
iNTRODUCTION
Hepatocyte growth factor (HGF), also known as scatter factor, is a pleiotropic polypeptide growth factor with a number of biological activities, including cell scattering, stimulation of cell motility, mitogenesis, morphogenesis, angiogenesis, and cellular invasiveness [1-3]. HGF was identified in 1984 [4, 5] and was subsequently determined to be a heterodimeric molecule composed of an alpha and beta chain [6]. The importance of HGF in organ development is demonstrated by HGF null mutation mice, which exhibit embryonic lethality [7]. In vivo, HGF and its receptor, the c-MET proto-oncogene product, are thought to be involved in embryogenesis, tissue reorganization, and tumor progression [8]. Overproduction of HGF by tumor cells or tumor-associated stromal cells and increased expression of the c-Met protein are two mechanisms that contribute to aberrant stimulation of this pathway. Consequences of dysregulated HGF and c-Met expression include tumor cell migration, proliferation, and protection from apoptosis [9]. Overexpression of HGF and c-Met has been detected in numerous cancer types and is associated with a worse prognosis [10].
A bilateral collaborative effort of normal epithelial cells and components of the stromal compartment maintain the integrity of a normal physiological system. The tumor microenvironment contains a diverse array of cell types, including endothelial cells, pericytes, smooth muscle cells, macrophages, mesenchymal stem cells and stromal fibroblasts, among others [11-13]. The stromal cells were crosstalk not only with tumor cells but with each other [14]. Tumorigenesis has classically been viewed as a largely cell-autonomous process involving genetically transformed cancer cells. Recently, many investigations support the notion that tumor stromal cells play important roles in tumor initiation, progression, and metastasis [15-17]. Fibroblasts are the most frequent component of tumor microenvironment, especially in colorectal, breast, ovarian and pancreatic cancers [18, 19]. Fibroblasts are often identified by their spindle-shaped morphology, their ability to adhere to plastic, and their lack of markers indicating other cell types [20]. Cancer associated fibroblasts (CAFs) stimulate malignant cell proliferation by providing different types of growth factors and cytokines in a context-dependent manner [21], such as HGF [22], members of the epidermal growth factor family [23], fibroblast growth factor (FGF) [24], Wnt families [25], and IL-6 [26]. CAFs are phenotypically and functionally distinguishable from their normal counterparts in their increased rate of proliferation. CAFs secrete a wide variety of growth factors, chemokines, collagens, and matrix-modifying enzymes, collectively supplying a communication network and altered three-dimensional ECM scaffold that governs the proliferation of cancer cells, tumor invasion, and metastasis across tissue types [27].
Despite the large repertoire of therapies available and the continuing efforts to incorporate new drugs into clinical practice, innate and acquired resistances remain intractable problems in clinical care. While the investigation of therapeutic resistance has not surprisingly centered on tumor cell-intrinsic mechanisms to date, recent findings have uncovered novel roles for the tumor microenvironment in modulating therapeutic efficacy [28]. Cancer cells are in contact with their surrounding cells by paracrine and autocrine mechanisms. The identification of resistance mechanisms has revealed a recurrent theme-the engagement of survival signals redundant to those transduced by the targeted kinase [29]. It is possible that evaluation of the HGF/c-MET signaling axis in the tumor may indicate the susceptibility of that tumor to show resistance through this pathway [30]. Indeed, higher expression of the c-MET receptor in different cancer cell lines correlated with their resistance to targeted therapy stimulated by HGF [31-33]. Based on these premises, the purpose of this study is to understand the role of fibroblasts-derived HGF in mediating the chemoresistance phenotype of cancer cells and the mechanism by which this occurs.
In the present study, we demonstrated that HGF secreted from fibroblast promotes chemoresistance of CRC. Thus, we have focused on evaluating the clinical significance of HGF/c-Met axis in cancer chemotherapy. The identification of stromal HGF as a mediator of CPT-11 resistance has important translational potential. In addition to this, we investigated the effects of anti-HGF monoclonal antibody on cancer chemoresistance.
Results
CM from colonic fibroblast rescues colorectal and lung cancer cells from growth suppressive activity of CPT-11
We postulated that innate drug resistance might be caused at least in part by factors secreted by the tumor stromal cells. While growth and metastasis-promoting effects of the stromal cells have been well reported [34], a role in drug resistance has only been partially described [35, 36]. To determine if the rescue effect was activated by the secretion factors, we tested the ability of CM from fibroblasts to recapitulate the resistance effect. For this purpose, cancer cells were treated with CPT-11 at concentrations ranging from 1.25 to 20 μM with or without CM from fibroblasts for 48 h. As shown in Figure 1A, cell proliferation was significantly suppressed by CPT-1 in all the human cell lines as compared with the vehicle control. Under these experimental conditions, cells became highly resistant to CPT-11 in the presence of CM from fibroblasts. CM from fibroblasts rescued human cancer cells from CPT-11, indicating that the rescue was due to a factor secreted by the fibroblasts. Similar results were obtained when we determined apoptosis of cells by annexin-V/PI staining after incubation of cells with CPT-11 in the presence of CM from fibroblasts (Figure 1B). CPT-11 caused apoptosis in DLD-1 and HCT116 cells; however, CM from fibroblast significantly attenuated apoptotic cell death in response to CPT-11. Figure 1 indicates that CM from fibroblasts enhanced the resistance against CPT-11 in cancer cells and suppressed apoptotic cell death, indicating that secreted factors from fibroblasts are crucial for sustaining cancer cell survival.
