition of tumor angiogenesis [2]. However, the antitumor mechanisms of sorafenib may involve complex interactions in cellular signaling pathways, which are independent of the inhibitory effects of sorafenib on Raf/MEK/ERK activities [3–6]. Clarification of these “off-target effects” of sorafenib will facilitate identification of predictive biomarkers for treatment efficacy and combination therapy design for HCC [3].
Growth arrest DNA damage-inducible gene 45 (GADD45) family proteins have been reported to play essential roles in cellular stress response, survival, senescence, and apoptosis regulation [7]. Both tumor-promoting and tumor-suppressing effects of GADD family proteins have been reported, depending on the cell types tested, the types of oncogenic stresses, and the interaction with other cellular signaling pathways [7]. GADD45 family proteins are frequently underexpressed in various types of cancers including HCC [8, 9]. The induction of GADD45 expression in the liver may facilitate the regulation of liver regeneration after hepatectomy or chemically induced liver injury [10, 11]. The role of GADD45 expression in hepatocarcinogenesis remains undetermined.
GADD45 family proteins are also crucial mediators of genotoxic stress-induced apoptosis and transforming growth factor-β-induced apoptosis [12, 13]. Induction of GADD45 expression can enhance the therapeutic efficacy of cytotoxic agents in cancer cells [14, 15]. We previously reported that GADD45β expression was induced in HCC cells after sorafenib treatment, and this induction was an important predictor of sorafenib sensitivity in HCC cells [16]. We hypothesized that GADD45 family proteins may serve as predictive biomarkers for efficacy of molecular targeted therapy for HCC.
In the present study, we explored the biological significance of GADD45γ expression in HCC tumor tissues, and the potential predictive value of GADD45γ induction in HCC cells on the efficacy of sorafenib treatment. GADD45γ has been reported to be a tumor suppressor in multiple cancer types, and it can induce growth arrest and apoptosis in response to environmental stress [17]. Furthermore, GADD45γ expression was suppressed in HCC, but the clinical significance was unclear [18]. The regulatory mechanisms of GADD45γ expression in response to sorafenib treatment was also explored.
RESULTS
Induction of GADD45γ expression in HCC cells by sorafenib correlated with sorafenib efficacy
In vitro studies on the effects of GADD45γ expression on sorafenib-sensitive (Huh-7 and HepG2, IC50 6–7 μM), acquired sorafenib-resistant (Huh-7R and HepG2R, IC50 12–15 μM), and intrinsic sorafenib-resistant (Hep3B, IC50 12 μM) HCC cells are summarized in Figure 1. The baseline GADD45γ expression did not vary signficantly between sorafenib-sensitive and sorafenib-resistant HCC cells (Figure 1A). However, GADD45γ induction was more prominent in sorafenib-sensitive HCC cells than in sorafenib-resistant HCC cells and was independent of the MEK/ERK signaling in HCC cells because the RAF inhibitor, ZM336372, or the MEK inhibitor, U0126, could not induce GADD45γ expression (Figure 1A and Supplementary Figure S1).
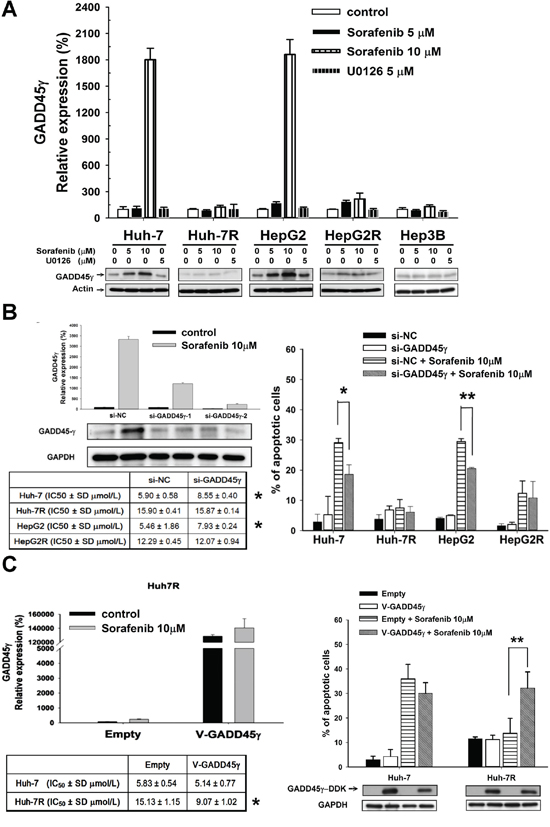
Figure 1: In vitro studies on the effects of GADD45γ expression on sorafenib efficacy. A. GADD45γ mRNA and protein induction after sorafenib, which was independent of cellular RAF/ERK activity, correlated with sorafenib efficacy. HCC cells were treated with sorafenib or U0126 for 24 hours. GADD45γ mRNA and protein levels were assessed using real-time qRT-PCR and Western blotting. Whole cell lysates after drug treatment were examined using Western blotting. B. GADD45γ knockdown increased the resistance of sorafenib-sensitive HCC cells to sorafenib. In the left panel, efficacy of GADD45γ knockdown was measured using qRT-PCR and Western blotting. Huh-7 cells were transfected with siRNA directed against GADD45γ (si-GADD45γ-1 and si-GADD45γ-2) or a negative-control (si-NC) siRNA and treated with sorafenib (10 μM) for 48 hours. IC50 of sorafenib with or without GADD45γ knockdown were assessed using MTT analysis. *p < 0.05 compared with si-NC. In the right panel, effects of GADD45γ knockdown on sorafenib-induced apoptosis were assessed using Sub-G1 analysis. Each value is the mean ± SD of three independent experiments. *, p < 0.05; **, p < 0.01, compared with si-NC + sorafenib 10 μM. C. GADD45γ over-expression could reverse sorafenib resistance. In the left panel, efficacy of GADD45γ overexpression was measured using quantitative RT-PCR and Western blotting. Huh-7 and Huh-7R cells were transfected with GADD45γ (V-GADD45γ) or empty vectors and treated with sorafenib (10 μM) or a control. IC50 of sorafenib with or without GADD45γ overexpression were assessed using MTT analysis. *p < 0.05 compared with empty vectors. Huh-7 and Huh-7R cells were transfected with GADD45γ or empty vectors and treated with sorafenib for 72 hours. In the right panel, proportions of apoptotic cells are indicated by the percentage of cells in the sub-G1 fraction. Columns, mean of three independent experiments; bars, SD. *, p < 0.05; **, p < 0.01, compared with Empty + Sorafenib 10 μM.
