
Introduction
Cancer is the most devastating and intractable human disease. There are numerous ways to kill cancer cells, but most of them do not translate into cancer therapy because they kill normal cells and tissues as well. Thus selective and efficient targeting of cancer cells is the foremost goal in the development of anti-cancer therapeutics[1]. Most of the early anti-cancer therapies have some selectivity for cancer cells by inhibiting essential process like DNA replication and cell division in rapidly dividing cancer cells[1]. Recently developed anti-cancer drugs often have specific molecular targets that are deregulated in cancer cells[2]. The critical barrier in the use of early anti-cancer drugs is the development of resistance in cancer cells and the severe toxicity to normal tissues[3, 4]. It was believed that these impediments could be significantly reduced in the new generation of drugs that target specific molecular pathways in cancer tissues[2, 4]. However, it has been found that the new drugs often suffer from the same drawbacks[5].
Due to the side effect or toxicity in normal tissues, the therapeutic window of most anti-cancer agents is very narrow[6]. As a result, the anti-cancer drugs cannot be administered at the dosages where they can eradicate all the cancer cells. This is in part responsible for the selection of resistant cancer cells, ultimately leading to growth of tumors that are refractory to subsequent anti-cancer therapy[6]. Although, the significance of resistance to anti-cancer drugs has been widely recognized and extensively studied[7, 8], the research on the mechanisms responsible for the toxicity to normal tissues has generally lagged far behind. The identification of molecular targets that reduce toxicity to normal tissues without blocking the anti-cancer effects has the potential to significantly improve the efficacy of chemotherapy.
Cisplatin and Nephrotoxicity
Cisplatin is one of the oldest, highly effective and most commonly used anti-cancer drugs[9-11]. Cisplatin is a platinum based inorganic compound that is believed to cross-link DNA, leading to inhibition of essential processes like DNA replication and transcription[11]. Higher rate of DNA replication in rapidly proliferative cancer cells make them particularly sensitive to cisplatin-induced DNA damage. Depending on the amount of DNA damage, cancer cells either repair/tolerate the DNA damage or undergo apoptosis in case of extensive damage[8]. Identification of resistant cells that have higher DNA repair or reduced DNA damage response/apoptosis have provided the basis that DNA damage is the major mechanism of cisplatin-induced tumor cell death[8, 12].
Along with its effectiveness in killing cancer cells, cisplatin has a wide range of side-effects in normal tissues, among which nephrotoxicity is most notable due to its potentially fatal nature[13, 14]. Cisplatin treatment can lead to severe kidney damage resulting in acute kidney injury, which has a very high rate of mortality[14]. Extensive hydration in patients can partially reduce the extent of kidney injury, however nephrotoxicity remains a main threat[13, 14]. Development of novel drugs derived from cisplatin like carboplatin and oxaloplatin have reduced toxicity, but these drugs are not as widely effective as cisplatin[13]. New strategies that reduce kidney injury during cisplatin treatment could have significant impact on the overall efficiency of chemotherapy.
Extensive studies have been conducted in the last decade to decipher the patho-physiological basis of cisplatin nephrotoxicity[13-17]. The major pathological feature of cisplatin-induced kidney injury is the cell death of renal tubular cells in the form of apoptosis and necrosis[13]. The specific sensitivity of tubular cells to cispatin is partly attributed to the fact that they accumulate many-fold higher amounts of cisplatin than other cells and tissues. Depending on the amount of cisplatin exposed, renal tubular cells undergo necrosis or apoptosis[18]. Along with renal cell death, an inflammatory component is also responsible for aggravating kidney injury[16, 19-22]. Injured kidney cells activate inflammatory processes, which further increase renal cell death leading to extensive kidney injury and acute renal failure[19]. Genetic and pharmacological strategies that inhibit both renal apoptotic pathways and inflammation in renal tissues provide significant protection during cisplatin treatment[13]. Other pharmacological/natural compounds that inhibit these processes indirectly also reduce cisplatin-induced kidney injury[13]. These studies have thus provided insight into the complex molecular mechanisms responsible for cisplatin nephrotoxicity. At the same time, it has been proposed that these strategies could have potential renoprotective effects during cisplatin-based chemotherapy.