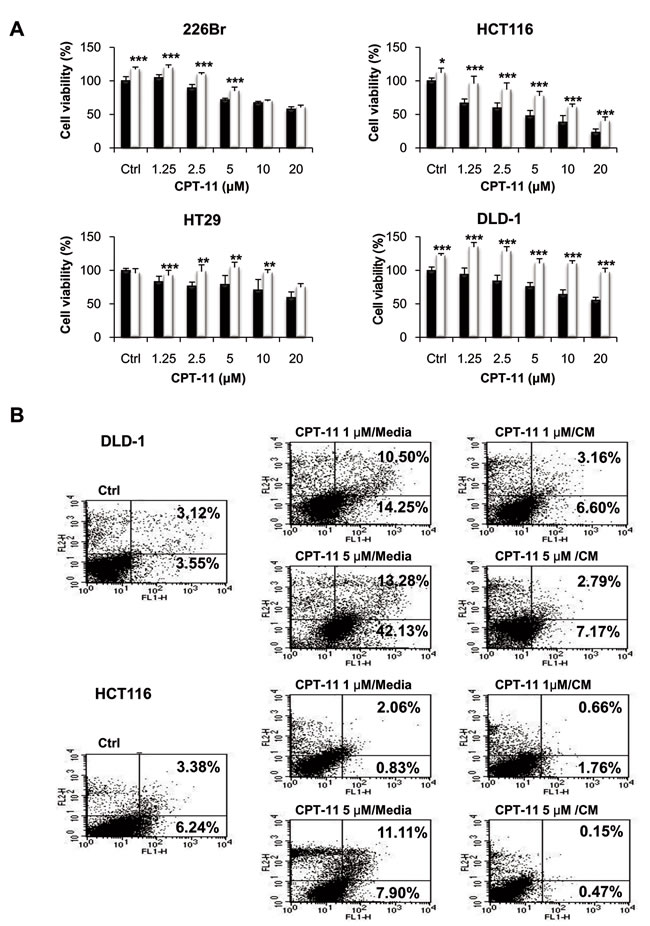
Figure 1: Conditioned media (CM) from fibroblasts enhances CPT-11 resistance in cancer cells. A. Human lung and colorectal cancer cells were plated into 96-well plates at a density of 3 x 103 cells/well on day 0. Under serum free culture media (black bar), cells were incubated with CCD-18co colonic fibroblast CM (white bar) and/or CPT-11 for 48 h. Inhibition of cell proliferation was determined by MTT assay. Significant differences were evaluated using an unpaired two-tailed Student’s t-test. (Error bars denote the standard deviation [SD]) (*p < 0.05, **p < 0.01 and ***p < 0.001). B. Induction of apoptosis in colorectal cancer cells. Cells were stained with PI and Annexin V-FITC, followed by flow cytometry analysis. Cells were treated with CPT-11 under serum free media or CCD-18co colonic fibroblast CM. The apoptotic cells were determined by Annexin V-FITC positive staining.
HGF derived from fibroblasts activates c-Met receptor in cancer cells
To determine whether fibroblast-derived HGF is responsible for CPT-11 resistance, we measured the amount of HGF in the CM from fibroblasts. HGF is not expressed in epithelial cells and therefore c-Met receptor requires HGF production by surrounding stromal cells for ligand-dependent activation [37]. As shown in Figure 2A, the ELISA assay demonstrated that HGF production is only shown in CM from fibroblast. The c-Met receptor tyrosine kinase activation induces pleiotropic biological effects in a wide variety of cells, including mitogenic, motogenic, morphogenic, and anti-apoptotic activities [10, 38]. To determine whether fibroblast-derived HGF activates the c-Met receptor resulting in CPT-11 resistance, we treated cells with CM from fibroblasts. We detected c-Met activation by treatment of fibroblasts-derived CM (Figure 2B). To mimic the tumor microenvironment, we performed co-culture experiments with fibroblasts and cancer cells. We investigated whether fibroblasts in co-culture adopted compensatory mechanisms such as the activation of the c-Met receptor and increased resistance to CPT-11 by cancer cells. As shown in Figure 2C and 2D, co-culture with fibroblasts induced resistance to CPT-11 and activated the c-Met receptor in cancer cells. To determine whether HGF is directly implicated in the activation of c-Met and the resistance to CPT-11, we knocked-down HGF by siRNA and measured cell viability in the presence of CPT-11. The HGF siRNA significantly suppressed HGF expression in CCD-18co cells (Figure 2E). As expected, CM from HGF siRNA treated cells failed to rescue cancer cells from the apoptosis by CPT-11 (Figure 2F). These results confirm the importance of fibroblast-derived HGF in CPT-11 resistance of cancer cells and indicate that HGF might be a therapeutic target for overcoming resistance to CPT-11.
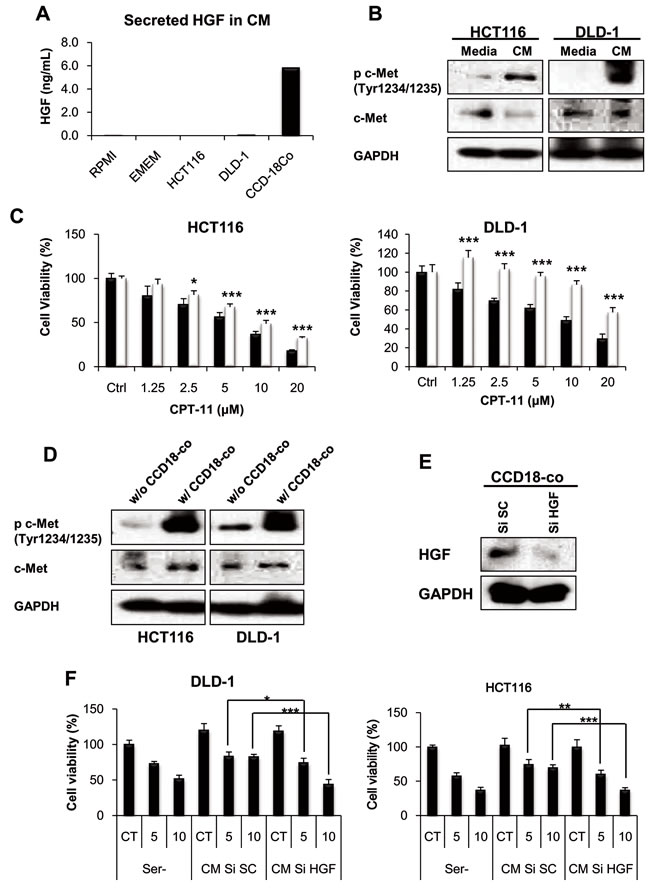
Figure 2: fibroblast-derived HGF activates c-MET receptor and induces CPT-11 resistance in colorectal cancer cell. A. HGF secreted by cancer cells (HCT-116 and DLD-1) and colonic fibroblasts (CCD-18co) were measured. Cells were cultured with serum free medium for 24 h and HGF concentrations were determined by ELISA. B. CM from fibroblast activates c-MET receptors. HCT-116 and DLD-1 cells were cultured with serum free media or CCD-18co CM for 1 h. Cells were collected, and the indicated proteins were detected by western blotting. C. Colonic fibroblast cells promote CPT-11 resistance of colorectal cancer cells (HCT-116 and DLD-1). Cancer cells were cultured with (white bar) or without (black bar) CCD-18co cells, in the presence or absence of CPT-11 (1.25-20 μM) for 48 h, and inhibition of cell proliferation was determined by MTT assay. Significant differences were evaluated using an unpaired two-tailed Student’s t-test. (Error bars denote the standard deviation [SD]) (*p < 0.05 and ***p < 0.001). D. Co-culture with colonic fibroblast CCD-18co cells increases c-MET receptor activation in colorectal cancer cells. HCT-116 and DLD-1 cells were co-cultured with CCD-18co cells for 24 h. Lysates were analyzed for c-MET activation by western blotting. E. Inhibition of HGF production from fibroblast by transfection with HGF siRNA. Colonic fibroblast cells were transfected with 10 nM HGF siRNA or scramble siRNA. After transfection, cells were collected and lysates were submitted to Western blotting to quantify HGF. F. HCT-116 and DLD-1 cells were cultured with CM from HGF siRNA transfected fibroblast for 48 h in the presence or absent of CPT-11. Cell viability was determined by MTT assay. Significant differences were evaluated using an unpaired two-tailed Student’s t-test. (Error bars denote the standard deviation [SD]) (*p < 0.05, **p < 0.01 and ***p < 0.001).