The suppression of GADD45γ expression by using siRNA increased the resistance to sorafenib in sorafenib-sensitive HCC cells; this was evident with the increased IC50 and reduced sorafenib-induced apoptosis (Figure 1B and Supplementary Figure S2A). Overexpression of GADD45γ in acquired sorafenib-resistant HCC cells significantly enhanced the suppression of proliferation and sorafenib-induced apoptosis (Figure 1C and Supplementary Figure S2B).
In vivo studies determining the significance of GADD45γ induction in sorafenib-induced apoptosis are summarized in Figure 2. Sorafenib treatment (10 mg/kg/day) inhibited tumor growth in Huh-7 but not in Huh-7R xenografts (Figure 2A). Inhibition of tumor growth in Huh-7 xenografts was associated with the induction of GADD45γ expression, increased tumor cell apoptosis (TUNEL assay), and reduced tumor angiogenesis (microvessel density) (Figure 2B and Supplementary Figure S3A and S3C). GADD45γ overexpression in Huh-7R xenografts, achieved using adenoviral transfer (Figure 2C), did not demonstrate tumor-suppressing effects by itself. However, significant sensitization of the Huh-7R xenograft to sorafenib, increased tumor cell apoptosis (TUNEL assay), and reduced tumor angiogenesis were observed (Figure 2D and Supplementary Figure S3D and S3F). The body weight of animals with or without sorafenib treatment or GADD45γ overexpression did not vary significantly (Supplementary Figure S3B and S3E). This indicated that GADD45γ induction may act in a synthetic-lethal manner to enhance the antitumor activity of sorafenib in HCC cells.
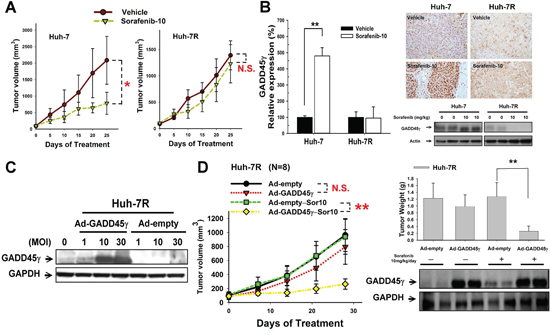
Figure 2: In vivo studies on the effects of GADD45γ induction on sorafenib efficacy. Huh-7 or Huh-7R cells were injected subcutaneously into male BALB/c athymic nude mice. Mice were treated daily by gavage as indicated (Vehicle or sorafenib 10 mg/kg/day (Sor-10)). A. Difference in tumor growth between Huh-7 and Huh-7R xenografts after sorafenib treatment (n = 5 in each group). B. Difference in GADD45γ mRNA and protein expression measured using real-time qRT-PCR, immunohistochemical staining, and Western blotting. GADD45γ mRNA was expressed relative to endogenous AFP expression. C. Efficacy of GADD45γ induction by infection with adenoviral vectors. Huh-7R cells were infected with Ad-GADD45γ and a control virus (Ad-empty) at the indicated multiplicity of infection (MOI). Expression of GADD45γ was measured using Western blotting. D. GADD45γ induction reversed sorafenib resistance in vivo. Huh-7R cells were infected with GADD45γ-expressing or control (10 MOI) adenoviruses and were implanted subcutaneously into BALB/c athymic (nu+/nu+) mice 24 hours after adenoviral infection. Mice were treated with sorafenib (10 mg/kg/day) or a vehicle (n = 8 in each group). Tumors were harvested after 28 days of sorafenib treatment. Difference in tumor growth and difference in tumor weight and GADD45γ expression at the end of sorafenib treatment are shown. **, p < 0.01, compared with the control group.
Survivin is a crucial downstream mediator of GADD45γ induction to reverse sorafenib resistance
To explain the antitumor enhancement between GADD45γ induction and sorafenib, we assessed the molecules that have been linked to sorafenib resistance, including survivin, DR5/FADD (the TRAIL pathway), and Mcl-1, by using Western blotting [19–21]. Survivin was selected as the candidate mediator to explain the apoptosis-enhancing effects of GADD45γ because the variation in survivin expression was compatible with the enhanced antitumor efficacy between sorafenib and GADD45γ induction (Figure 3A and 3B). The baseline survivin expression did not vary signficantly between sorafenib-sensitive and acquired sorafenib-resistant HCC cells (Figure 3A and Supplementary Figure S4A). Survivin overexpression reduced the apoptosis-inducing effects of GADD45γ overexpression combined with sorafenib in the sorafenib-sensitive and sorafenib-resistant HCC cells (Figure 3C, Supplementary Figure S4A–S4C, and S5A). However, suppression of survivin expression by using siRNA knockdown further increased the apoptosis-inducing effects between GADD45γ overexpression and sorafenib (Figure 3D and Supplementary Figure S5B). Mcl-1overexpression, which has been reported as an essential mediator of antitumor effects of sorafenib in HCC, resulted in a similar trend of negating the enhancement between GADD45γ and sorafenib (Supplementary Figure S6). The preceding data supports the role of survivin as an important downstream mediator of antitumor enhancement between GADD45γ overexpression and sorafenib.
