
INTRODUCTION
CIC is an HMG box-containing transcriptional repressor evolutionarily conserved from nematodes to humans [1]. CIC preferentially binds to TG/CAATGA/GA sequences within promoters and enhancers of target genes in Drosophila and mammals [2], and a bacterial one-hybrid screen for DNA binding motifs of Drosophila transcription factors has revealed that the consensus sequence of CIC binding motifs is 5’-YYCATTSA-3’ [3, 4]. At least two CIC isoforms exist in Drosophila and mammals, CIC-L and CIC-S, which differ in their amino-terminal regions. The longer isoform CIC-L contains a unique amino-terminal region of approximately 900 amino acids in length in mammals [2]. In mammals, CIC was identified as an interacting protein of ATXN1, the causative protein of SCA1 neurodegenerative disease [5]. Haploinsufficiency of CIC partially rescues ataxia phenotypes in Atxn1154Q knock-in mice, suggesting that CIC facilitates pathogenesis of SCA1 [6]. It is also known that Cic hypomorphic (Cic-L-/-) mice have defects in lung alveolarization and bile acid homeostasis [7, 8].
Several previous findings have implicated that CIC might be involved in tumorigenesis and/or cancer progression in humans. First, dozens of mutations in CIC have been identified in patients with various types of cancers [9-11]. Second, a chromosomal translocation generating a CIC-DUX4 fusion was identified in Ewing-like sarcomas [12]. Third, the best known target genes of CIC include PEA3 group genes, ETV1/ER81, ETV4/PEA3, and ETV5/ERM, which are frequently overexpressed in several different types of cancers [13, 14]. Despite the findings supporting the potential role of CIC in cancer, it has not been clear whether CIC deficiency or mutations indeed contribute to cancer progression. In this study, we show that CIC functions as a negative regulator of prostate cancer progression.
RESULTS
CIC is down-regulated in prostate cancer cells
Given that several ETS transcription factor genes (ERG and PEA3 group genes) are frequently overexpressed due to chromosomal translocations in prostate cancer cells, thereby contributing to prostate cancer pathogenesis [15], we hypothesized that CIC might suppress prostate cancer progression through repressing expression of PEA3 group genes. To test this hypothesis, we first examined expression of CIC in mouse prostate cells by immunocytochemistry. We found that CIC is expressed in the nucleus of both basal and luminal cells of mouse prostate glands (Supplementary Figure 1). As a control, a marked decrease in fluorescence signal in thymus sections from Cic-L-/- mice [6, 7] compared with wild-type (WT) littermates verified the suitability of our antibody [8] in evaluating CIC expression (Supplementary Figure 2). We next determined patterns of CIC expression in prostate cancer patient specimens. Consistent with the results from mouse (Supplementary Figure 1), CIC is apparently expressed in the nuclei of both basal and luminal cells of non-cancerous human prostate glands (Figures 1A~1C’’’). Intriguingly, however, the proportion of cells with nuclear CIC expression was markedly decreased in prostatic intraepithelial neoplasia (PIN) (Figures 1D~1F’’’). Moreover, the nuclear expression of CIC disappeared in advanced prostatic adenocarcinoma with complete loss of basal cells (Figures 1G~1I’’’). These results were consistently observed in prostate tissue specimens from 13 different prostate cancer patients (Supplementary Table 1) and the average values for the proportion of cells with nuclear CIC expression in non-cancerous prostate glands, PIN, and prostatic adenocarcinoma are presented in Figure 1J. Consistent with these findings, CIC protein levels were lower in prostate cancer cell lines than in PNT2 normal prostate epithelial cell (Supplementary Figure 3). Notably, CIC levels were the lowest in PC-3, the most aggressive type of prostate cancer cell [16], among the tested prostate cell lines (Supplementary Figure 3). Taken together, these data demonstrate that prostate cancer progression is accompanied with down-regulation of CIC expression.
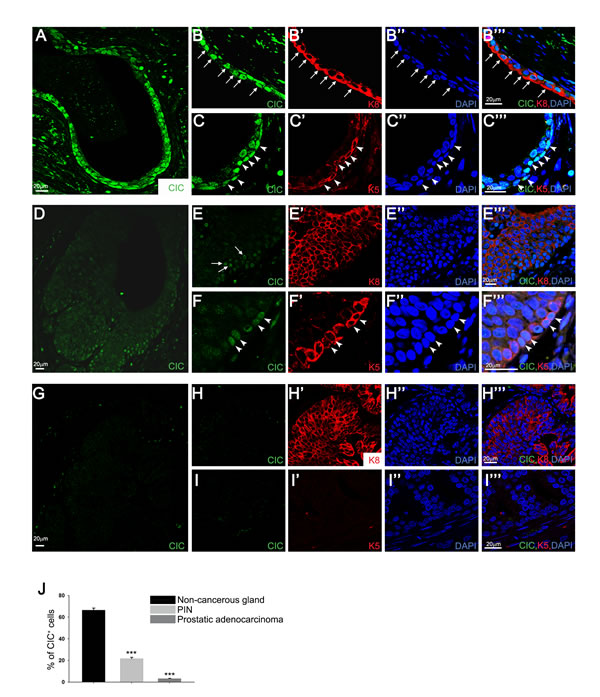
Figure 1: Decreased nuclear expression of CIC in prostate cancer cells. A.-C.’’’ Immunocytochemistry for CIC, keratin 8 (K8, luminal cell marker), and keratin 5 (K5, basal cell marker) in prostate cancer patient specimens. CIC is markedly expressed in the nucleus of non-cancerous prostate glands. Arrows and arrow heads indicate luminal and basal cells, respectively. Images were taken from the specimen of patient #3. D.-F.’’’ Expression patterns of CIC in prostatic intraepithelial neoplasia (PIN). The number of cells with the nuclear expression of CIC was apparently reduced in PIN compared with the non-cancerous prostate glands. Arrows and arrow heads indicate luminal and basal cells, respectively. Images were taken from the specimen of patient #3. G.-I.’’’ Absence of nuclear expression of CIC in prostatic adenocarcinoma. Images were taken from the specimen of patient #2. J. Quantitative analysis for the proportion of cells with nuclear expression of CIC in non-cancerous prostate glands, PIN, and prostatic adenocarcinoma. Four to seven regions of non-cancerous prostate glands, PIN, and prostatic adenocarcinoma per each specimen were randomly selected from all the tested patient samples, and the proportion of nuclear CIC signal-positive cells was examined. ***P < 0.001. All error bars show s.e.m.
CIC suppresses cell proliferation, invasion and migration in prostate cancer cells
We then examined whether the decrease in CIC levels is necessary for promotion of prostate cancer progression. We overexpressed CIC in PC-3 and LNCaP cells by infection with lentivirus expressing either mouse CIC-S or CIC-L (Figure 2A), and checked cell proliferation, invasion, and migration. Clonogenic and BrdU labeling assays demonstrated that overexpression of CIC suppresses prostate cancer cell proliferation (Figures 2B and Supplementary Figure 4). Moreover, cell invasion and migration were markedly inhibited in PC-3 and LNCaP cells overexpressing CIC (Figure 2C and Supplementary Figure 5A). We also tested whether deficiency of CIC could promote prostate cancer progression. To this end, we generated prostate cancer cell lines that stably express three different shRNAs targeting CIC (shCIC-1~3). These CIC shRNAs showed different knock-down efficiency in each cell line: shCIC-3 most dramatically decreased CIC levels in PC-3, while such was the case for shCIC-2 in LNCaP (Figure 2D). Both clonogenic and BrdU labeling assays demonstrated that reduction in CIC levels significantly increases cell proliferation in PC-3 and LNCaP cells (Figure 2E and Supplementary Figure 6). We also found that invasive property of cells was markedly enhanced by knock-down of CIC in both LNCaP and PC-3 cells (Figure 2F) and that cell migration was significantly increased in the CIC knock-down PC-3 cells (Supplementary Figure 5B). The increases in cell proliferation, invasion, and migration were apparently correlated with CIC knock-down efficiency, suggesting that these results were certainly due to a decrease in CIC levels and not owing to the off-target effect of CIC shRNAs. Taken together, these data demonstrate that CIC could function as a negative regulator in prostate cancer progression.