Targeting of fibroblast-derived HGF abrogates HGF stimulated CPT-11 resistance in colorectal cancer cells
To test whether HGF directly contributes to the effect on overcoming CPT-11 resistance in cancer cells, anti-HGF antibody was added to the CM to neutralize the HGF activity. The viability of cancer cells in the CM from fibroblasts was significantly decreased by the addition of 200 ng/ml of the anti-HGF antibody (Figure 3A). To determine the inhibitory effect of anti-HGF antibody in conditions mimicking a tumor microenvironment, co-cultures of fibroblasts and cancer cells were subjected to MTT assay (Figure 3B). As shown in Figure 3B, neutralization of fibroblast-derived HGF maintained the reduction in cell viability induced by CPT-11 indicating that HGF targeting significantly enhances CPT-11 stimulated anti-cancer activity. Taken together, our findings suggest that the fibroblast-derived HGF may play an important role in cancer cell survival and that the interference with its effect in tumor cells may represent a novel strategy for the treatment of colorectal cancers.
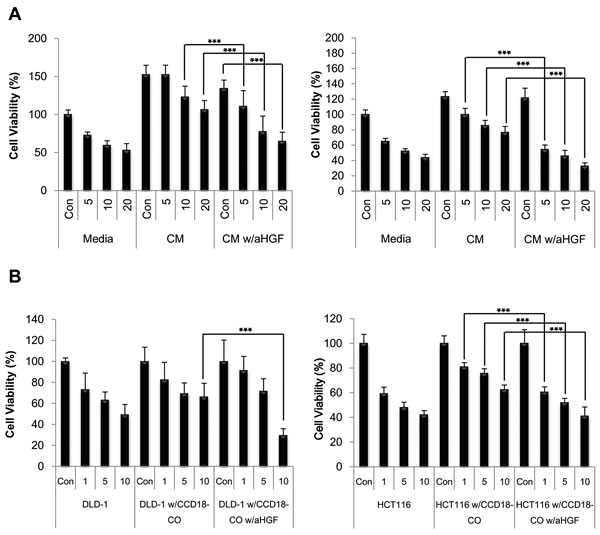
Figure 3: Humanized anti-HGF antibodies attenuated HGF activated c-Met signaling pathway and enhanced apoptotic cell death induced by CPT-11 in colorectal cancer cells. A. HCT-116 (right panel) and DLD-1 (left panel) cells were treated with or without CPT-11 and/or CCD-18co CM in the presence or absence of humanized anti-HGF antibody (200 ng/ml) for 48 h. B. HCT-116 (right panel) and DLD-1(left panel) cells were cultured with CCD-18co cells with or without CPT-11 in the presence or absence of humanized anti-HGF antibody (200 ng/ml) for 48 h. The inhibition of cell proliferation was determined by MTT assay. Significant differences were evaluated using an unpaired two-tailed Student’s t-test. (Error bars denote the standard deviation [SD]) (***p < 0.001).
Humanized anti-HGF antibody attenuated HGF activated c-Met signaling pathway and enhanced apoptotic cell death induced by CPT-11 in colorectal cancer cells
To examine the effects of anti-HGF antibody on cell signaling and apoptosis in cancer cells, we performed Western blot analysis. Anti-HGF antibody (200 ng/ml) effectively suppressed c-Met, AKT, and ERK activation induced by recombinant HGF (50 ng/ml) in HCT116 cancer cells (Figure 4A). Anti-HGF antibody (200 ng/ml) enhanced cleavage of PARP and caspase-3 which are apoptotic molecules induced by CPT-11 (Figure 4B upper panel). Anti-HGF antibody also enhanced cleavage of PARP and caspase-3 induced by CPT-11 in HCT116 cells co-cultured with CCD-18C human colon fibroblast cells (Figure 4B, lower panel). These results indicate that HGF-induced CPT-11 resistance is activated by HGF/c-Met signaling and is abrogated by treatment with anti-HGF antibody. Furthermore, we examined whether anti-HGF antibody augmented apoptotic cell death inhibited by recombinant HGF. We found that CPT-11 induced significant apoptotic cell death, recombinant HGF inhibited that apoptosis, and anti-HGF antibody reversed such inhibition. For example when cells were treated with CPT-11 alone, 5 μM of CPT-11 increased apoptotic cell death in HCT116 and DLD-1 cells by 67.75 % and 39.73 %, respectively. When cells were treated with recombinant HGF after treatment with 5 μM CPT-11, apoptotic activity was suppressed by 32.55 % and 10.19 % in HCT116 and DLD-1, respectively. The rescue effect stimulated by recombinant HGF was attenuated by anti-HGF antibody (200 ng/ml) (43.85 % and 20.52 % in HCT116 and DLD-1, respectively). These results indicated that CPT-11 resistance is induced by fibroblast-derived HGF/c-Met signaling and is attenuated by treatment with anti-HGF antibody in colorectal cancer cells.