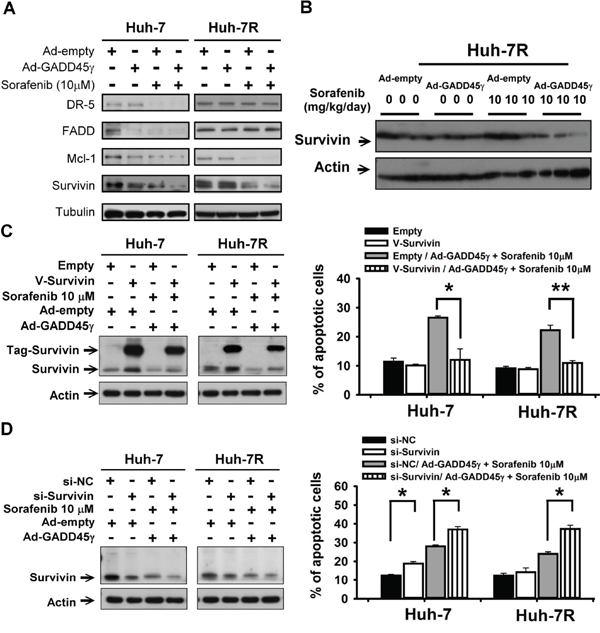
Figure 3: Survivin is a crucial downstream mediator of GADD45γ induction to reverse sorafenib resistance. A. Effects of GADD45γ induction to reverse sorafenib resistance on apoptosis-related proteins in Huh-7R cells. Huh-7 and Huh-7R cells were infected with GADD45γ-expressing (Ad-GADD45γ) or control (Ad-empty) adenoviruses and sorafenib (10 μM) for 48 hours. Whole cell lysates were subjected to Western blotting. B. Expression of survivin in xenograft experiments described in Figure 2D. C. Survivin was overexpressed by transfecting pCMV6-Myc-DDK-survivin (V-Survivin) into Huh-7 and Huh-7R cells. The cells were then treated with sorafenib (10 μM) and GADD45γ-expressing (Ad-GADD45γ) or control (Ad-empty) adenoviruses. D. Survivin knockdown enhanced the efficacy of GADD45γ induction combined with sorafenib. Huh-7 and Huh-7R cells were transfected with si-survivin or scrambled siRNA (si-NC) for 12 hours. The cells were then treated with sorafenib (10 μM) and GADD45γ-expressing or control adenoviruses. In C. and D. whole-cell lysates were collected for Western blotting after 48-hour drug treatment. The percentages of apoptotic cells were measured using flow cytometry after 72-hour drug treatment. Columns, mean of three independent experiments; bars, SD. *, p < 0.05; **, p < 0.01.
CCAAT/enhancer binding protein as a potential regulator of GADD45γ induction by sorafenib
We initially analyzed luciferase reporter activities of GADD45γ promoter plasmids (with serial deletions in the 5′-flanking region) because our data indicated that sorafenib induced GADD45γ expression at the transcriptional level. The region of -449/-82 in the 5′-flanking region was crucial for GADD45γ induction by sorafenib (Figure 4A). Two binding sites for the CCAAT/enhancer binding protein (C/EBP) transcriptional factor were identified in this region (-340/-330 and -103/-93) by using the TFSEARCH program (Supplementary Figure S7). Reporters with mutations at these C/EBP binding sites demonstrated no GADD45γ transcriptional activity after sorafenib treatment, further supporting the role of C/EBP in mediating GADD45γ induction by sorafenib (Figure 4B).
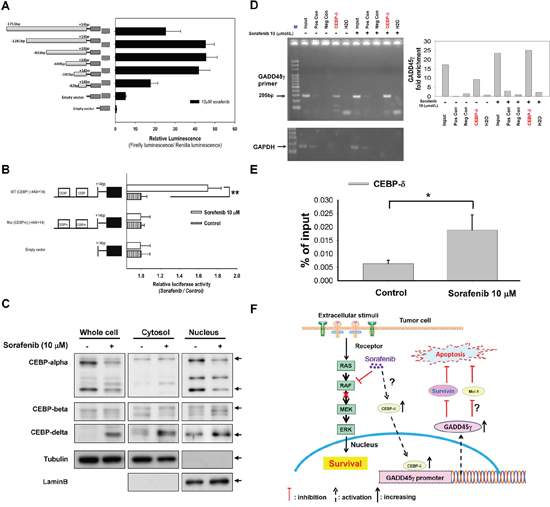
Figure 4: C/EBP as a potential regulator of GADD45γ induction by sorafenib. A. Localization of the transcriptional regulatory region of the GADD45γ promoter by using 5′-deletion analysis. The 5′-deletion constructs of the GADD45γ promoter (shown on the left) were transfected into Huh-7 cells, and the relative luciferase activity of each promoter fragment after sorafenib treatment is shown on the right. B. The GADD45γ promoter constructs with mutations in the C/EBP binding sites (shown on the left) were transfected into Huh-7 cells. The relative luciferase activities of each construct after sorafenib treatment are shown on the right. **, p < 0.01, compared with the control group. The results were representative data from three independent experiments. C. Analysis of C/EBP isoforms in total cell lysates and nuclear and cytoplasmic fragmentation. Huh-7 cells were treated with sorafenib for 24 hours; lysates were then collected and analyzed by Western blotting. D. Chromatin immunoprecipitation assay of the C/EBP-δ association with the GADD45γ promoter-enhancer in Huh-7 cells. Left panel, negative control (Neg Con) was chromatin immunoprecipitated with a normal mouse IgG. Positive control (Pos Con) was chromatin immunoprecipitated with an antiRNA polymerase II antibody. The input was 0.1% of the sonicated chromatin before immunoprecipitation. Right panel, the band intensities were quantified using AutoChemi imaging system (UVP, Cambridge, UK) and signal intensities, representing the DNA content, were normalized to Lane 5 from H2O. E. The amount of C/EBP-δ binding to the GADD45γ promoter-enhancer was assessed using real-time PCR in chromatin immunoprecipitation assay. *, p < 0.05, compared with control. F. Proposed mechanisms of the apoptosis-enhancing effects of GADD45γ induction by sorafenib in HCC cells.