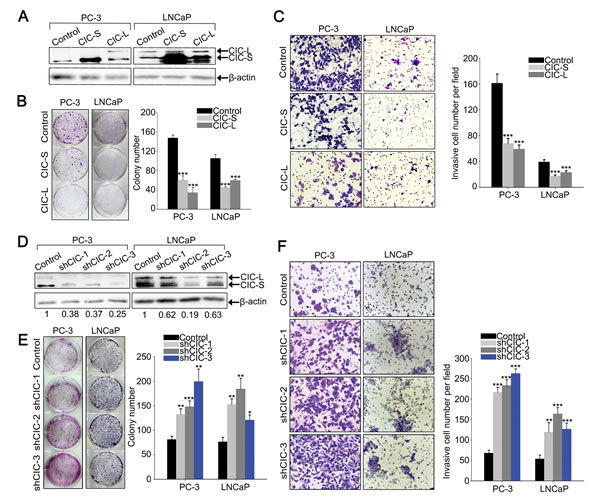
Figure 2: CIC suppresses cell proliferation and invasion in PC-3 cells. A. Western blot analysis for ectopic expression of CIC-S and CIC-L in PC-3 and LNCaP cells. B. Clonogenic assay showing inhibition of cell growth by overexpression of CIC in PC-3 and LNCaP cells. The right panel is a bar graph for quantitative analysis on colony numbers. Three independent experiments were performed. ***P < 0.001. All error bars show s.e.m. C. Matrigel invasion assay showing inhibition of cell invasion by overexpression of CIC in PC-3 and LNCaP cells. The right panel is a bar graph for quantitative analysis on invasive cell numbers. Three independent experiments were performed. ***P < 0.001. All error bars show s.e.m. D. Western blot analysis to examine CIC knock-down efficiency of three different shRNAs against CIC (shCIC-1~3) in PC-3 and LNCaP cells. Relative CIC levels were calculated based on band intensities of CIC and β-actin, and indicated below the images. Two and three independently obtained western blot images for samples of PC-3 and LNCaP cells, respectively, were subjected to the quantitative analysis for the relative CIC levels. E. Clonogenic assay showing promotion of cell growth by knock-down of CIC in PC-3 and LNCaP cells and its quantification. Three independent experiments were performed. *P < 0.05, **P < 0.01 and ***P < 0.001. All error bars show s.e.m. F. Matrigel invasion assay showing promotion of cell invasion by knock-down of CIC in PC-3 and LNCaP cells and its quantification. Three independent experiments were performed. **P < 0.01 and ***P < 0.001. All error bars show s.e.m.
Derepression of ETV5 contributes to the increases in cell proliferation and invasion in the CIC knock-down LNCaP cells
Given that PEA3 group genes are regulated by CIC and that altered expression of these genes is associated with the pathogenesis of various types of cancers including prostate cancer [17], we assessed levels of PEA3 group genes in the CIC knock-down cell lines by qRT-PCR. Significant up-regulation of ETV5 levels was found in CIC knock-down LNCaP cells (Figure 3A), while unexpectedly, levels of ETV1, ETV4, and ETV5 were comparable among control and CIC knock-down PC-3 cell lines (Supplementary Figure 7), suggesting a cell-type specific regulation of PEA3 group genes by CIC. We then examined whether the derepression of ETV5 contributed to promotion of cell proliferation and invasion in the CIC knock-down LNCaP cells. We treated the shCIC-1 and shCIC-2 LNCaP cells with siRNA against ETV5 (siETV5) to make the level of ETV5 similar to that in control cells (Figure 3B), and assessed cell proliferation and invasion. The RNAi against ETV5 significantly suppressed cell proliferation and invasion in the CIC knock-down LNCaP cells (Figures 3C and 3D and Supplementary Figure 8), indicating that ETV5 is a critical target of CIC in LNCaP cells in terms of regulation of cancer progression.
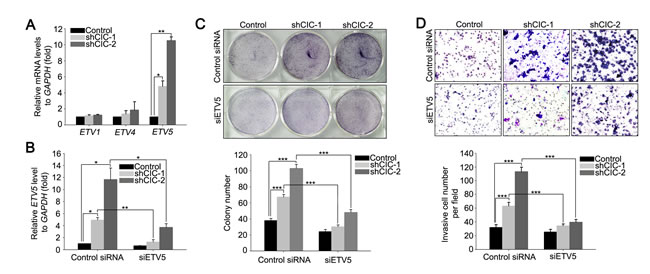
Figure 3: Derepression of ETV5 contributes to the increased cell proliferation and invasion in CIC knock-down LNCaP cells. A. qRT-PCR analysis for ETV1, ETV4 and ETV5 levels in the CIC knock-down LNCaP cells. Three independent experiments were performed. *P < 0.05 and **P < 0.01. All error bars show s.e.m. B. qRT-PCR analysis for ETV5 levels in the CIC knock-down LNCaP cells transfected with either control or ETV5 siRNA. Three independent experiments were performed. *P < 0.05 and **P < 0.01. All error bars show s.e.m. C. Clonogenic assay using control, shCIC-1 and shCIC-2 LNCaP cells treated with either control or ETV5 siRNA. Suppression of ETV5 expression reduced colony formation in the shCIC-1 and shCIC-2 LNCaP cells. The lower panel is a bar graph for quantitative analysis on colony numbers. Three independent experiments were performed. ***P < 0.001. All error bars show s.e.m. D. Matrigel invasion assay using control, shCIC-1 and shCIC-2 LNCaP cell lines treated with either control or ETV5 siRNA. Suppression of ETV5 expression reduced the number of invasive cells. The lower panel is a bar graph for quantitative analysis on invasive cell numbers. Three independent experiments were performed. ***P < 0.001. All error bars show s.e.m.
Overexpression of CRABP1 contributes to the increases in cell proliferation and invasion in the CIC knock-down PC-3 cells
Given the comparable expression of PEA3 group genes in control and CIC knock-down PC-3 cells (Supplementary Figure 7), we investigated which gene expression changes upon knock-down of CIC promoted cell proliferation and invasion in PC-3 cells. To this end, we analyzed gene expression profiles in control and CIC knock-down (shCIC-3) PC-3 cells. The high throughput mRNA sequencing analysis revealed that a total of 262 genes (159 up-regulated, and 103 down-regulated) was differentially expressed (Fold change (log2) > 2 and P-value < 0.05) in the shCIC-3 PC-3 cells compared with control cells (Supplementary Table 2). Among the differentially expressed genes, cellular retinoic acid binding protein 1 (CRABP1) primarily caught our attention, because of the highest fold increase upon knock-down of CIC in PC-3 cells (Supplementary Table 2) and its previously known pro-tumorigenic and pro-metastatic activity in mesenchymal tumors [18]. Moreover, it is known that CRABP1 is overexpressed in castration-resistant prostate cancer cells [19]. Consistent with this, we observed that CRABP1 levels are much higher in PC-3 cells, which are independent of androgen for their growth [16], than in LNCaP and PNT2 cells (Supplementary Figure 9). We confirmed the overexpression of CRABP1 at protein level in the CIC knock-down PC-3 cells by western blot analysis (Figure 4A). To verify that the overexpression of CRABP1 was certainly due to deficiency of CIC, we carried out RNAi against CIC in PC-3 cells using siRNA duplexes (siCIC) and examined the expression of CRABP1 by qRT-PCR. The treatment with siCIC also up-regulated CRABP1 levels in PC-3 cells (Supplementary Figure 10), suggesting that CIC indeed negatively regulates CRABP1 expression. On the other hand, we found that CRABP1 levels were not significantly altered in CIC knock-down LNCaP cells (Supplementary Figure 11), indicating that CIC regulates CRABP1 expression in a cell-type dependent manner.