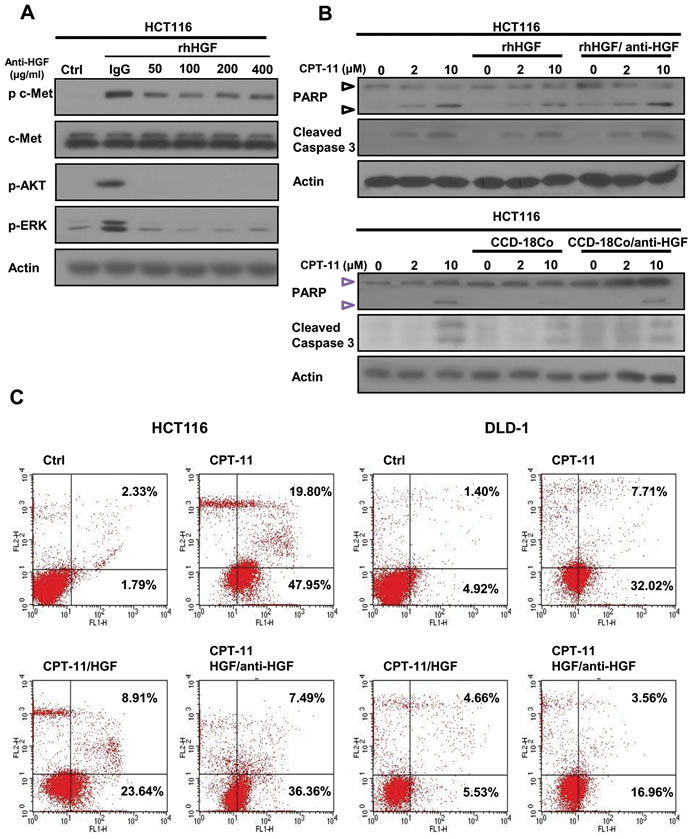
Figure 4: Humanized anti-HGF antibodies inhibit HGF-activated c-MET signaling pathways and enhanced apoptotic cell death induced by CPT-11. A. Humanized anti-HGF antibody inhibited c-MET signaling pathways. HCT-116 cells were treated with or without recombinant HGF (50 ng/ml) in the presence or absence of humanized anti-HGF antibody for 24 h. Cells were lysed, and the indicated proteins were detected by western blotting. B. Protein lysates were collected and subjected to western blotting to detect pro-apoptotic molecules such as cleaved PARP and cleaved caspase-3. C. Cells were stained with PI and Annexin V-FITC followed by flow cytometry analysis. The apoptotic cells were determined by Annexin V-FITC positive staining.
Fibroblast-derived HGF induces CPT-11 resistance in cancer cells and anti-HGF antibody restores the sensitivity of tumor growth to CPT-11 in vivo
To examine the possible induction of CPT-11 resistance by fibroblast-derived HGF and the efficacy of combined treatment with anti-HGF antibody and CPT-11 on colorectal cancer in vivo, we generated xenografts by inoculating HCT116 human colorectal cancer cells, with or without fibroblast CCD-18co cells, into NOD/SCID mice subcutaneously. We found that the mice injected with HCT116 plus CCD-18co cells had tumors that grew significantly faster than those in mice injected with HCT116 cells alone. CPT-11 treatment suppressed the growth of tumors in mice grafted with HCT116 plus CCD-18co cells. Interestingly, the combination of CPT-11 and anti-HGF antibody treatment induced the strongest suppression of tumors. The in vivo tumor growth analysis showed that injecting mice with HCT116 and CCD-18co cells led to significantly faster tumor growth as compared with only injecting HCT116 cells (2653.04 ± 396.62 mm3 versus 1775.46 ± 257.53 mm3 p < 0.001). Marginal suppression was observed with CPT-11 treatment (1775.46 ± 257.53 mm3 versus 850.38 ± 183.15 mm3, p < 0.05). The combination treatment with CPT-11 plus anti-HGF significantly inhibited tumor growth as compared to the control group, HCT-116 plus CCD-18co group and the HCT-116 plus CCD-18co treated with CPT-11 groups (1775.46 ± 257.53 mm3 versus 467.57 ± 81.90 mm3, p < 0.001; 2653.04 ± 396.62 mm3 versus 467.57 ± 81.90 mm3, p < 0.001; 850.38 ± 183.15 mm3 versus 467.57 ± 81.90 mm3, p < 0.05, respectively). At the end of the experiment, tumor weight in the control, HCT-116 plus CCD-18co, HCT-116 plus CCD-18co with CPT-11, and HCT-116 plus CCD-18co with CPT-11 plus anti-HGF groups were 1.15 ± 0.27 g, 1.88 ± 0.31 g, 0.61 ± 0.20 g and 0.35 ± 0.15 g, respectively. These results suggest that anti-HGF antibody abrogates fibroblast-derived HGF-induced CPT-11 resistance in vivo (Figure 5A and 5B). To determine whether anti-HGF antibody decrease HGF/c-Met signaling, we examined the expression of phospho-c-MET by immunofluorescence and western blotting (Figure 5C and 5D). Our data revealed that c-Met signaling was suppressed in the anti-HGF antibody treatment group when compared to HCT-116 plus CCD-18co and HCT-116 plus CCD-18co with CPT-11 groups. In addition, a fibroblast marker (ER-TR7) was detected in HCT116 plus CCD-18co tumors. Whereas, the tumors in mice injected with HCT116 alone did not produce a detectable level of ER-TR7 (Figure 5C). As expected, tumors in the anti-HGF antibody and CPT-11 treatment group contained a significantly higher number of TUNEL positive and cleaved caspase-3 positive cells as compared with HCT-116 plus CCD-18co and HCT-116 plus CCD-18co with CPT-11 groups (Figure 5D and 5E). Taken together, these results suggests that cancer associated fibroblasts could change the nature of cancer cells and generating resists to chemotherapy.