Sorafenib treatment increased total and nuclear C/EBP-δ levels (Figure 4C). Increased binding of C/EBP-δ to the GADD45γ promoter was confirmed using chromatin immunoprecipitation (ChIP) (Figure 4D and 4E). The C/EBP-δ RNA levels in HCC cells were independent of sorafenib treatment, suggesting that sorafenib increases C/EBP-δ at the protein level (Supplementary Figure S8). This indicates that C/EBP-δ may help regulate the GADD45γ induction by sorafenib. The potential effects of GADD45γ induction on sorafenib-induced apoptosis in HCC cells was depicted in Figure 4F.
GADD45γ expression in HCC tumors facilitated the prediction of survival in patients with HCC who had undergone curative surgery
GADD45γ mRNA was more suppressed in HCC tumor tissue than in the adjacent nontumor liver tissue, and the median T to non-T ratio of GADD45γ mRNA was 33.8% (0.7%–974.0%) (Figure 5A). The expression levels of GADD45γ did not correlate with any clinicopathological factors, including age, sex, underlying chronic viral hepatitis, presence of cirrhosis, tumor grade and stage, serum α-fetoprotein levels, and the presence of vascular invasion (Table 1). Univariate analysis revealed that low GADD45γ expression was associated with short overall survival (Figure 5B and Supplementary Table S2). After controlling the potential confounding factors by using multivariate analysis, low GADD45γ expression became an independent predictor of poor overall survival, in addition to age and the presence of vascular invasion (Table 1). The hazard ratio of death was 2.94 (95% CI 1.27–6.77, p = 0.012) for patients with low GADD45γ expression (tumor to nontumor ratio of < 33.8%). The potential prognostic value of GADD45γ expression was supported by data from the cBio Cancer Genomics Portal (http://cbioportal.org) [22, 23], which revealed a consistent trend of superior survival in patients with GADD45γ overexpression (Supplementary Table S3).
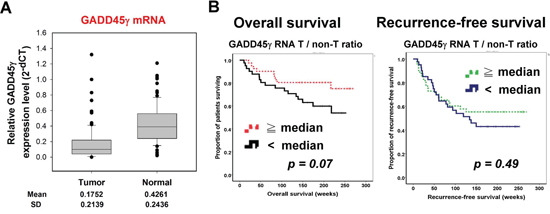
Figure 5: Low GADD45γ expression in HCC tumors predicted poor survival in patients with HCC who had undergone curative surgery. A. GADD45γ expression was more suppressed in the HCC tumor tissue than in the adjacent normal liver tissue. The expression levels were represented by the relative amount of the target gene (GADD45γ) vs. control gene (HPRT). 2 − ΔCT, where ΔCT = CT (target gene) − CT (control gene). B. Kaplan–Meier survival curves of overall survival and recurrence-free survival for patients with HCC whose tumors expressed high vs. low levels of GADD45γ. *, log-rank test (univariate analysis).
Table 1: GADD45γ expression and survival of patients with HCC who had undergone curative surgery
GADD45γ expression and clinical characteristics* |
|||
GADD45γ RNA T/non-T ratio |
P value |
||
< median (n = 41) |
≧ median (n = 41) |
||
Age (years) (median/range) |
59.0/37–85 |
65.0/38–86 |
0.17 |
Male/female |
29/12 |
28/13 |
0.81 |
HBV/HCV |
24/17 |
17/24 |
0.12 |
Cirrhosis yes/no |
19/22 |
16/25 |
0.50 |
AFP (ng/mL) (median/range) |
97.4/2.6 - 173900 |
22.3/2.2 - 181200 |
0.64 |
Stage I/III/IIIA |
17/18/6 |
21/12/8 |
0.38 |
MVI yes/no |
18/23 |
17/24 |
0.82 |
Tumor grade 1/2/3 |
0/27/14 |
3/29/9 |
0.12 |
Multi-variate analysis of overall survival and recurrence-free survival** |
|||
Coefficient |
HR (95% C.I.) |
P value |
|
Overall survival |
|||
MVI (-) vs. (+) |
−1.11 |
0.33 (0.15–0.74) |
0.007 |
Age < 65 y vs. ≥ 65 y |
−1.37 |
0.25 (0.11–0.59) |
0.001 |
GADD45γ RNA T/non-T ratio ≥ median vs. < median |
−1.08 |
0.34 (0.15–0.78) |
0.012 |
Recurrence-free survival |
|||
MVI (-) vs. (+) |
−1.01 |
0.37 (0.16–0.58) |
0.004 |
Age < 65 y vs. ≥ 65 y |
−0.89 |
0.41 (0.25–0.88) |
0.007 |
*Continuous variables were compared using one-way ANOVA; categorical variables were compared using the chi-square and Fisher's exact tests.
**The variables analyzed in the regression model included age, sex, tumor stage (stage IIIA vs. stage I/II), tumor grade (grade 2 vs. 0–1), virus infection (hepatitis B vs. C), α-fetoprotein levels, presence of cirrhosis, and the presence of macrovascular invasion (MVI).
DISCUSSION
In this study, we demonstrated that GADD45γ expression in HCC cells was associated with the sorafenib sensitivity of HCC cells. The induction of GADD45γ expression can reverse the resistance of HCC cells to sorafenib. In addition, suppression of GADD45γ expression in HCC tumor tissue was observed to be an independent predictor of poor overall survival in patients with HCC who had undergone curative surgery. The results indicate that GADD45γ may act as a prognostic factor and as a marker for response to sorafenib in HCC.