We then explored whether CRABP1 could regulate cell proliferation and invasion in prostate cancer cells. Lentivirus-mediated overexpression of FLAG-CRABP1 increased cell proliferation as well as invasion in PC-3 cells (Figures 4B and 4C and Supplementary Figure 12), suggesting that CRABP1 can function as a positive regulator of prostate cancer progression. Finally, we examined whether the up-regulation of CRABP1 levels contributed to the increased cell proliferation and invasion in the CIC knock-down PC-3 cells. We carried out RNAi against CRABP1 using two different CRABP1 siRNAs (siCRABP1-1 and siCRABP1-2) in the CIC knock-down PC-3 cells to make the level of CRABP1 comparable to that in control cells (Figure 4D). Under this condition, we performed clonogenic, cell growth, and invasion assays, and found that transfection with CRABP1 siRNAs reduced cell proliferation and invasion in the CIC knock-down PC-3 cells (Figures 4E and 4F and Supplementary Figure 13), demonstrating that the increase in CRABP1 levels contributed to the promotion of cancer progression in the CIC knock-down PC-3 cells.
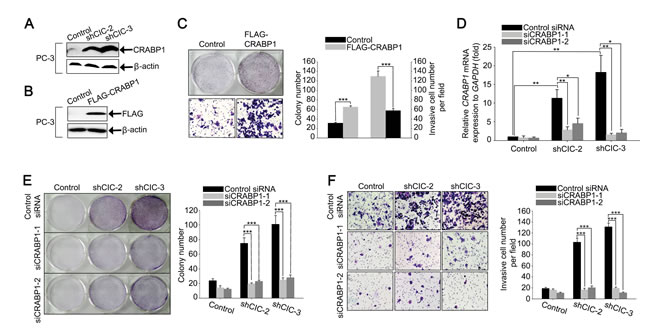
Figure 4: Overexpression of CRABP1 promotes cancer progression in CIC knock-down PC-3 cells. A. Western blot analysis for endogenous CRABP1 expression in control and CIC knock-down PC-3 cell lines. B. Western blot analysis for ectopic expression of FLAG-CRABP1 in PC-3 cells. C. Clonogenic and matrigel invasion assays showing promotion of cell growth and invasion, respectively, by overexpression of CRABP1 in PC-3 cells, and its quantification. Three clonogenic assays and five matrigel invasion assays were performed independently. ***P < 0.001. All error bars show s.e.m. D. qRT-PCR analysis for CRABP1 levels to check suppression of CRABP1 expression by siRNAs against CRABP1 (siCRABP1-1 and siCRABP1-2) in control and CIC knock-down PC-3 cell lines. Seven independent experiments were performed. *P < 0.05 and **P < 0.01. All error bars show s.e.m. E. Clonogenic assay using control and CIC knock-down PC-3 cell lines treated with two different CRABP1 siRNAs. Suppression of CRABP1 expression reduced colony formation in the CIC knock-down PC-3 cell lines. The right panel is a bar graph for quantitative analysis on colony numbers. Four independent experiments were performed. ***P < 0.001. All error bars show s.e.m. F. Matrigel invasion assay using control and CIC knock-down PC-3 cell lines treated with two different CRABP1 siRNAs. Suppression of CRABP1 expression reduces the number of invasive cells. The right panel is a bar graph for quantitative analysis on invasive cell numbers. Three independent experiments were performed. ***P < 0.001. All error bars show s.e.m.
CRABP1 is a direct target gene of CIC in PC-3 cells
We next set out to determine the molecular basis of CRABP1 overexpression by CIC deficiency. Given that CIC functions as a transcriptional repressor and that the levels of CRABP1 are increased by CIC deficiency, we hypothesized that CRABP1 might be a target gene of CIC. To test this hypothesis, we first searched for CIC binding motifs in CRABP1 promoter region and found a putative CIC binding motif within 3kb region upstream from the transcriptional start site of CRABP1 (Supplementary Figure 14). We then examined whether CIC binds to the CRABP1 promoter region containing the CIC binding motif. To this end, we carried out chromatin immunoprecipitation (ChIP) using anti-FLAG antibody in PC-3 cells expressing FLAG-CIC-S followed by qPCR for CRABP1 promoter regions. The ChIP-qPCR analyses indicate that DNA fragments corresponding to the CRABP1 promoter region with the CIC binding motif are significantly enriched in the immunoprecipitates of FLAG-CIC-S (Figure 5A), suggesting that CIC might repress CRABP1 expression through a direct binding to the promoter region in PC-3 cells. To determine that CIC indeed represses CRABP1 promoter activity, we generated luciferase expression vector driven by CRABP1 promoter (pGL3-CRABP1 pro) and carried out dual luciferase assay using this reporter construct. When CIC-S and ATXN1, the latter of which has been known to enhance the transcriptional repressor activity of CIC [5, 20], were co-expressed in PC-3 cells, luciferase activity was significantly decreased, indicating suppression of CRABP1 promoter activity by CIC (Figure 5B). Moreover, mutagenesis at the putative CIC binding motif abolished the suppressive effect of CIC on CRABP1 promoter activity (Figure 5B), demonstrating that CIC represses transcription of CRABP1 through the CIC binding motif. Taken together, our data indicate that CRABP1 expression is directly regulated by CIC in PC-3 cells.
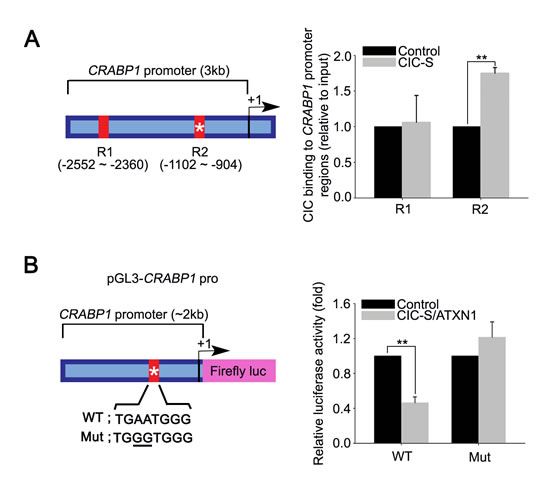
Figure 5: CRABP1 is a direct target of CIC in PC-3 cells. A. ChIP-qPCR analyses for CIC occupancy of CRABP1 promoter in PC-3 cells. PC-3 cells infected with control or FLAG-CIC-S expressing lentivirus were subjected to ChIP using anti-FLAG antibody followed by qPCR for CRABP1 promoter regions. The left panel is schematic illustration showing the loci of the putative CIC binding motif (asterisk) and PCR amplified regions (R1 and R2) within 3kb region upstream from the CRABP1 transcriptional start site. Four independent experiments were performed. **P < 0.01. All error bars show s.e.m. B. Luciferase assay showing that CIC represses CRABP1 promoter activity through the CIC binding motif in CRABP1 promoter. The left panel is a schematic illustration for the luciferase reporter construct harboring the CRABP1 promoter region (pGL3-CRABP1 pro) and mutated sequences of the putative CIC binding motif. Four independent experiments were performed. **P < 0.01. All error bars show s.e.m.