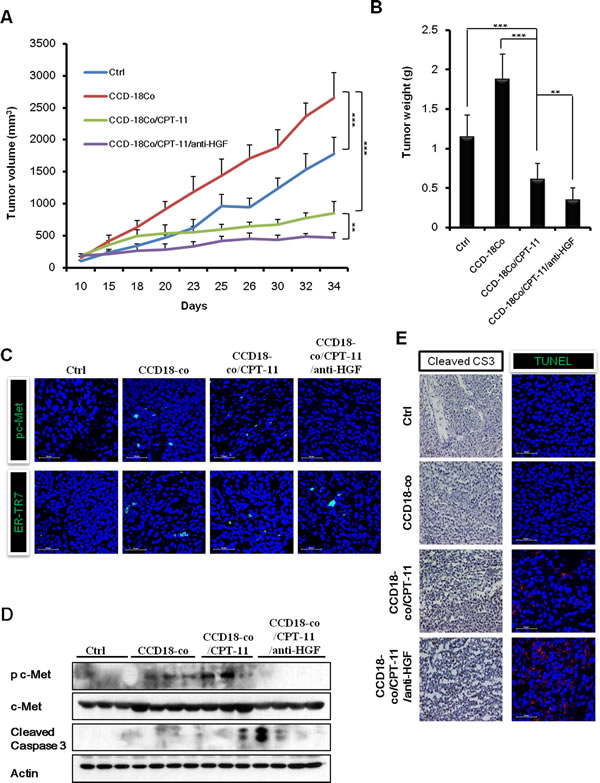
Figure 5: Combination of humanized anti-HGF antibody and CPT-11 inhibits tumor growth in vivo HCT-116 cells with or without CCD-18co cells were inoculated subcutaneously into NOD/SCID mice. Mice in the control group of received PBS. Mice in the treatment groups received CPT-11 (10 mg/kg; 3 times/wk; intraperitoneally) with or without humanized anti-HGF antibody (10 mg/kg; 2 times/wk; intraperitoneally). A. Tumor volume was measured every 3 or 4 days and calculated. B. Average tumor weights were measured on day 34. The differences were statically significant between the combination treatments versus single treatment. Significant differences were evaluated using an unpaired two-tailed Student’s t-test. (Error bars denote the standard deviation [SD]) (**p < 0.001 and ***p < 0.001). All four treatment groups were well tolerated, as total mouse body weights remained unchanged (<10 % fluctuation). C. Immunofluoresence images of phospho-c-Met receptors and a fibroblast marker (ER-TR7) in tumors. Co-staining with DAPI was done to show nuclei. Frozen tumor specimens were subjected to immunofluorescence analysis using anti-phospho-c-Met antibody (green; upper panel) or anti-ER-TR7 antibody (green; lower panel). D. Western blot analysis for cleaved caspases-3 and phospho-c-Met receptors. E. Immunohistochemistry and TUNEL assays were performed to observe apoptotic cell death in tumors. Immunohistochemical analysis in tumors was performed using an antibody against cleaved caspase-3 (left panel). Tumor sections were stained with hematoxylin for counterstaining. The red fluorescent staining indicates positive apoptotic cells and DAPI staining was used as a nuclear counterstaining (right panel).
Discussion
Irinotecan (CTP-11) is a semi-synthetic analog of camptothecin, which was originally isolated from Camptotheca acuminate [39]. CTP-11 inhibits topoisomerase-1 (Topo-I) activity by trapping Topo-I-DNA cleavage complexes, leading to lethal replication-mediated double strand breaks [40]. CPT-11 is used as a first- and second-line chemotherapy for advanced or recurrent colorectal cancer and has shown activity in patients with advanced colorectal cancer resistant to leucovorin (LV) and 5-fluorouracil (5-FU) [41, 42]. Although treatment of advanced CRC patients with CPT-11 as a single agent has shown response rates of approximately 25 %, these rates can reach 60 % when used in combination with other agents [43, 44]. However, the efficacy of CPT-11 is strongly limited by the development of drug resistance. Classically, drug resistance may be due to modulated drug influx/efflux, gene amplification and subsequent overexpression of the target molecule or alteration in drug target specificity drug detoxification by increased levels of drug target, enhanced DNA repair in the case of DNA-damaging drugs or NF-kB activation [39]. Recent studies have proposed that microenvironment could be associated with chemoresistance [45]. Stromal cells could potentially induce chemoresistance acquisition in tumor cells, including cell-cell and cell-matrix interactions, local release of soluble factors and so on [46-48]. We hypothesized that fibroblasts generate soluble factors that could stimulate resistance to chemotherapy. The fibroblast cell line CCD-18co conditioned medium can support the survival of colorectal cancer cells against CPT-11. Using co-culture experiments in vitro and in vivo, we found that a minority of CCD-18co cells could rescue the colorectal cancer cells from CPT-11 treatment.
These results indicate that a novel strategy, targeting the crosstalk between tumor cells and stromal fibroblasts, may be important for circumventing the drug resistance of colorectal cancers. By using colonic fibroblast cell line CCD-18co and chemotherapy drug CPT-11, we demonstrate that fibroblasts play a functional role in chemoresistance of colorectal cancer cells presumably through fibroblast-derived HGF in tumors via c-MET signaling. Our results revealed that CPT-11 resistance of colorectal cancer cells were enhanced upon exposure to CCD-18co CM and co-culture with CCD-18co cells in vitro. Furthermore, CCD-18co cells produced and secreted HGF in CM and c-Met (receptor tyrosine kinase of HGF) was activated upon exposure to CCD-18co CM. HGF neutralizing antibody effectively reduces the activation of HGF/c-MET signaling, which blocks its ability to promote CPT-11 resistance in colorectal cancer cells. Our finding indicates that HGF is involved in CRC chemoresistance. The administration of anti-HGF antibody significantly reverses the chemoresistance effect of fibroblast-derived HGF in vivo and in vitro.
Multiple biological actions of HGF, mediated via c-Met tyrosine phosphorylation, are outlined. These activities depend on downstream adaptor molecules recruited by c-Met tyrosine-phosphorylated multi-docking sites [10]. These phosphotyrosines stimulate high-affinity interactions with various src homology region 2-containing cytoplasmic effectors, including Gab1, PI3-kinase, Shc, and Grb2. We demonstrate here that HGF activates Akt and ERK signaling pathways which inhibited apoptosis processing in CRC. Different studies suggest that HGF triggers anti-apoptotic signals [49-51]. The present study demonstrates that fibroblast-derived HGF can protect cells from apoptosis by means of both Akt and ERK signaling. Our data indicate that fibroblast-derived HGF rescues cells from apoptosis and that the anti-HGF antibody sensitizes cells to apoptosis and abolishes HGF protection.