However, the most critical issue that has not been addressed is the effect of these protective strategies on the anti-cancer efficacy of cisplatin. Indeed, some of the signaling pathways, for example the DNA damage response leading to p53 activation, observed in renal cells, also contribute to cisplatin mediated cancer cell death[23]. Identification of targets that can reduce renal toxicity without blocking the anti-cancer efficacy of cisplatin could have important clinical significance.
Identification of PKCδ as target for renoprotection during cisplatin-based chemotherapy
Protein kinase C (PKC) comprises of a family of highly conserved serine/threonine kinases that influence a wide range of cellular processes[24]. PKCδ is a member of the novel PKC sub-family, that are activated in a calcium-independent, but diacylglycerol-dependent manner[25, 26]. PKCδ is widely expressed in multiple tissues and has been implicated in a plethora of cellular processes including proliferation and apoptosis[25, 27, 28]. Knockout of PKCδ does not affect have overt effects on murine development, suggesting that PKCδ is not essential for normal mammalian development[29].
PKCδ is not only activated by cofactors, such as diacylglycerol and phorbol esters, but its activation is also dependent on post-translational modifications, especially phosphorylation[29]. Several tyrosine kinases, including growth factor receptors, Src family tyrosine kinases and c-Abl, have been implicated in PKCδ phosphorylation[29]. In addition PKCδ can be cleaved by caspases to generate a constitutively active catalytic fragment[25]. Numerous studies have thus implicated PKCδ activation as a pro-apoptotic mechanism during various stimuli, including treatment with anti-cancer agents[29]. Intriguingly, PKCδ can also function as an anti-apoptotic factor and confer resistance to anticancer drugs. Furthermore, PKCδ is pro-survival factor in several cancers[29]. Thus, depending on the treatment and cellular context, PKCδ may play contrasting roles in the decision of cell death or survival. The mechanisms underlying the dual roles of PKCδ remain unclear, but the localization, phosphorylation status and downstream substrates may be involved.
The lack of severe phenotype of PKCδ-deficient mice suggested that PKCδ might be dispensable for normal development and tissue maintenance[29]. However, PKCδ may play regulatory roles in cell stress and pathological conditions including treatment with anti-cancer drugs. Indeed our recent study[30] has identified a novel role of PKCδ during cisplatin-induced kidney injury. Genetic ablation and pharmacological inhibition of PKCδ provided significant renal protection during cisplatin treatment in in vitro cell cultures and in vivo murine models[30]. Mechanistically, PKCδ was shown to be activated in a Src-dependent manner leading to downstream activation of MAPK pathway.
While our study suggested a renal protective strategy during cisplatin chemotherapy by targeting PKCδ, we were however concerned that PKCδ inhibition might attenuate the anti-cancer efficacy of cisplatin in tumors. To address this key issue, we conducted a series of in vitro and in vivo experiments to determine the effect of PKCδ inhibition on the anti-cancer efficacy of cisplatin. Intriguingly, we found that PKCδ inhibition did not reduce the effect of cisplatin in cancer cells and in some cases PKCδ inhibition actually increased the chemotherapy efficacy. Importantly, we developed new in vivo mouse models in which both the anti-cancer efficacy of cisplatin and its toxicity or side effects in kidneys could be directly monitored. These experiments demonstrated unambiguously that PKCδ inhibition reduced cisplatin toxicity in normal tissues without diminishing its anti-cancer effects. As a matter of fact, in ovarian and breast tumor models, PKCδ inhibitors enhanced the cancer therapy effect of cisplatin (Figure 1).
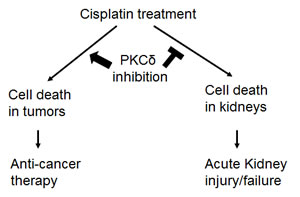
Figure 1: PKCδ inhibition enhances anti-cancer therapy while protecting kidneys during cisplatin treatment. Cisplatin induces cell death in both cancer and kidney cells, resulting in chemotherapy in tumors and acute kidney injury and kidney failure. Genetic or pharmacologic inhibition of PKCδ enhances the chemotherapy effect of cisplatin in tumors and diminishes cisplatin-induced side effect in kidneys.