miR-93, miR-106b, and miR-375 cooperatively down-regulate CIC levels
Since CIC levels were markedly decreased in prostate cancer cells, we set out to identify a potential underlying mechanism. It is known that several microRNAs (miRNAs) are abnormally expressed in prostate cancer cells [21, 22], implicating their potential roles in tumorigenesis and/or cancer progression. In this regard, it would be conceivable that overexpression of a subset of miRNAs targeting CIC might down-regulate CIC levels to promote cancer progression in the prostate. Thus, we sought to identify miRNAs targeting CIC among the miRNAs known to be overexpressed in patients with prostate cancer. Based on previous work [21, 23-27], we searched for miRNAs that have been reported to be up-regulated in samples from prostate cancer patients by at least two independent studies. We also looked for miRNAs potentially targeting CIC using miRNA target prediction databases [28, 29]. Comparative analysis on the selected miRNAs identified five miRNAs, miR-20a, miR-25, miR-93, miR-106b, and miR-375, which not only potentially target CIC, but are also known to be frequently overexpressed in prostate cancer cells (Supplementary Figure 15A). Since the seed sequences of miR-20a, miR-93, and miR-106b are identical, they are classified as the same miRNA family (miR-17 family). There are two putative binding sites for miR-20a/miR-93/miR-106b, one for miR-25 and another for miR-375 in the 3’UTR of CIC (Supplementary Figure 15B). Of the five miRNAs, we initially chose to evaluate miR-93, miR-106b, and miR-375, considering the number of putative binding sites for each miRNA in the 3’UTR of CIC and their frequency of overexpression in prostate cancer patients, and tested whether these miRNAs can down-regulate CIC levels. Transfection with each individual miRNA duplex did not significantly affect CIC levels in PC-3 cells (Figures 6A and 6C). However, co-transfection with all three miRNA duplexes markedly down-regulated CIC levels in PC-3 cells (Figures 6B and 6C), indicating that miR-93, miR-106b, and miR-375 cooperatively regulate CIC levels. We did not observe such effect when different combinations of two miRNA duplexes were co-transfected (Figure 6B), suggesting that miR-93 and miR-106b may not function redundantly to regulate CIC levels, although they share the same seed sequences. In fact, among the two putative miR-17 family miRNA binding sites in the CIC 3’UTR, the first site is predicted to be more preferentially targeted by miR-93, whereas the second one by miR-106b, according to the miRNA target prediction databases (Supplementary Figure 15B). Moreover, it has been known that nucleotide sequences outside of seed sequences also contribute to determination of target mRNA binding specificity of miRNAs [30, 31]. We also examined whether CIC expression is regulated by endogenous miR-93, miR-106b, and miR-375 in PC-3 cells. Inhibition of miR-375, but not miR-93 and miR-106b, significantly increased CIC levels (Figure 6D), suggesting that, among the three miRNAs, miR-375 is the most critical endogenous miRNA for regulation of CIC levels in PC-3 cells. To verify that miR-93, miR-106b, and miR-375 directly target the 3’UTR of CIC, we constructed luciferase reporter gene linked to the CIC 3’UTR (pGL3-CIC 3’UTR WT), and carried out dual luciferase assays. Co-transfection with three miRNAs decreased luciferase activity in PC-3 cells (Figure 6E), suggesting that miR-93, miR-106b, and miR-375 down-regulate CIC levels through the 3’UTR of CIC. Moreover, disruption of the putative miRNA binding sites in the 3’UTR of CIC abrogated suppression of luciferase activity by the three miRNAs (Figure 6E), demonstrating that miR-93, miR-106b, and miR-375 directly target the 3’UTR of CIC to regulate CIC levels. On the other hand, the three miRNAs still slightly repressed luciferase activity derived from the pGL3-CIC 3’UTR Mut compared with control vector (Figure 6E), implying that there might be other binding sites for miR-93, miR-106b, and miR-375 in the 3’UTR of CIC, or that the three miRNAs might also be able to repress CIC expression indirectly.
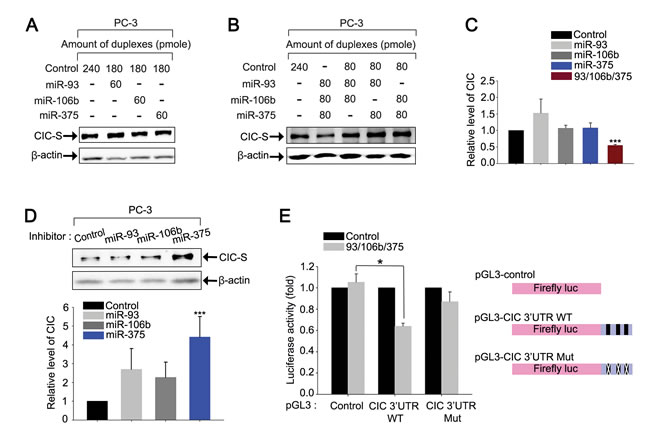
Figure 6: miR-93, miR-106b, and miR-375 cooperatively down-regulate CIC levels in PC-3 cells. A. Western blot analysis for CIC levels in PC-3 cells transfected with control, miR-93, miR-106b, or miR-375 duplexes. B. Western blot analysis for changes in CIC levels by overexpression of miR-93, miR-106b, and miR-375 in PC-3 cells. Co-expression of three miRNAs significantly decreased levels of CIC. C. Bar graph for quantitative analysis on the level of CIC based on western blot images. More than three independent experiments were carried out. ***P < 0.001. All error bars show s.e.m. D. Western blot analysis for changes in CIC levels by inhibition of endogenous miR-93, miR-106b or miR-375 in PC-3 cells and its quantification. Inhibition of miR-375 significantly increased levels of CIC. More than three independent experiments were carried out. ***P < 0.001. All error bars show s.e.m. E. Luciferase assay showing that the 3’UTR of CIC is responsible for the miRNAs-mediated down-regulation of CIC expression in PC-3 cells. Three independent experiments were carried out. *P < 0.05. All error bars show s.e.m.
miR-93, miR-106b, and miR-375 cooperatively regulate CIC-CRABP1 axis to promote prostate cancer progression
To determine the impact of the miRNAs-mediated down-regulation of CIC on prostate cancer progression, we assessed cell proliferation and invasion in PC-3 cells transfected with either control, miR-93/miR-106b/miR-375 or siCIC duplexes. Overexpression of miR-93, miR-106b, and miR-375 increased cell proliferation and invasion (Figures 7B and 7C and Supplementary Figure 16), accompanied with down-regulation of CIC levels (Figure 7A), suggesting the cancer promoting property of these three miRNAs in prostate cancer cells. To verify that the miRNAs-mediated down-regulation of CIC levels contributed to the enhanced cell proliferation and invasion, we restored CIC levels in PC-3 cells co-transfected with the three miRNA duplexes by mouse CIC-S expressing lentiviral infection (Figure 7A), and conducted clonogenic, cell growth, and invasion assays. The miRNAs-mediated increases in cell proliferation and invasion were partially abolished by recovery of CIC levels in PC-3 cells (Figures 7B and 7C and Supplementary Figure 16), suggesting that miR-93, miR-106b, and miR-375 promote prostate cancer progression in part by down-regulation of CIC expression. We also measured CRABP1 levels in the same set of cells, and found that co-expression of miR-93, miR-106b, and miR-375 resulted in up-regulation of CRABP1, which was restored by overexpression of CIC in PC-3 cells (Figure 7D). Taken together, these data demonstrate that miR-93, miR-106b, and miR-375 function cooperatively to regulate the CIC-CRABP1 axis in promoting prostate cancer progression.