Regulation of GADD45 expression, which involves complex interaction between cellular growth control and stress response, has been extensively studied [24, 25]. Although suppression of GADD45 expression in cancer cells is commonly noted, the clinical significance is unclear. In this study we demonstrated that GADD45γ expression was not associated with other clinicopathological features of HCC, and the suppression of GADD45γ expression was correlated with poor overall survival in patients with HCC who had undergone curative surgery. Our data suggest that GADD45 family proteins may serve as tumor suppressors in carcinogenesis, which is different from the survival-promoting effects of GADD45 in liver regeneration [26, 27]. Because GADD45 family proteins may be regulated by similar cellular mechanisms, it is of value to explore the potential interaction between GADD45γ and other GADD45 family proteins in HCC progression and their prognostic values [28].
Validation of the predictive value of GADD45γ induction in response to sorafenib treatment is challenging. In our studies, the difference in GADD45γ induction after sorafenib treatment varied significantly between sorafenib-sensitive and sorafenib-resistant HCC cells. Therefore, comparison of GADD45γ between pre- and posttreatment tumor samples is essential to validate the concept. Tumor specimens for biomarker analysis were difficult to obtain because of the prevalent clinical diagnosis of HCC [1]. An additional difficulty was obtaining pre- and posttreatment biopsy specimens from patients with advanced HCC because the majority of patients have a high bleeding risk due to the underlying cirrhosis. Development of novel and noninvasive technology may help detect the molecular changes after drug therapy and facilitate the validation of predictive biomarkers for HCC [29, 30].
A major finding in this study is that while GADD45γ overexpression alone had no evident effects on growth or apoptosis induction in HCC cells, GADD45γ overexpression can significantly reverse the resistance of HCC cells to sorafenib in vitro and in vivo. This finding is consistent with previous studies, which suggest that GADD45γ can enhance the efficacy of cytotoxic therapy and provide opportunities for synthetic lethality-based development of new therapeutic targets [31]. The frequently reported downstream mediators of the proapoptotic effects of GADD family proteins include stress-related mitogen-activated protein kinases (p38 and MKK4) and the proapoptotic proteins Bim and PUMA [32, 33]. The possibility of using GADD45 expression as a marker for drug screening should be explored. A more comprehensive exploration of the signaling network pertinent to the proapoptotic effects of GADD45γ in HCC will facilitate identifying new druggable targets for HCC treatment.
Our results indicate that survivin and Mcl-1 are possible downstream mediators of the antitumor enhancement between GADD45γ and sorafenib. Survivin, which facilitates the integration of the cellular signals determining cellular proliferation, survival, and drug resistance, has been reported as a key regulator of the resistance of HCC cells to various molecular targeted agents [21, 34–36]. GADD45γ may act as a transcriptional coactivator or interact with other cellular signaling pathways, including cell cycle control and stress response, to regulate cell survival. Therefore, it may inhibit survivin expression through both transcriptional and posttranslational mechanisms [37, 38]. On the other hand, survivin expression is regulated by a complex intracellular signaling network and sorafenib may inhibit survivin expression through the downregulation of a mammalian target of rapamycin activity, independent of GADD45γ [39]. Mcl-1 is also a crucial antiapoptotic protein for cell survival maintenance and drug resistance regulation [40–42]. Therapeutic strategies targeting survivin or Mcl-1 have been developed, and additional studies based on these strategies alone or in combination with sorafenib in patients with HCC are warranted [43, 44].
In this study, we demonstrated that the transcriptional factor C/EBP-δ may increase the induction of GADD45γ by sorafenib. Previous studies have indicated that C/EBP-δ has contrasting effects at different stages of carcinogenesis [45]. However, C/EBP-δ can suppress tumor growth by inducing apoptosis, augmenting DNA damage response, and inhibiting cell cycle progression [46, 47]. Furthermore, C/EBP-δ can induce lymphangiogenesis and tumor metastases in response to hypoxia [48, 49]. GADD45γ can also be induced by other transcriptional factors and by the methylation status of the promoter region [50]. Cumulative evidence indicates that GADD45 family proteins may facilitate the integration of cellular response and environmental stress by interacting with different signaling pathways [7]. An improved understanding of the mechanisms regulating GADD45 family protein expression in HCC cells will facilitate clarification on whether GADD45 family proteins are suitable targets for new drug development for HCC.
In conclusion, our study indicates that GADD45γ suppression is a poor prognostic factor in patients with HCC. The induction of GADD45γ expression contributes to sorafenib-induced apoptosis in HCC cells and may serve as a biomarker for the development of new targeted therapy for HCC.
MATERIALS AND METHODS
Cell culture
The HCC cell lines, HepG2 and Hep3B, were obtained from the American Type Culture Collection, and the Huh-7 cell line was from the Health Science Research Resources Bank. In this study Huh-7 and HepG2 cells (sorafenib IC50 6–7 μM) were classified as sorafenib-sensitive because previous pharmacokinetic studies have determined the maximal plasma concentration of sorafenib in patients treated by the recommended dosage by Food Drug Administration (400 mg twice daily), was between 5 and 10 μM [51, 52]. Cell lines with acquired resistance to sorafenib, Huh-7R and HepG2R, were generated through continuous sorafenib treatment (up to 10 μM). Cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, penicillin (100 units/mL), streptomycin (100 μg/mL), L-glutamine (2 mmol/L), and sodium pyruvate (1 mmol/L) at 37°C in a humidified incubator containing 5% CO2.