The observed different effects of PKCδ inhibitors in kidneys and tumors remain enigmatic. Earlier studies implicated a pro-apoptotic role of PKCδ in both normal and cancer cells[29], leading to the suggestion that PKCδ may function as a tumor suppressor. However, direct in vivo evidence for this theory is lacking and PKCδ-deficient mice do not display any propensity for carcinogenesis under normal conditions. In a recent study[31] the role of PKCδ in K-ras-dependent lung tumorigenesis was examined by using a mouse carcinogen model. Surprisingly, the incidence of urethane-induced lung tumors was significantly reduced in PKCδ-deficient mice compared with wild-type mice. PKCδ-KO tumors were smaller and showed significantly reduced proliferation. It is suggested that PKCδ may act as a tumor promoter downstream of oncogenic K-ras during lung carcinogenesis. Unexpectedly, these studies indicate that the function of PKCδ in tumor cells may depend on specific oncogenic context, as loss of PKCδ suppressed growth only in the cells that depend on oncogenic K-ras for proliferation and survival. Consistently, a pro-survival role of PKCδ has been documented in several cancer cell lines[29].
How can these seemingly contradictory findings be reconciled? We speculate that PKCδ may not have a critical pro-apoptotic or pro-survival role in normal mammalian development or physiological conditions. However, the cancer cells in some tumor types require PKCδ for survival and proliferation. Such scenario goes by the principle of ‘non-oncogenic addiction’[6] (Figure 1). According to this model, activation of oncogenes and rapid proliferation in cancer cells induce a stress phenotype that is particularly sensitive to cellular stress [6]. As a result, the cancer cells become dependent on certain pathways for survival, which are non-essential in normal cells and tissues. These non-oncogenic pathways are not important for initial cancer development, but are critical for the survival and proliferation of cancer cells in tumors. PKCδ seems to be a good candidate protein for ‘non-oncogenic addiction’ in cancer cells, which becomes essential for their survival under oncogenic stresses such as ras activation. If this is true, PKCδ could turn out to be an important target for anti-cancer therapy. Our study demonstrated a role of PKCδ in mediates the toxic effects of cisplatin in the kidneys and hence inhibition of PKCδ could reduce the toxicity and at the same time increase the anti-cancer efficacy of Cisplatin (Figure 2).
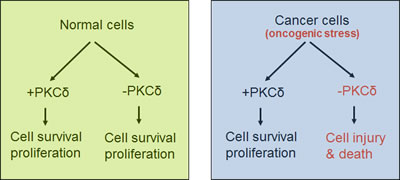
Figure 2: PKCδ in survival of normal versus cancer cells. PKCδ is not required for the survival normal cells. However, it is required for the survival and proliferation of cancer cells with an oncogenic stress phenotype and, as a result, suppression of PKCδ leads to cell injury and death in tumors.
Conclusions and Future Directions
Our recent studies have shed light on a hitherto under-appreciated yet important aspect of cancer therapy. Since the side effect or toxicity to normal tissues is one of the key determinants in the success of anti-cancer drugs, identification of targets and development of normal tissue protective strategies could have significant implications in the treatment of cancer. Several strategies have been developed to selectively kill certain types of cancer cells, while simultaneously protecting normal cells [32-44]. By including normal tissue specific-protective agents in chemotherapy, these strategies may increase the therapeutic window. In line with this idea, PKCδ has now been demonstrated to be a critical component in nephrotoxicity during cisplatin treatment. The fact that PKCδ might be a “non-oncogenic addiction” factor for cancer cell survival provides an immense opportunity to increase the efficacy of chemotherapy by reducing the side effect in normal tissues and increasing the anti-cancer effect in tumors. Studies in the future should further elucidate the differential functions of PKCδ and other potential ‘non-oncogenic addiction’ genes in normal versus cancer tissues, opening new avenues to improve the efficacy of cancer therapy.
Acknowledgements
The work was supported by grants from the National Institutes of Health and Veterans Administration of the U.S.A.
References
1. Chabner BA, Roberts TG, Jr.: Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 2005; 5:65-72.
2. Cao Y, DePinho RA, Ernst M, Vousden K: Cancer research: past, present and future. Nat Rev Cancer 2011;11:749-754.
3. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000; 100:57-70.
4. Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011;144:646-674.
5. Verheul HM, Pinedo HM: Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer 2007; 7:475-485.
6. Luo J, Solimini NL, Elledge SJ: Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136:823-837.
7. Stewart DJ: Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol 2007; 63:12-31.
8. Siddik ZH: Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22:7265-7279.
9. Jamieson ER, Lippard SJ: Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev 1999; 99:2467-2498.
10. Cohen SM, Lippard SJ: Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol 2001; 67:93-130.
11. Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ: Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 1999; 399:708-712.