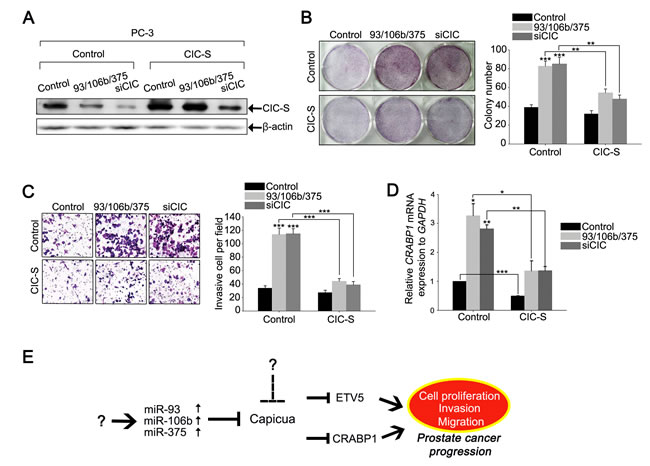
Figure 7: miR-93, miR-106b, and miR-375 co-regulate CIC-CRABP1 axis to promote cancer progression in PC-3 cells. A. Western blot analysis for CIC levels in PC-3 cells treated with different combinations of three miRNA duplexes or siCIC, and control or CIC-S-expressing lentivirus. B. Clonogenic assay for the PC-3 cells used in A. and its quantification. Three independent experiments were carried out. **P < 0.01 and ***P < 0.001. All error bars show s.e.m. C. Matrigel invasion assay for the PC-3 cells used in A. and its quantification. Three independent experiments were carried out. ***P < 0.001. All error bars show s.e.m. D. qRT-PCR analysis for CRABP1 levels in the PC-3 cells used in A.. Three independent experiments were carried out. *P < 0.05, **P < 0.01 and ***P < 0.001. All error bars show s.e.m. E. The proposed model that describes how prostate cancer progression is regulated by miRNAs, CIC, and its target genes, based on the findings in this study. The question marks mean unidentified factors or mechanisms.
DISCUSSION
Our study demonstrates that CIC negatively regulates prostate cancer progression and raises a possibility that CIC could be a novel tumor suppressor for prostate cancer. According to The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/), the frequencies of deletional and truncating CIC gene mutations found in 258 prostate cancer patient samples are 3.1% (8 cases) and 0.4% (1 case), respectively. Examination of how those mutations affect the activity of CIC would be critical to further evaluate the potential role of CIC as a tumor suppressor in the prostate.
In this study, we identified CRABP1 as a novel CIC target, and provided a molecular basis of how CIC regulates prostate cancer progression. Although a few studies have shown that CRABP1 could promote cancer progression in some types of cancer [18, 32], we now for the first time show that CRABP1 has cancer promoting property in prostate cancer cells. However, it remains unknown how CRABP1 facilitates cell proliferation and invasion in prostate cancer cells. In fact, the binding of retinoic acid, which stimulates differentiation and inhibits proliferation of cells, is the only known function of CRABP1 [33]. On the other hand, it has also been reported that the cancer promoting activity of CRABP1 is independent of the retinoic acid binding activity [18]. Therefore, in-depth molecular characterization of CRABP1 will be required for understanding of the molecular mechanism underlying the promotion of prostate cancer progression by CRABP1.
We found that CIC represses expression of ETV5 and CRABP1 in a cell-type dependent manner. This conclusion is further supported by our findings that expression of ETV5 and CRABP1 is not up-regulated by knock-down of CIC in DU145 cells whereas levels of ETV5, but not CRABP1, are significantly up-regulated in CIC knock-down LNCaP-LN3 cells (Supplementary Figure 17). Given that PC-3 cells are highly metastatic and independent of androgen [16], the CIC-CRABP1 axis might play a critical role in regulation of prostate cancer progression at metastatic and castration-resistant stages. The mRNA sequencing analysis also reveals that several genes, such as NUDT11, CCNA1, and HNF1B, known to promote prostate cancer progression [34, 35], are significantly up-regulated in the CIC knock-down PC-3 cells (Supplementary Table 2). In fact, knock-down of CIC more dramatically increased cell proliferation and invasion in PC-3 cells than overexpression of CRABP1 (Figures 2E, 2F and 4C), suggesting that not only CRABP1, but other genes regulated by CIC are also involved in promotion of cancer progression by CIC deficiency. Identification of CIC target genes and elucidation of CIC-mediated gene expression regulatory networks at different stages of prostate cancer pathogenesis will uncover CIC-mediated molecular pathways regulating cancer progression in the prostate.
Our data indicate that CIC levels are drastically decreased in prostatic carcinoma. We consistently observed disappearance of the nuclear CIC expression in prostatic adenocarcinoma in all of the tested patient specimens. Moreover, overexpression of CIC markedly suppressed cell proliferation, invasion, and migration in PC-3 cells. Therefore, CIC could be considered as a therapeutic target as well as a molecular marker of prostate cancer. We identified miRNAs targeting CIC from the miRNAs known to be overexpressed in prostate cancer tissues, and proposed that miR-93, miR-106b, and miR-375 could potentially contribute to the down-regulation of CIC levels in the process of prostate cancer progression. Comparative miRNA profiling of prostate carcinomas with increasing tumor stages has revealed that levels of miR-375 and miR-106b gradually increase from normal to lymph node metastasizing tumors, whereas miR-93 increases from normal to extracapsular growing tumors [36], suggesting that these miRNAs are likely to participate in the gradual decrease in CIC levels during prostate cancer progression.
In sum, our findings suggest that miR-93/miR-106b/miR-375-CIC-CRABP1 is a novel regulatory axis in prostate cancer progression (Figure 7E). Identification of other components involved in this regulatory axis and further studies on their functions will not only advance the understanding of pathogenesis of prostate cancer, but may also uncover novel therapeutic targets that can be used in the design of treatment modalities for prostate cancer.
MATERIALS AND METHODS
Human tissue samples
This study was approved by the Asan Medical Center Institutional Review Board (2011-499). Patients who underwent radical prostatectomy between January 2006 and December 2012 at Asan Medical Center were selected. We randomly chose 13 prostate cancer patients’ tissues to detect CIC expression by immunofluorescence staining. According to the value of the Gleason score, the patient’s samples were divided into patients with Gleason score with 5-9.
Immunofluorescence staining
Immunofluorescence staining was performed using standard protocols. Briefly, paraffin sections were dewaxed using xylene for 30 min, and antigen retrieval was performed by boiling in 10mM sodium citrate for 15 min. The sections were treated with rabbit anti-CIC [8] (1:100), chicken anti-Keratin 5 (1:100) (COVANCE, Hertfordshire, England), mouse anti-Cytokeratin 8 (1:500) (COVANCE, Hertfordshire, England), or mouse anti-Cytokeratin 14 (1:100) (BioGenex, CA, USA) antibodies overnight at 4°C, and then incubated with secondary antibodies, Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, NY, USA), Alexa Fluor 594 goat anti- mouse IgG (Invitrogen, NY, USA) and goat anti-chicken IgY-TR (Santa Cruz Biotechnology, TX, USA) for 1 h at room temperature with 4,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, MO, USA). Immunofluorescence staining was imaged using ZEISS Axioplan2 fluorescence microscope or ZEISS LSM700 confocal microscope.