Quantitative reverse transcriptase polymerase chain reaction
RNA extraction, cDNA synthesis, and cDNA quantification were performed as described previously [16]. The primers for the GADD45γand CEBPδ genes were purchased from Applied Biosystems (ABI TaqMan assay ID: Hs00198672_m1 and Hs00270931_s1). The primers for the hypoxanthine phosphoribosyltransferase gene were used as endogenous controls (see Supplementary Table S1 for the primer sequences). The conditions for PCR were as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s (denaturation), and 60°C for 1 min (annealing and extension). The relative mRNA amount of the target and control genes was calculated using the ΔCt (threshold cycle) method. Relative expression = 2 −ΔCt, where ΔCt = Ct (target gene) − Ct (control gene).
Small interfering RNA knockdown
The GADD45γ, Mcl-1, and scrambled nonspecific (negative control) siRNAs were purchased from Ambion (Austin, TX, USA) (see Supplementary Table S1 for the sequences). The si-survivin (catalog number L-003459-00-0005) and scrambled nonspecific (negative control) siRNA (catalog number D-001810-10-20) were purchased from Thermo Scientific (Dharmacon Division) as previously described [34]. siRNA were transfected using the siPORT NeoFx siRNA transfection reagent (Ambion). The transfected HCC cells were subsequently treated with sorafenib (10 μM) for 48 hours and collected for subsequent Western blot or quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis.
Chromatin immunoprecipitation assay
Huh-7 cells (approximately 5 × 106) with or without sorafenib treatment were used for ChIP, employing an EZ-ChIP assay kit (Millipore, Billerica, MA, USA). The PCR amplification was performed using primers spanning the CEBP sites on the GADD45γ promoter from nucleotides -443 to -61 (see Supplementary Table S1 for sequences), as described previously [16]. The band intensities were quantified using AutoChemi imaging system (UVP, Cambridge, UK), and signal intensities representing DNA content were normalized to H2O. The conditions for quantitative PCR were as follows: 50°C for 2 min, 95°C for 10 min, 50 cycles of 95°C for 20 s, and 60°C for 1 min. The relative DNA amount of the target and 0.1% input GADD45γ was calculated using the ΔCt (threshold cycle) method as previously described.
Tumor xenograft experiments
The protocol for the xenograft experiments was approved by the Institutional Animal Care and Use Committee of the College of Medicine, National Taiwan University. The animal studies were performed according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. Male BALB/c athymic (nu+/nu+) mice were inoculated subcutaneously with Huh-7 cells or Huh-7R cells (approximately 1 × 106). Huh-7R cells infected with the Ad-GADD45γ or Ad-empty virus were used to determine the effects of GADD45γ overexpression on treatment efficacy. When the tumor volume reached approximately 100 mm3 (volume [mm3] = [width]2 × length × 0.5), the mice were randomized to receive sorafenib (10 mg/kg/day) or vehicle treatment (n ≥ 5 in each group). Drug treatments were given daily by gavage. Tumor volume and body weight were recorded every 5 days. Tumor samples, freshly frozen after drug treatment, were collected to measure the levels of pertinent mRNA and proteins by using real-time PCR and Western blot, respectively. The total RNA was extracted from cells of tumor specimens, which was collected using laser capture microdissection [15]. Formalin-fixed and paraffin-embedded tumor samples after drug treatment were collected for immunohistochemical analysis of pertinent protein expression and tumor angiogenesis and TUNEL assay to measure tumor cell apoptosis, as described previously [16].
Tumor specimens from patients with HCC
Total RNA was extracted from HCC tumors and adjacent liver tissues, which were obtained from the tissue bank of Taiwan Liver Cancer Network (TLCN). Eighty-two patients with American Joint Committee on Cancer (AJCC) stage I to IIIA disease who had undergone curative surgery were included in this study. An informed consent for tissue collection was obtained from each patient prior to surgery. Data of clinicopathological features at surgery and postoperational follow-up were provided by TLCN. Recurrence-free survival time was calculated from the date of surgery to the date of documented HCC tumor recurrence, and overall survival time was calculated from the date of surgery to the date of death or last follow-up. The primers for the GADD45γ gene were purchased from Applied Biosystems (ABI TaqMan assay Hs00198672_m1). Quantification of GADD45γ expression was using the ΔCt (threshold cycle) method described previously and expressed as a tumor ΔCt/nontumor ΔCt (T/non-T) ratio.
Statistical analysis
For in vitro and xenograft experiments, data were representative of three independent experiments. Quantitative data were expressed as mean ± standard deviation (SD). Comparisons were analyzed using the Student t test and one-way analysis of variance (ANOVA). Survival time for the human HCC tissue studies was calculated using the Kaplan–Meier method and compared in different subgroups of patients by using the log-rank test. The prognostic significance of individual clinicopathological factors and GADD45γ expression was analyzed using the Cox proportional hazards model. Significance was defined as p < 0.05.
ACKNOWLEDGMENTS
We thank Taiwan Liver Cancer Network (TLCN) for providing the hepatocellular carcinoma tissue samples and related clinical data (all are anonymous) for our study. This network currently includes five major medical centers (National Taiwan University Hospital; Chang-Gung Memorial Hospital-Linko; Veteran General Hospital, Taichung; Chang-Gung Memorial Hospital-Kaohsiung; and Veteran General Hospital-Kaohsiung). TLCN has been supported by grants from National Science Council since 2005 (NSC 100-2325-B-182-006) and National Health Research Institutes, Taiwan.
CONFLICTS OF INTEREST
Dr. Ann-Lii Cheng is a consultant for and a member of the speaker's bureau of Bayer Schering Pharma. Dr. Chiun Hsu is a member of the speaker's bureau of Bayer Schering Pharma. The other authors have no conflict of interest to disclose.
FINANCIAL SUPPORT
NSC 101-2325-B-002-039, NSC 101-2321-B-002-014, NSC 102-2325-B-002-038, NSC 100-2314-B-002-058-MY3, NSC 102-2314-B-002-142-MY3, and NSC 103-2314-B-002-112-MY3 from National Science Council, Taiwan, and a research grant from Liver Disease Prevention and Treatment Foundation, Taiwan.