12. Siddik ZH: Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat Res 2002; 112:263-284.
13. Pabla N, Dong Z: Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008; 73:994-1007.
14. Arany I, Safirstein RL: Cisplatin nephrotoxicity. Semin Nephrol 2003; 23:460-464.
15. Hodeify R, Megyesi J, Tarcsafalvi A, Safirstein RL, Price PM: Protection of cisplatin cytotoxicity by an inactive cyclin-dependent kinase. Am J Physiol Renal Physiol 2010; 299:F112-120.
16. Ramesh G, Reeves WB: TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 2002;110:835-842.
17. Cummings BS, Schnellmann RG: Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther 2002; 302:8-17.
18. Pabla N, Murphy RF, Liu K, Dong Z: The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol 2009; 296:F505-511.
19. Miller RP, Tadagavadi RK, Ramesh G, Reeves WB: Mechanisms of Cisplatin nephrotoxicity. Toxins (Basel) 2010; 2:2490-2518.
20. Ramesh G, Reeves WB: TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 2003; 285:F610-618.
21. Ramesh G, Zhang B, Uematsu S, Akira S, Reeves WB: Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am J Physiol Renal Physiol 2007; 293:F325-332.
22. Zhang B, Ramesh G, Norbury CC, Reeves WB: Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int 2007; 72:37-44.
23. Palab N, Huang S, Mi QS, Daniel R, Dong Z: ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem 20082; 83:6572-6583.
24. Parker PJ, Murray-Rust J: PKC at a glance. J Cell Sci 2004; 117:131-132.
25. Kikkawa U, Matsuzaki H, Yamamoto T: Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem 2002;132:831-839.
26. Liu WS, Heckman CA: The sevenfold way of PKC regulation. Cell Signal 1998;10:529-542.
27. DeVries-Seimon TA, Ohm AM, Humphries MJ, Reyland ME: Induction of apoptosis is driven by nuclear retention of protein kinase C delta. J Biol Chem 2007; 282:22307-22314.
28. Humphries MJ, Ohm AM, Schaack J, Adwan TS, Reyland ME: Tyrosine phosphorylation regulates nuclear translocation of PKCdelta. Oncogene 2008; 27:3045-3053.
29. Jackson DN, Foster DA: The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J 2004; 18:627-636.
30. Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z: Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 2011;121:2709-2722.
31. Symonds JM, Ohm AM, Carter CJ, Heasley LE, Boyle TA, Franklin WA, Reyland ME: Protein kinase C delta is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res 2011; 71: 2087-2097.
32. Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res. 2001; 61: 4301-4305.
33. Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell Cycle. 2002; 1: 375-382.
34. Blagosklonny MV, Robey R, Bates S, Fojo T.Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000; 105:533-539.
35. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005; 65: 1918-1924.
36. Demidenko ZN, Halicka D, Kunicki J, McCubrey JA, Darzynkiewicz Z, Blagosklonny MV. Selective killing of adriamycin-resistant (G2 checkpoint-deficient and MRP1-expressing) cancer cells by docetaxel. Cancer Res. 2005; 65: 4401-4407.
37. Demidenko ZN, An WG, Lee JT, Romanova LY, McCubrey JA, Blagosklonny MV. Kinase-addiction and bi-phasic sensitivity-resistance of Bcr-Abl- and Raf-1-expressing cells to imatinib and geldanamycin. Cancer Biol Ther. 2005; 4: 484-490.
38. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2: 222-233.
39. Darzynkiewicz Z. Novel strategies of protecting non-cancer cells during chemotherapy: are they ready for clinical testing? Oncotarget. 2011; 2: 107-108.
40. van Leeuwen IM, Laín S. Pharmacological manipulation of the cell cycle and metabolism to protect normal tissues against conventional anticancer drugs. Oncotarget. 2011; 2: 274-276.
41. Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106: 3964-3969.
42. Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle. 2010; 9: 4474-4476.
43. Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget. 2010; 1: 639-650.
44. Blagosklonny MV. NCI’s provocative questions on cancer: some answers to ignite discussion. Oncotarget. 2011; 2: 1352-1367.