Cell culture
PNT2, PC-3, LNCaP, DU145 and LNCaP-LN3 cells were maintained in RPMI 1640 (HyClone, UT, USA) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (WELGENE, Daegu, Republic of Korea). All cell lines were maintained at 37℃ in a humid atmosphere containing 5% CO2. Instead of trypsin, cell scrapers were used to detach PC-3 cells during subculture of the cells.
Transfection of siRNA and miRNA duplexes and miRNA inhibitor
miRNA inhibitors, miRNA mimic duplexes, and siRNAs targeting CRABP1, CIC, and ETV5 were purchased from Bioneer (Daejun, Republic of Korea). Sequences are as follows: miR-93 sense; 5’-CAAAGUGCUGUUCGUGCAGGUAG-3’, and antisense; 5’-ACCUGCACGAACAGCACUUUAUU-3’. miR-106b sense; 5’-UAAAGUGCUGACAGUGCAGAU-3’, and antisense; 5’-CUGCACUGUCAGCACUUUGUU-3’. miR-375 sense; 5’-UUUGUUCGUUCGGCUCGCGUGA-3’, and antisense; 5’-ACGCGAGCCGAACGAACAAAUU-3’. siETV5 sense; 5’-CACAAGCUUAGAUUCUCUA-3’, and antisense; 5’- UAGAGAAUCUAAGCUUGUG-3’. siCIC sense; 5’-CAGAACGGCUACACACAGU-3’, and antisense; 5’-ACUGUGUGUAGCCGUUCUG-3’. siCRABP1-1 sense; 5’-GUAUCCCUAGUGCUCCAUA-3’, and antisense; 5’-UAUGGAGCACUAGGGAUAC-3’. siCRABP1-2 sense; 5’-CGAAGUCAUUAAACUGGUU-3’, and antisense; 5’-AACCAGUUUAAUGACUUCG-3’. Transfection of siRNA duplexes, miRNA duplexes, or miRNA inhibitors was conducted using Dharmafect 2 (Dharmcon, CO, USA) according to the manufacturer’s instructions. The cells were transfected with 120 pmole of siRNA duplexes or 60 pmole of miRNA inhibitors, and incubated for 72~96 h.
Clonogenic assay
For clonogenic assay of CIC knock-down PC-3 or LNCaP cells, 2 × 103 or 4 × 103 cells were seeded in six well plates and incubated for 7~8 days or 14~15 days, respectively. For clonogenic assay of PC-3 or LNCaP cells overexpressing CIC, 2 × 103 or 4 × 103 cells were seeded in six well plates and incubated for 14~15 days or 10~11 days, respectively. For clonogenic assay of PC-3 cells overexpressing FLAG-CRABP1, 2 × 103 cells were seeded in six well plates and incubated for 5 days. The cells were stained with formalin/0.1% crystal violet solution. For clonogenic assay of siRNAs-treated CIC knock-down PC-3 or LNCaP cells, 2 × 103 or 4 × 103 cells were seeded in six well plates a day before transfection, respectively, and then siRNAs were transfected using Dhamafect 2. The cells were stained with formalin/0.1% crystal violet solution after 72 h incubation. For clonogenic assay of PC-3 cells treated with miRNA duplexes and CIC-S expressing lentivirus, 5 × 103 PC-3 cells were seeded in six well plates a day before transfection, and then co-transfected with miR-93, miR-106b, and miR-375 duplexes using Dhamafect 2. After 24 h, the cells were infected with lentivirus expressing CIC-S for 3 sequential days. The cells were stained with formalin/0.1% crystal violet solution and analyzed using Olympus CKX41 microscope and Adobe Photoshop CS6 software. Analyzed cell number is listed in Table S3.
Cell growth assay
For cell growth assay of PC-3 cells overexpressing FLAG-CRABP1, 1 × 103 cells were seeded in 24 well plates and incubated for 6 days. The cells were trypsinized, stained with Trypan Blue (Sigma-Aldrich, MO, USA), and counted for the number of viable cells using hemacytometer. For cell growth assay of siRNAs-treated CIC knock-down PC-3 or LNCaP cells, 1 × 103 cells were seeded in 24 well plates a day before transfection, and then siRNAs were transfected using Dhamafect 2 and set as day “0”. The cells were trypsinized, stained with Trypan Blue (Sigma-Aldrich, MO, USA), and counted for the number of viable cells using hemacytometer every other day for 6 days. For cell growth assay of PC-3 cells treated with miRNA duplexes and CIC-S expressing lentivirus, 1 × 103 PC-3 cells were seeded in 24 well plates a day before transfection, and then co-transfected with miR-93, miR-106b, and miR-375 duplexes using Dhamafect 2 and set as day “0”. After 24 h, the cells were infected with lentivirus expressing CIC-S for 3 sequential days. The cells were trypsinized, stained with Trypan Blue (Sigma-Aldrich, MO, USA), and counted for the number of viable cells using hemacytometer over 4 days.
Invasion assay
For invasion assay of CIC knock-down PC-3 or LNCaP cells, 5 × 103 cells were suspended in serum-free medium and seeded into the inserts of a 24-well BioCoat Matrigel Invasion Chamber (BD falcon, CA, USA). For invasion assay of PC-3 or LNCaP cells overexpressing CIC, 1 ×104 or 4 × 103 cells were seeded into the inserts, respectively. For invasion assay of PC-3 cells overexpressing CRABP1, 5 × 103 cells were seeded into the inserts. For invasion assay of siRNAs-treated CIC knock-down PC-3 or LNCaP cells, 2 × 103 or 3 × 103 cells were seeded into the inserts, respectively. For invasion assay of PC-3 cells treated with miRNA duplexes and CIC-S expressing lentivirus, 2 × 103 cells were seeded into the inserts. The inserts were co-cultured in a well of 10% FBS-containing media and incubated for 24h. Inserts were then removed and the upper surface of the membrane was rubbed off to remove non-migrating cells with cotton swabs and washed with PBS. Then, inserts were stained with formalin/0.1% crystal violet solution and analyzed under ZEISS Axioplan2 microscope. Multiple 15~20 images per insert were acquired, and average counts were calculated. Analyzed cell number is listed in Supplementary Table 3.
Wound healing assay
For the wound-healing assay, cells were seeded into 6-well plates and cultured at full confluency. A sterile 1ml pipette tip was used to scratch the cells to form a wound. The cells were washed with PBS and cultured in serum-free medium. Wound closure was visualized with ZEISS AxioCamICc1 microscope. Multiple 9 images per well were acquired and average counts were calculated. Analyzed cell number is listed in Supplementary Table 3.
BrdU labeling assay
For BrdU labeling assay of CIC knock-down PC-3 or LNCaP cells, 2 × 103 or 4 × 103 cells were seeded in six well plates and incubated for 7~8 days and 14~15 days, respectively. For BrdU labeling assay of PC-3 or LNCaP cells overexpressing CIC, 2 × 103 or 6 × 103 cells were seeded in six well plates and incubated for 14~15 days, respectively. BrdU (Sigma-Aldrich, MO, USA) was added in cell culture media to a final concentration of 100mM and incubated for 2 h at 37℃ with 5% CO2. The cells were fixed with 4% paraformaldehyde, incubated with mouse anti-BrdU (1:200) (DSHB, Iowa, USA) overnight at 4°C, and then with Alexa Fluro 594 goat anti-mouse IgG (Invitrogen, NY, USA).