When human colorectal cancer cells were inoculated with colonic fibroblast cells in immunodeficiency mice, they enhanced drug resistance to CPT-11. This finding is consistent with other reports that have shown that acquired resistance to chemotherapy reagents is a problem [52-54]. Indeed, our research has shown that anti-HGF antibody can reverse the fibroblast-derived resistance to CPT-11. This phenomenon, besides highlighting the importance of the tumor microenvironment, further confirms that fibroblast-derived HGF/c-MET signaling is involved in resistance to chemotherapy reagents. In our in vivo models, the combination of CPT-11 with anti-HGF antibody showed a potent anti-cancer effect on human colorectal cancer, as evidenced by the strong inhibition of tumor growth and interference with c-MET signal transduction. We also demonstrated that combination treatment was significantly associated with the induction of apoptotic cell death. Consistently, our research suggests that the combination treatment of CPT-11 with anti-HGF could interfere with the process of acquired. In summary, HGF appears to play a substantial role in the complex mechanisms involved in the induction of chemoresistance of colorectal cancer by fibroblasts.
In summary, we identified fibroblast-derived HGF activated CPT-11 resistance through c-MET signaling pathways. Our findings point to the micro-environment as an important factor but this does not preclude the likely involvement of other growth factors and/or cytokines. Moreover, our results suggest that such resistance mechanisms can be uncovered through the systematic dissection of interactions between tumors and their micro-environment. Future studies should therefore seek to identify such resistance mechanisms for all of the drugs that are in development, potentially leading to mechanism-based combination regimens such as the CPT-11 and c-MET inhibitor combination proposed here.
Materials and methods
Cell culture and reagents
The human colorectal cancer cell lines, HCT116, HT29 and DLD-1 and lung cancer cell line 226Br were cultured in RPMI1640 (Welgene, Seoul, Korea) containing 10% fetal bovine serum (FBS; Welgene) and antibiotics (100 mg/L penicillin and 100 mg/L streptomycin; GIBCO-BRL Life Technologies; Gaithersburg, MD, USA). Human colonic fibroblasts (CCD-18co) were cultured in DMEM (Welgene, Seoul, Korea) containing 10 % FBS and antibiotics Cells were incubated in a humidified thermostat under 5 % CO2 at 37 °C. A predesigned siRNA targeting HGF and scramble siRNA were purchased from Shanghai Genepharma (Shanghai, China). Recombinant Human HGF was purchased from R&D system (Minneapolis, MN, USA). Humanized anti-HGF antibody was kindly provided by YooYoung pharmaceutical Co. Ltd (Seoul, Korea) and iBIO Inc (Seoul, Korea). Irinotecan (CPT-11) was obtained from Sigma Aldrich (St. Louis, MO, USA). Antibodies to c-MET, phosphorylated-c-Met, phosphorylated-ERK, phosphorylated-AKT, PARP, and cleaved Caspase-3 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody against HGF, actin and fibroblast marker (ER-TR7) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Conditioned medium preparation
Conditioned medium (CM) were generated from 70-80 % confluent CCD-18co human colonic fibroblasts cultured with serum free RPMI1640 for 48h. All cellular debris was removed by filtration (0.4 μM). The level of human HGF in CM was determined by using Human HGF ELISA KIT (R&D system, Minneapolis, MN, USA) according to the manufacturer’s protocol. For HGF knockdown studies, early-passage CCD-18co cells were transfected with 10 nM siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. Conditioned medium from HGF siRNA transfected CCD-18co cells were used to assess colorectal cancer cell proliferation assays.
Assay for cell proliferation
Cells were seeded into 96-well plates (SPL, Seoul. Korea) and cultured 24 h before exposure for 48 h to various concentrations of CPT-11, recombinant HGF, or anti-HGF antibody, as indicated. Following this, 20 μl of 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) solution (5 mg/ml) was added to each well and the plate was incubated at 37 °C for 2 h. The medium was then discarded, and the purple-blue MTT formazan precipitate was dissolved in 100 μl dimethyl sulfoxide (DMSO). The plates were mixed and absorbance at 570 nm was measured by an ELISA reader (Bio-Rad Laboratories, Hercules, CA, USA).
Western blotting
Western blotting was performed, as described previously [55]. Briefly, whole-cell lysates were prepared in a modified RIPA buffer containing proteinase inhibitors and phosphatase inhibitors, as described elsewhere [56]. Equivalent amounts of protein (20-80 µg) were loaded onto 10 % or 12 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred by blotting to polyvinylidene fluoride membranes. The blot was incubated with primary antibodies for over nigh at 4 °C and then Horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperrature. The protein-antibody complexes were detected using enhanced chemiluminescence (Amersham, Arlington Heights, IL, USA), according to the manufacturer’s recommended protocol.
Apoptosis analysis
Apoptosis was determined by FACS analysis using propidium iodide (PI) and Annexin-V-FITC (Santa Cruz, CA, USA). Briefly, cancer cells were plated in 6 well plates with or without CM derived from CCD-18co cells. Cells were treated with different concentrations of CPT-11 with or without anti-HGF antibody the next day. After 48 h, cells were collected, re-suspended in binding buffer, and incubated with PI and Annexin-V-FITC for 10 min at 4 °C. Cells were then analyzed by flow cytometry, using a FITC signal detector with an excitation wavelength of 488 nm and an emission of 530 nm (to detect Annexin-V -FITC), and a PI signal detector with an excitation wavelength of 496 nm and an emission of 615 nm (to detect PI). We defined “apoptotic cell death” as the population of cells that were positive for annexin-V-FITC staining.
In vivo xenograft models
For xenograft model, 5-6 week-old NOD/SCID mice (NOD/LtSz-Prkdcscid/J) were purchased from KRIBB (Cheongwon-gun, Korea). HCT116 human colorectal cancer cells 5 x 106 in 100ul of PBS were injected subcutaneously (s.c.) in the left flank. HCT116 human colorectal cancer cells 4.5 x 106 and 5 X 105 CCD-18co cells were injected in the right flank. Tumor volumes were calculated as length × width2 × 0.5. Mice were weighed and tagged and when the tumor reached the standard volume of approximately 200 mm3, the mice was randomly assigned to four experimental groups (n = 5): Control; CPT-11treatment; humanized anti-HGF Ab treatment; CPT-11/humanized anti-HGF Ab combination treatment. CPT-11 (10 mg/kg, three times a week) and humanized anti-HGF (40 mg/kg/day, twice a week) were administered intra-peritoneally for 3 weeks. Tumors were measured twice weekly using digital calipers. All animal experiments were approved by the Institutional Animal Care and Use Committee of the National Cancer Center.