AUTHORS' CONTRIBUTIONS
Concept and design: DL Ou, C Hsu; Methodology development: DL Ou, SK Shyue, LI Lin, ZR Feng, JY Liou, HH Fan, BS Lee; Writing and review: DL Ou, C Hsu; Study supervision: C Hsu, AL Cheng.
REFERENCES
1. Bruix J, Sherman M. American Association for the Study of Liver D. Management of hepatocellular carcinoma: an update. Hepatology. 2011; 53:1020–1022.
2. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Molecular cancer therapeutics. 2008; 7:3129–3140.
3. Berasain C. Hepatocellular carcinoma and sorafenib: too many resistance mechanisms? Gut. 2013.
4. Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43–9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. Journal of biological chemistry. 2005; 280:35217–35227.
5. Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ, Chen KF. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. Journal of hepatology. 2011; 55:1041–1048.
6. Wang WQ, Liu L, Xu HX, Sun HC, Wu CT, Zhu XD, Zhang W, Xu J, Liu C, Long J, Ni QX, Tang ZY, Yu XJ. The combination of HTATIP2 expression and microvessel density predicts converse survival of hepatocellular carcinoma with or without sorafenib. Oncotarget. 2014; 5:3895–3906.
7. Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B. Gadd45 stress sensors in malignancy and leukemia. Critical reviews in oncogenesis. 2011; 16:129–140.
8. Qiu W, David D, Zhou B, Chu PG, Zhang B, Wu M, Xiao J, Han T, Zhu Z, Wang T, Liu X, Lopez R, Frankel P, Jong A, Yen Y. Down-regulation of growth arrest DNA damage-inducible gene 45beta expression is associated with human hepatocellular carcinoma. The American journal of pathology. 2003; 162:1961–1974.
9. Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Ambinder R, Tao Q. The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clinical cancer research. 2005; 11:6442–6449.
10. Ozawa S, Gamou T, Habano W, Inoue K, Yoshida M, Nishikawa A, Nemoto K, Degawa M. Altered expression of GADD45 genes during the development of chemical-mediated liver hypertrophy and liver tumor promotion in rats. The Journal of toxicological sciences. 2011; 36:613–623.
11. Tian J, Huang H, Hoffman B, Liebermann DA, Ledda-Columbano GM, Columbano A, Locker J. Gadd45beta is an inducible coactivator of transcription that facilitates rapid liver growth in mice. Journal of clinical investigation. 2011; 121:4491–4502.
12. Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, Fornace AJ Jr. Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer research. 2002; 62:7305–7315.
13. Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr., Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. Journal of biological chemistry. 2003; 278:43001–43007.
14. Jinawath N, Vasoontara C, Yap KL, Thiaville MM, Nakayama K, Wang TL, Shih IM. NAC-1, a potential stem cell pluripotency factor, contributes to paclitaxel resistance in ovarian cancer through inactivating Gadd45 pathway. Oncogene. 2009; 28:1941–1948.
15. Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. GADD45 proteins: central players in tumorigenesis. Current molecular medicine. 2012; 12:634–651.
16. Ou DL, Shen YC, Yu SL, Chen KF, Yeh PY, Fan HH, Feng WC, Wang CT, Lin LI, Hsu C, Cheng AL. Induction of DNA damage-inducible gene GADD45beta contributes to sorafenib-induced apoptosis in hepatocellular carcinoma cells. Cancer research. 2010; 70:9309–9318.
17. Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou JR, Libermann TA. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:13618–13623.
18. Sun L, Gong R, Wan B, Huang X, Wu C, Zhang X, Zhao S, Yu L. GADD45gamma, down-regulated in 65% hepatocellular carcinoma (HCC) from 23 chinese patients, inhibits cell growth and induces cell cycle G2/M arrest for hepatoma Hep-G2 cell lines. Molecular biology reports. 2003; 30:249–253.
19. Huber S, Oelsner M, Decker T, zum Buschenfelde CM, Wagner M, Lutzny G, Kuhnt T, Schmidt B, Oostendorp RA, Peschel C, Ringshausen I. Sorafenib induces cell death in chronic lymphocytic leukemia by translational downregulation of Mcl-1. Leukemia. 2011; 25:838–847.
20. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, Roberts JD, Fisher PB, Grant S, Dent P. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clinical cancer research. 2008; 14:5385–5399.
21. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC, Thung S, Friedman SL, Llovet JM. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. Journal of hepatology. 2012; 56:1343–1350.
22. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–404.
23. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:l1.
24. Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D'Adamio L, Franzoso G. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nature cell biology. 2004; 6:146–153.
25. Zerbini LF, de Vasconcellos JF, Czibere A, Wang Y, Paccez JD, Gu X, Zhou JR, Libermann TA. JunD-mediated repression of GADD45alpha and gamma regulates escape from cell death in prostate cancer. Cell cycle. 2011; 10:2583–2591.
26. Papa S, Zazzeroni F, Fu YX, Bubici C, Alvarez K, Dean K, Christiansen PA, Anders RA, Franzoso G. Gadd45beta promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. Journal of clinical investigation. 2008; 118:1911–1923.
27. Su AI, Guidotti LG, Pezacki JP, Chisari FV, Schultz PG. Gene expression during the priming phase of liver regeneration after partial hepatectomy in mice. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:11181–11186.
28. Ou DL, Feng ZR, Liao SC, Fan HH, Hsu C, Cheng AL. Prognostic value of DNA-damage-inducible 45 beta/gamma expression for hepatocellular carcinoma patients who underwent curative surgery. 7th International Liver Cancer Association (ILCA) Annual Conference. 2013; abstr # P-14.
29. Sun YF, Xu Y, Yang XR, Guo W, Zhang X, Qiu SJ, Shi RY, Hu B, Zhou J, Fan J. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013; 57:1458–1468.