Cloning
To construct pGL3-CIC 3’UTR, entire human CIC 3’UTR sequences (606nt) were amplified from cDNA of MCF7 cells using Pfu-X DNA polymerase (SolGent, Daejun, Republic of Korea) and cloned into the pGL3-control vector (Promega, WI, USA). To construct pGL3-CRABP1 pro, human CRABP1 promoter region (-1927bp ~ +2bp) was amplified from MCF7 genomic DNA using Pfu-X DNA polymerase and cloned into the pGL3-basic vector (Promega, WI, USA). To make pHAGE-FLAG-CIC-S, and CIC-L, mouse Cic-S and Cic-L coding sequences were amplified from cDNA of NIH3T3 cells using Pfu-X DNA polymerase and cloned into the pHAGE-FLAG lentiviral vector. To make pHAGE-FLAG-CRABP1, human CRABP1 coding sequences were amplified from cDNA of MCF7 cells using Pfu-X DNA polymerase and cloned into the pHAGE-FLAG lentiviral vector.
Site-directed mutagenesis
Site-directed mutagenesis was performed using QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, CA, USA) according to the manufacturer’s instructions. The primers used for mutagenesis at miR-93, miR-106b, and miR-375 binding sites within the CIC 3’UTR are as follows: 93-106b mut-1sense;5’-GTGGGGGCTCCTGCGTCTTGCCACAGGCACGGGGAGGGTT -3’, 93-106b mut-1 antisense; 5’- AACCCT CCCCGCCTGTGGCAAGACGCAGGAGCCCCCAC - 3’, 93-106b mut-2 sense;5’GTGACCTTCAGAGCTTTTCGTCTTATGCAAAATGGCTCCT-3’, 93-106b mut-2 antisense; 5‘- AGGAGC CATTTTGCATAAGACGAAAAGCTCTGAAGGTCAC - 3’, 375 mutsense;5’-CTTGCCCCCTTCCCCAGATGTAAACATGTTGATCATGTGC-3’, and 375 mut antisense; TCAACATGTTTACATCTGGGGAAGGGGGCAAG-3’.The primers used for mutagenesis at the CIC binding motif within CRABP1 promoter are as follows: sense;5’-GGGTTAATCAAATCTTGCCCACCCACGAAAGCCCATCTTTATGC-3’, antisense; 5’- GCATAAAGATGG GCTTTCGTGGGTGGGCAAGATTTGATTAACCC-3’.
Dual luciferase assay
To examine the effect of miRNAs on the 3’UTR of CIC, PC-3 cells seeded in 24-well plates were transfected with pGL3-control, CIC 3’UTR WT or CIC 3’UTR Mut (60ng), together with pRL-TK (15ng), and control or miRNA duplexes using Lipofectamine2000 (Invitrogen, NY, USA) according to the manufacturer’s instruction. To examine the effect of CIC and ATXN1 on the CRABP1 promoter activity, PC-3 cells seeded in 24-well plates were transfected with pGL3-CRABP1 pro WT or Mut (120ng), together with pRL-TK (30ng), pHAGE-FLAG-CIC-S (60ng) and pcDNA3.1-FLAG-ATXN1 (60ng) [37] using Lipofectamine2000 (Invitrogen, NY, USA) according to the manufacturer’s instruction. The cells were lysed 48h later, and luciferase activities were analyzed using the Dual-Luciferase Reporter Assay System (Promega, WI, USA) according to the manufacturer’s protocol.
Lentivirus production and transduction
Non-silencing control shRNA (Cat no. RHS4346) and shCIC expressing lentiviral vectors (pGIPZ) were purchased from Open Biosystems. The clone IDs for shCIC-1, shCIC-2, and shCIC-3 are V3LHS-358902, V3LHS-408353, and V3LHS-358903, respectively. Lentivirus was produced by co-transfection of HEK-293T cells with pGIPZ-shCIC and plasmids for viral particles using FuGENE (Promega, WI, USA). Viral supernatants were collected at 48 h post-transfection and used to infect into the PC-3 and LNCaP cells pre-seeded with approximately 50% confluence. Puromycin was added to select drug-resistant pools at 48 h post-infection. For overexpression of CIC-S, CIC-L, and CRABP1, lentivirus was produced by the same protocol using the cloned pHAGE-FLAG-CIC-S, pHAGE-FLAG-CIC-L, pHAGE-FLAG-CRABP1, or pHAGE-FLAG control plasmid. Viral supernatants were collected at 48 h post-transfection and used to infect into the PC-3 cells for 3 sequential days. The cells were used for further biochemical assays as specified in each experiment.
Western blot
Cells were lysed with RIPA buffer (50mM Tris (pH 7.4), 150mM NaCl, 0.5% Sodium deoxycholate, 0.1% SDS, 1% Triton X-100) containing protease inhibitor cocktail tablets (Roche, CA, USA). Western blot analysis was performed as described previously [8] using primary antibodies as follows: rabbit anti-CIC (1:1000) [8], rabbit anti-CRABP1 (1:200) (Cell Signaling Technology, MA, USA), mouse anti-β-actin (1:2500) (Santa Cruz Biotechnology, TX, USA), and rabbit anti-FLAG (1:1000) (Sigma-Aldrich, MO, USA). The western blot images were obtained using ImageQuant LAS 4000 (GE Healthcare Life Science, PA, USA) and quantified with Image J software.
RNA extraction and quantitative RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen, NY, USA) and subjected to cDNA synthesis using random hexamer and GoScript™ Reverse Transcription System (Promega, WI, USA) according to the manufacturer’s instructions. SYBR Green real-time PCR master mix (TOYOBO, NY, USA) was used for PCR reactions. Primers used for qRT-PCR are as follows: CRABP1 forward; 5’-GCAGCAGCGAGAATTTCGAC-3’, and reverse; 5’-CGTGGTGGATGTCTTGATGTAGA-3’. ETV1 forward; 5’-ACACCTGTGTTGTCCCAGAA-3’, and reverse; 5’-GTTGGTATGTGGGTCCTTCC-3’. ETV4 forward; 5’-GATGAAAGCCGGATACTTGGAC-3’, and reverse; 5’-TTCGCGCAAGCTCCCATTT-3’. ETV5 forward; 5’-CATCCTACATGAGAGGGGGTTA-3’, and reverse; 5’-AAGTATAATCGGGGATCTTTTTCA-3’. GAPDH forward; 5’-AGCCACATCGCTCAGACAC-3’, and reverse; 5’-GCCCAATACGACCAAATCC-3’. 18S rRNA forward; 5’-ATCAACTTTCGATGGTAGTCG-3’, and reverse; 5’-ACTCATTCCAATTACAGGGC-3’.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed manually. Briefly, control and FLAG-CIC-S expressing PC-3 cells were fixed in 1% formaldehyde. The cross-linked DNA was then sheared into about 200–1000 base pairs in length with sonication. Ten percent of the sheared DNA was set aside as an input control. The rabbit anti-FLAG antibody (Sigma-Aldrich, MO, USA) was incubated with sheared DNA at 4 °C overnight with rotation, then the protein G agarose (Millipore, Darmstadt, Germany) was added. After that, protein/DNA complexes were eluted from the agarose. The cross-linking DNA was reversed to free DNA, and purified DNA was analyzed by qPCR using the primers that amplify CRABP1 promoter region containing the CIC-binding site (R2). The primers that amplify CRABP1 promoter region without CIC-biding sites (R1) were used as a negative control. The primer sequences are as follows: R1 forward; 5’-CAGAGCCAGACCCTGTC-3’, and reverse; 5’-CAGATGAAGGTGTCCACTC-3’. R2 forward; 5’-AAATAATCACAGTTTAGGAAAC-3’, and reverse; 5’-CACCTCAGCCAAACTGTAC-3’.