Immunohistochemistry and TUNEL staining
The tumors were fixed in 10 % neutral buffered formalin and embedded in paraffin. Immunohistochemical assays and terminal deoxynucleotidyl transferase-mediated UTP end-labeling (TUNEL) staining was done as previously described [57]. For immunohistochemistry, sections were incubated with cleaved caspases-3 antibodies overnight at 4 °C. Antibody detection was performed using the EnVision detection system (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s protocol. Another part of the tumors was sectioned for TUNEL staining for the measurement of apoptosis. TUNEL assay was performed as previously described [57]. For apoptosis analysis, we counted all nuclei in the field as well as all TUNEL-positive cells in three to five randomly selected fields of view at a magnification of ×200.
Statistical analysis
All experiments were done in duplicate or triplicate. A two-tailed Student’s t-test was used for statistical analysis of comparative data using Microsoft Excel software. Values of p < 0.05 were considered statically significant and indicated by asterisks in the figures. For graphical representation of data, y-axis error bars indicate the standard deviation (s.d.) of the data for each point marked on the graph.
Acknowledgments
This work was supported by a grant from the Global Core Research Center (GCRC) grant (No. 2011-0030678) from NRF, MEST, Republic of Korea
Conflicts of interest
The authors declare that they have no conflict of interest.
References
1. Yoshida S, Harada T, Iwabe T, Taniguchi F, Fujii A, Sakamoto Y, Yamauchi N, Shiota G and Terakawa N. Induction of hepatocyte growth factor in stromal cells by tumor-derived basic fibroblast growth factor enhances growth and invasion of endometrial cancer. The Journal of clinical endocrinology and metabolism. 2002; 87:2376-2383.
2. Felix AS, Edwards RP, Stone RA, Chivukula M, Parwani AV, Bowser R, Linkov F and Weissfeld JL. Associations between hepatocyte growth factor, c-Met, and basic fibroblast growth factor and survival in endometrial cancer patients. British journal of cancer. 2012; 106:2004-2009.
3. Felix AS, Stone RA, Chivukula M, Bowser R, Parwani AV, Linkov F, Edwards RP and Weissfeld JL. Survival outcomes in endometrial cancer patients are associated with CXCL12 and estrogen receptor expression. International journal of cancer Journal international du cancer. 2012; 131:E114-121.
4. Nakamura T, Teramoto H, Tomita Y and Ichihara A. L-proline is an essential amino acid for hepatocyte growth in culture. Biochemical and biophysical research communications. 1984; 122:884-891.
5. Luetteke NC and Michalopoulos GK. Partial purification and characterization of a hepatocyte growth factor produced by rat hepatocellular carcinoma cells. Cancer research. 1985; 45:6331-6337.
6. Funakoshi H and Nakamura T. Hepatocyte growth factor: from diagnosis to clinical applications. Clinica chimica acta; international journal of clinical chemistry. 2003; 327:1-23.
7. Sengupta S, Gherardi E, Sellers LA, Wood JM, Sasisekharan R and Fan TP. Hepatocyte growth factor/scatter factor can induce angiogenesis independently of vascular endothelial growth factor. Arteriosclerosis, thrombosis, and vascular biology. 2003; 23:69-75.
8. Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT and Day ML. Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. The American journal of pathology. 1995; 147:386-396.
9. Stella MC, Trusolino L and Comoglio PM. The Slit/Robo system suppresses hepatocyte growth factor-dependent invasion and morphogenesis. Molecular biology of the cell. 2009; 20:642-657.
10. Birchmeier C, Birchmeier W, Gherardi E and Vande Woude GF. Met, metastasis, motility and more. Nature reviews Molecular cell biology. 2003; 4:915-925.
11. Bissell MJ, Radisky DC, Rizki A, Weaver VM and Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation; research in biological diversity. 2002; 70:537-546.
12. Tripathi M, Billet S and Bhowmick NA. Understanding the role of stromal fibroblasts in cancer progression. Cell adhesion & migration. 2012; 6:231-235.
13. Ferrucci A, Moschetta M, Frassanito MA, Berardi S, Catacchio I, Ria R, Racanelli V, Caivano A, Solimando AG, Vergara D, Maffia M, Latorre D, Rizzello A, Zito A, Ditonno P, Maiorano E, et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014; 20:5796-5807.
14. Castells M, Thibault B, Mery E, Golzio M, Pasquet M, Hennebelle I, Bourin P, Mirshahi M, Delord JP, Querleu D and Couderc B. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer letters. 2012; 326:59-68.
15. Tlsty TD and Coussens LM. Tumor stroma and regulation of cancer development. Annual review of pathology. 2006; 1:119-150.
16. Trimmer C, Sotgia F, Whitaker-Menezes D, Balliet RM, Eaton G, Martinez-Outschoorn UE, Pavlides S, Howell A, Iozzo RV, Pestell RG, Scherer PE, Capozza F and Lisanti MP. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: a new genetically tractable model for human cancer associated fibroblasts. Cancer biology & therapy. 2011; 11:383-394.
17. Erez N, Truitt M, Olson P, Arron ST and Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010; 17:135-147.
18. De Boeck A, Hendrix A, Maynard D, Van Bockstal M, Daniels A, Pauwels P, Gespach C, Bracke M and De Wever O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics. 2013; 13:379-388.
19. Mao Y, Keller ET, Garfield DH, Shen K and Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews. 2013; 32:303-315.
20. Sugimoto H, Mundel TM, Kieran MW and Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer biology & therapy. 2006; 5:1640-1646.
21. Bhowmick NA, Neilson EG and Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004; 432:332-337.
22. Li Q, Wang W, Yamada T, Matsumoto K, Sakai K, Bando Y, Uehara H, Nishioka Y, Sone S, Iwakiri S, Itoi K, Utsugi T, Yasumoto K and Yano S. Pleural mesothelioma instigates tumor-associated fibroblasts to promote progression via a malignant cytokine network. The American journal of pathology. 2011; 179:1483-1493.
23. Yan XL, Fu CJ, Chen L, Qin JH, Zeng Q, Yuan HF, Nan X, Chen HX, Zhou JN, Lin YL, Zhang XM, Yu CZ, Yue W and Pei XT. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast cancer research and treatment. 2012; 132:153-164.
24. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M and Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PloS one. 2009; 4:e4992.
25. Fu L, Zhang C, Zhang LY, Dong SS, Lu LH, Chen J, Dai Y, Li Y, Kong KL, Kwong DL and Guan XY. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011; 60:1635-1643.
26. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC and Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer research. 2001; 61:5911-5918.
27. Turner N and Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010; 10:116-129.
28. Olson OC and Joyce JA. Microenvironment-mediated resistance to anticancer therapies. Cell research. 2013; 23:179-181.
29. Engelman JA and Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Current opinion in genetics & development. 2008; 18:73-79.
30. Natan S, Tsarfaty G, Horev J, Haklai R, Kloog Y and Tsarfaty I. Interplay Between HGF/SF-Met-Ras Signaling, Tumor Metabolism and Blood Flow as a Potential Target for Breast Cancer Therapy. Oncoscience. 2014; 1:30-38.
31. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012; 487:505-509.
32. Hack SP, Bruey JM and Koeppen H. HGF/MET-directed therapeutics in gastroesophageal cancer: a review of clinical and biomarker development. Oncotarget. 2014; 5:2866-2880.
33. Mariani M, McHugh M, Petrillo M, Sieber S, He S, Andreoli M, Wu Z, Fiedler P, Scambia G, Shahabi S and Ferlini C. HGF/c-Met axis drives cancer aggressiveness in the neo-adjuvant setting of ovarian cancer. Oncotarget. 2014; 5:4855-4867.
34. Joyce JA and Pollard JW. Microenvironmental regulation of metastasis. Nature reviews Cancer. 2009; 9:239-252.
35. McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC and Mitsiades CS. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nature medicine. 2010; 16:483-489.
36. Shekhar MP, Santner S, Carolin KA and Tait L. Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. The American journal of pathology. 2007; 170:1546-1560.
37. Sonnenberg E, Meyer D, Weidner KM and Birchmeier C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. The Journal of cell biology. 1993; 123:223-235.
38. Matsumoto K and Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. International journal of cancer Journal international du cancer. 2006; 119:477-483.
39. Xu Y and Villalona-Calero MA. Irinotecan: mechanisms of tumor resistance and novel strategies for modulating its activity. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2002; 13:1841-1851.
40. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nature reviews Cancer. 2006; 6:789-802.
41. Shimada Y, Yoshino M, Wakui A, Nakao I, Futatsuki K, Sakata Y, Kambe M, Taguchi T and Ogawa N. Phase II study of CPT-11, a new camptothecin derivative, in metastatic colorectal cancer. CPT-11 Gastrointestinal Cancer Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1993; 11:909-913.
42. Ikeguchi M, Arai Y, Maeta Y, Ashida K, Katano K and Wakatsuki T. Topoisomerase I expression in tumors as a biological marker for CPT-11 chemosensitivity in patients with colorectal cancer. Surgery today. 2011; 41:1196-1199.
43. Saridaki Z, Androulakis N, Vardakis N, Vamvakas L, Kabouraki E, Kalbakis K, Hatzidaki D, Voutsina A, Mavroudis D, Georgoulias V and Souglakos J. A triplet combination with irinotecan (CPT-11), oxaliplatin (LOHP), continuous infusion 5-fluorouracil and leucovorin (FOLFOXIRI) plus cetuximab as first-line treatment in KRAS wt, metastatic colorectal cancer: a pilot phase II trial. British journal of cancer. 2012; 107:1932-1937.
44. Kim S, Dobi E, Jary M, Monnien F, Curtit E, Nguyen T, Lakkis Z, Heyd B, Fratte S, Cleau D, Lamfichekh N, Nerich V, Guiu B, Demarchi M and Borg C. Bifractionated CPT-11 with LV5FU2 infusion (FOLFIRI-3) in combination with bevacizumab: clinical outcomes in first-line metastatic colorectal cancers according to plasma angiopoietin-2 levels. BMC cancer. 2013; 13:611.
45. Dayan D, Salo T, Salo S, Nyberg P, Nurmenniemi S, Costea DE and Vered M. Molecular crosstalk between cancer cells and tumor microenvironment components suggests potential targets for new therapeutic approaches in mobile tongue cancer. Cancer medicine. 2012; 1:128-140.
46. Castells M, Thibault B, Delord JP and Couderc B. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. International journal of molecular sciences. 2012; 13:9545-9571.
47. Blagosklonny MV. Rapalogs in cancer prevention: anti-aging or anticancer? Cancer biology & therapy. 2012; 13:1349-1354.
48. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, Blagosklonny MV and Lisanti MP. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. The American journal of pathology. 2012; 181:278-293.
49. Nakamura T, Matsumoto K, Kiritoshi A, Tano Y and Nakamura T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions. Cancer research. 1997; 57:3305-3313.
50. Uddin S, Hussain AR, Ahmed M, Al-Dayel F, Bu R, Bavi P and Al-Kuraya KS. Inhibition of c-MET is a potential therapeutic strategy for treatment of diffuse large B-cell lymphoma. Laboratory investigation; a journal of technical methods and pathology. 2010; 90:1346-1356.
51. Liu Y, Liu JH, Chai K, Tashiro S, Onodera S and Ikejima T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. The Journal of pharmacy and pharmacology. 2013; 65:1622-1642.
52. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M and Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. British journal of cancer. 2014; 110:469-478.
53. Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K and Boerner JL. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast cancer research : BCR. 2012; 14:R104.
54. Han W, Wang W, Mohammed KA and Su Y. Alpha-defensins increase lung fibroblast proliferation and collagen synthesis via the beta-catenin signaling pathway. The FEBS journal. 2009; 276:6603-6614.
55. Woo JK, Choi Y, Oh SH, Jeong JH, Choi DH, Seo HS and Kim CW. Mucin 1 enhances the tumor angiogenic response by activation of the AKT signaling pathway. Oncogene. 2012; 31:2187-2198.
56. Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T and Sessa WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circulation research. 2002; 90:866-873.
57. Oh SH, Woo JK, Yazici YD, Myers JN, Kim WY, Jin Q, Hong SS, Park HJ, Suh YG, Kim KW, Hong WK and Lee HY. Structural basis for depletion of heat shock protein 90 client proteins by deguelin. Journal of the National Cancer Institute. 2007; 99:949-961.