30. Schulze K, Gasch C, Staufer K, Nashan B, Lohse AW, Pantel K, Riethdorf S, Wege H. Presence of EpCAM-positive circulating tumor cells as biomarker for systemic disease strongly correlates to survival in patients with hepatocellular carcinoma. International journal of cancer. 2013; 133:2165–2171.
31. Weidle UH, Maisel D, Eick D. Synthetic lethality-based targets for discovery of new cancer therapeutics. Cancer genomics & proteomics. 2011; 8:159–171.
32. van der Heide LP, van Dinther M, Moustakas A, ten Dijke P. TGFbeta activates mitogen- and stress-activated protein kinase-1 (MSK1) to attenuate cell death. Journal of biological chemistry. 2011; 286:5003–5011.
33. Zhang P, Liu SS, Ngan HY. TAp3-mediated the activation of c-Jun N-terminal kinase enhances cellular chemosensitivity to cisplatin in ovarian cancer cells. PloS one. 2012; 7:e42985.
34. Ou DL, Lee BS, Lin LI, Liou JY, Liao SC, Hsu C, Cheng AL. Vertical blockade of the IGFR- PI3K/Akt/mTOR pathway for the treatment of hepatocellular carcinoma: the role of survivin. Molecular cancer. 2014; 13:2.
35. Chen KF, Chen HL, Shiau CW, Liu CY, Chu PY, Tai WT, Ichikawa K, Chen PJ, Cheng AL. Sorafenib and its derivative SC-49 sensitize hepatocellular carcinoma cells to CS-1008, a humanized anti-TNFRSF10B (DR5) antibody. British journal of pharmacology. 2013; 168:658–672.
36. Wang R, Chen DQ, Huang JY, Zhang K, Feng B, Pan BZ, Chen J, De W, Chen LB. Acquisition of radioresistance in docetaxel-resistant human lung adenocarcinoma cells is linked with dysregulation of miR-41/c-Myc-survivin/rad-1 signaling. Oncotarget. 2014; 5:6113–6129.
37. Cheung CH, Huang CC, Tsai FY, Lee JY, Cheng SM, Chang YC, Huang YC, Chen SH, Chang JY. Survivin - biology and potential as a therapeutic target in oncology. OncoTargets and therapy. 2013; 6:1453–1462.
38. Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: the cell-signalling perspective. Drug discovery today. 2011; 16:485–494.
39. Kim YS, Jin HO, Seo SK, Woo SH, Choe TB, An S, Hong SI, Lee SJ, Lee KH, Park IC. Sorafenib induces apoptotic cell death in human non-small cell lung cancer cells by down-regulating mammalian target of rapamycin (mTOR)-dependent survivin expression. Biochemical pharmacology. 2011; 82:216–226.
40. Lian J, Ni Z, Dai X, Su C, Smith AR, Xu L, He F. Sorafenib sensitizes (-)-gossypol-induced growth suppression in androgen-independent prostate cancer cells via Mcl-1 inhibition and Bak activation. Molecular cancer therapeutics. 2012; 11:416–426.
41. Rahmani M, Aust MM, Attkisson E, Williams DC Jr., Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012; 119:6089–6098.
42. Yeh YY, Chen R, Hessler J, Mahoney E, Lehman AM, Heerema NA, Grever MR, Plunkett W, Byrd JC, Johnson AJ. Up-regulation of CDK9 kinase activity and Mcl-1 stability contributes to the acquired resistance to cyclin-dependent kinase inhibitors in leukemia. Oncotarget. 2014.
43. Aoyama Y, Kaibara A, Takada A, Nishimura T, Katashima M, Sawamoto T. Population pharmacokinetic modeling of sepantronium bromide (YM155), a small molecule survivin suppressant, in patients with non-small cell lung cancer, hormone refractory prostate cancer, or unresectable stage III or IV melanoma. Investigational new drugs. 2013; 31:443–451.
44. Billard C. BH3 mimetics: status of the field and new developments. Molecular cancer therapeutics. 2013; 12:1691–1700.
45. Pelorosso FG, Balmain A. C/EBPdelta: friend or foe? a novel role for C/EBPdelta in metastasis. The EMBO journal. 2010; 29:4063–4065.
46. Pan YC, Li CF, Ko CY, Pan MH, Chen PJ, Tseng JT, Wu WC, Chang WC, Huang AM, Sterneck E, Wang JM. CEBPD reverses RB/E2F1-mediated gene repression and participates in HMDB-induced apoptosis of cancer cells. Clinical cancer research. 2010; 16:5770–5780.
47. Wang J, Sarkar TR, Zhou M, Sharan S, Ritt DA, Veenstra TD, Morrison DK, Huang AM, Sterneck E. CCAAT/enhancer binding protein delta (C/EBPdelta, CEBPD)-mediated nuclear import of FANCD2 by IPO4 augments cellular response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:16131–16136.
48. Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, Sterneck E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. The EMBO journal. 2010; 29:4106–4117.
49. Min Y, Ghose S, Boelte K, Li J, Yang L, Lin PC. C/EBP-delta regulates VEGF-C autocrine signaling in lymphangiogenesis and metastasis of lung cancer through HIF-1alpha. Oncogene. 2011; 30:4901–4909.
50. Zerbini LF, Libermann TA. GADD45 deregulation in cancer: frequently methylated tumor suppressors and potential therapeutic targets. Clinical cancer research. 2005; 11:6409–6413.
51. Awada A, Hendlisz A, Gil T, Bartholomeus S, Mano M, de Valeriola D, Strumberg D, Brendel E, Haase CG, Schwartz B, Piccart M. Phase I safety and pharmacokinetics of BAY 43–9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. British journal of cancer. 2005; 92:1855–1861.
52. Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, Schwartz B, Awada A, Voigtmann R, Scheulen ME, Seeber S. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43–9006 in patients with advanced refractory solid tumors. Journal of clinical oncology. 2005; 23:965–972.