RNA sequencing and data analysis
For isolation of mRNA from total RNA, Oligotex mRNA mini kit (QIAGEN, CA, USA) was used according to the manufacturer’s instructions. The cDNA was synthesized by random hexamer and Superscript III reverse transcriptase (Invitrogen, NY, USA). The library for the mRNA sequencing was constructed and sequenced on the Genome Analyzer IIx (Illumina, USA) using the protocol described earlier [38]. We mapped sequencing reads to the human reference genome (hg18 RefSeq) using Tophat (v 2.0.9). Reads Per Kilobase of exon per Million aligned tags (RPKM) values for each transcript were calculated by Cufflinks (v 2.2.1) as well as identification of the differential expressed genes. The GEO accession number of RNA sequencing data is GSE64025.
Statistical analysis
For statistical analysis, all experiments were performed more than three times independently. Data are presented as mean ± standard error. Student’s t-test was used to determine significance between groups. For all statistical tests, the 0.05 level of confidence was accepted for statistical significance.
ACKNOWLEDGMENTS and funding
We thank Dr. Rodney Samaco and the Lee lab members for helpful discussions and comments on the manuscript. This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean Ministry of Science, ICT and Future Planning (NRF-2012R1A1A1005631 and NRF-2015R1A1A1A05001401 to YL and NRF-2013R1A1A2063089 to NC), the POSTECH Basic Science Research Institute (4.0010906.01), and TJ Park Science Fellowship of POSCO TJ Park Foundation. NC, JP, J-SL, JY, GYP and EK were supported by the BK21 Plus Program (Program of Bio-Molecular Function, POSTECH).
CONFLICTs OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Jiménez G, Guichet A, Ephrussi A, Casanova J. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev 2000; 14:224–231.
2. Jiménez G, Shvartsman SY, Paroush Z. The Capicua repressor—a general sensor of RTK signaling in development and disease. J Cell Sci 2012; 125:1383–1391.
3. Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, Sinha S, Wolfe SA, Brodsky MH. FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 2011; 39:D111–117.
4. Shin D-H, Hong J-W. Capicua is involved in Dorsal-mediated repression of zerknüllt expression in Drosophila embryo. BMB Rep 2014; 47:518–523.
5. Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 2006; 127:1335–1347.
6. Fryer JD, Yu P, Kang H, Mandel-Brehm C, Carter AN, Crespo-Barreto J, Gao Y, Flora A, Shaw C, Orr HT, Zoghbi HY. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 2011; 334:690–693.
7. Lee Y, Fryer JD, Kang H, Crespo-Barreto J, Bowman AB, Gao Y, Kahle JJ, Hong JS, Kheradmand F, Orr HT, Finegold MJ, Zoghbi HY. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell 2011; 21:746–757.
8. Kim E, Park S, Choi N, Lee J, Yoe J, Kim S, Jung HY, Kim KT, Kang H, Fryer JD, Zoghbi HY, Hwang D, Lee Y. Deficiency of Capicua disrupts bile acid homeostasis. Sci Rep 2015; 5:8272.
9. Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268–274.
10. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010; 466:869–873.
11. Alentorn A, Sanson M, Idbaih A. Oligodendrogliomas: new insights from the genetics and perspectives. Curr Opin Oncol 2012; 24:687–693.
12. Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, Takizawa T, Nakamura T. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 2006; 15:2125–2137.
13. Kurpios NA, Sabolic NA, Shepherd TG, Fidalgo GM, Hassell JA. Function of PEA3 Ets transcription factors in mammary gland development and oncogenesis. J Mammary Gland Biol Neoplasia 2003; 8:177–190.
14. Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, Prescott AR, MacKintosh C. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicúa. Biochem J 2011; 433:515–525.
15. Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol 2009; 6:429–439.
16. Russell PJ, Kingsley EA. Human prostate cancer cell lines. Methods Mol Med 2003; 81:21–39.
17. Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta 2012; 1826:1–12.
18. Kainov Y, Favorskaya I, Delektorskaya V, Chemeris G, Komelkov A, Zhuravskaya A, Trukhanova L, Zueva E, Tavitian B, Dyakova N, Zborovskaya I, Tchevkina E. CRABP1 provides high malignancy of transformed mesenchymal cells and contributes to the pathogenesis of mesenchymal and neuroendocrine tumors. Cell Cycle 2014; 13:1530–1539.
19. Sirotnak FM, She Y, Khokhar NZ, Hayes P, Gerald W, Scher HI. Microarray analysis of prostate cancer progression to reduced androgen dependence: studies in unique models contrasts early and late molecular events. Mol Carcinog 2004; 41:150–163.
20. Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet 2010; 6:e1001021.
21. Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res 2008; 68:6162–6170.
22. Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 2008; 27:1788–1793.
23. Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TLJ, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res 2007; 67:6130–6135.
24. Tong AW, Fulgham P, Jay C, Chen P, Khalil I, Liu S, Senzer N, Eklund AC, Han J, Nemunaitis J. MicroRNA profile analysis of human prostate cancers. Cancer Gene Ther 2009; 16:206–216.
25. Schaefer A, Jung M, Mollenkopf H-J, Wagner I, Stephan C, Jentzmik F, Miller K, Lein M, Kristiansen G, Jung K. Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int J Cancer 2010; 126:1166–1176.
26. Wach S, Nolte E, Szczyrba J, Stöhr R, Hartmann A, Ørntoft T, Dyrskjøt L, Eltze E, Wieland W, Keck B, Ekici AB, Grässer F, Wullich B. MicroRNA profiles of prostate carcinoma detected by multiplatform microRNA screening. Int J Cancer 2012; 130:611–621.
27. Szczyrba J, Löprich E, Wach S, Jung V, Unteregger G, Barth S, Grobholz R, Wieland W, Stöhr R, Hartmann A, Wullich B, Grässer F. The microRNA profile of prostate carcinoma obtained by deep sequencing. Mol Cancer Res 2010; 8:529–538.
28. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15–20.
29. Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet 2005; 37:495–500.
30. Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 2007; 27:91–105.
31. Shin C, Nam J-W, Farh KK-H, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell 2010; 38:789–802.
32. Won JY, Nam E-C, Yoo SJ, Kwon HJ, Um SJ, Han HS, Kim SH, Byun Y, Kim SY. The effect of cellular retinoic acid binding protein-I expression on the CYP26-mediated catabolism of all-trans retinoic acid and cell proliferation in head and neck squamous cell carcinoma. Metab Clin Exp 2004; 53:1007–1012.
33. Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem 1999; 274:23695–23698.
34. Wegiel B, Bjartell A, Tuomela J, Dizeyi N, Tinzl M, Helczynski L, Nilsson E, Otterbein LE, Härkönen P, Persson JL. Multiple cellular mechanisms related to cyclin A1 in prostate cancer invasion and metastasis. J Natl Cancer Inst 2008; 100:1022–1036.
35. Grisanzio C, Werner L, Takeda D, Awoyemi BC, Pomerantz MM, Yamada H, Sooriakumaran P, Robinson BD, Leung R, Schinzel AC, Mills I, Ross-Adams H, Neal DE, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci USA 2012; 109:11252–11257.
36. Hart M, Nolte E, Wach S, Szczyrba J, Taubert H, Rau TT, Hartmann A, Grässer FA, Wullich B. Comparative microRNA profiling of prostate carcinomas with increasing tumor stage by deep sequencing. Mol Cancer Res 2014; 12:250–263.
37. Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci 2008; 11:1137–1139.
38. Choe MK, Hong C-P, Park J, Seo SH, Roh T-Y. Functional elements demarcated by histone modifications in breast cancer cells. Biochem Biophys Res Commun 2012; 418:475–482.