
INTRODUCTION
The epidermal growth factor receptor variant III (EGFRvIII) is an oncogenic variant of EGFR that is exclusively expressed by malignant tumors. EGFRvIII causes constitutive phosphorylation of tyrosine kinases and enhances the malignancy of gliomas [1-5]. The expression of EGFRvIII within a cell is associated with survival, invasion, angiogenesis and resistance against radiation and chemotherapy [6-9]. EGFRvIII is, therefore, an attractive target for cancer immunotherapy [10-13].
Chimeric antigen receptor (CAR)-based adoptive immunotherapy employs T lymphocytes that are genetically modified to express CARs that combine both specific targeting of antibodies and T-cells mediated immune responses, which enable T cells to target cell surface antigens independent of MHC restriction [14-16]. Typically, CARs are designed to consist of a single chain antibody fragment (scFv) that are fused to extracellular spacer and transmembrane domains, which can contain various combinations of cytoplasmic signaling moieties, such as CD3ζ, CD28, OX40 or 4-1BB [16-19]. The scFv are usually derived from monoclonal antibodies that are prepared to specifically direct against tumor-associated antigens (TAAs) expression on cancer cells membrane. Recently, a study reported a novel and universal CAR strategy that extends the recognition specificity potential of CAR-T cells by using a biotin-avidin system [20, 21]: First, the TAAs are pre-targeted with biotinylated molecules, such as monoclonal antibodies, peptides, ligands or enzyme substrates. Then, T lymphocytes expressing a CAR that contains avidin (avidin-CAR) instead of a scFv linking the intracellular T cell signaling domain are then adoptively transferred. In this study, we used this novel strategy to treat EGFRvIII expressing gliomas and evaluate the therapies progress through bioluminescent imaging: We first biotinylated a novel anti-EGFRvIII monoclonal antibody, 4G1 (biotin-4G1), and bioengineered the expression of avidin-CAR on T cells. Next, we pre-targeted xenograft EGFRvIII expressing gliomas with biotin-4G1 and then targeted biotin-4G1 with avidin-CAR-T cells. More importantly, in this study we firstly employed optical molecular imaging approach to evaluate the specificity of pre-target or target and determine the right time for T cells transfer, as well. We labeled biotin-4G1 with near-infrared dye to get biotin-4G1-dye and then injected it into EGFRvIII+ or EGFRvIII- gliomas to confirm the accumulation of biotin-4G1 in tumors and normal organs. The results indicated that our strategy offered a specific and efficient cytotoxicity against an EGFRvIII expressing gliomas, both in vitro and in vivo, and optical molecular imaging can facilitate the observation and evaluation of the therapy.
Results
Avidin-CAR was constructed and expressed on activated T cells
Lentiviral plasmid pITA-Avadin-CAR was constructed as shown in Figure 1A. Avidin-CAR encodes a fusion protein consisting of streptavidin, the hinge and transmembrane region of human CD8, the costimulatory signal transduction domain of CD28 and 4-1BB, and the intracellular signal domain of CD3ζ. After 12 d of lentiviral infection, avidin-CAR expression efficiency reached 100% and 93.5% as determined by B5F and anti-avidin antibody analysis, respectively (Figure 1B).
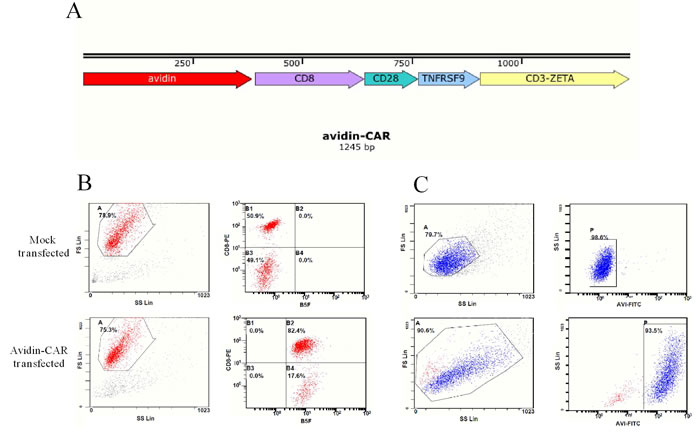
Figure 1: Plasmids construction and avidin expression on T cells. A. Schematic representations of pITA-Avadin-CAR. Avidin expression efficiency of mock or avidin-CAR infection was detected by flow cytometry with B5F B. or mouse anti-avidin IgG C..
Phenotypic and cytokine analysis
To determine the characteristics of T cells expressing avidin-CAR for adoptive transfer, immune-phenotypes of cells were determined after 12d of infection. Classic cytokine-induced killer (CIK) phenotypes were confirmed again in this study, which showed that 72.8% of the T cells expressed CD3+CD56+ [23] (Figure 2A). Furthermore, results showed that 77.1% of the population were CD8+CD3+ cells (cytotoxic T cells), whereas, CD3+CD4+ (helper T cells) and CD4+CD25+ [24] (regulatory T cells or activated CD4 T cells) cells only accounted for 9 and 0% of the population, respectively. This suggests that the cells would favor a cytotoxic function.
Cytokine release by avidin-CAR-T cells from No.1 samples was analyzed by Cytometric Bead Array. In comparison to the control, increased IFN-γ (2,961.1 vs. 24.4 pg/ml), IL-4 (45.3 vs. 7.7 pg/ml), IL-6 (48.5 vs. 4.9 pg/ml), IL-10 (21.1 vs. 6.8 pg/ml), TNF-ɑ (33.7 vs. 5.5 pg/ml) and IL-17 (162.1 vs. 43.4 pg/ml) were produced by the avidin-CAR-T cells, suggesting their activation was effective. Lower amounts of IL-2 (15,868.7 vs 21,120.4 pg/ml) were detected in the avidin-CAR-T cell culture, which may be the result IL-2 uptake by the activated T cells (Figure 2B). More sample testing results were summarized in Supplemental Table 1.
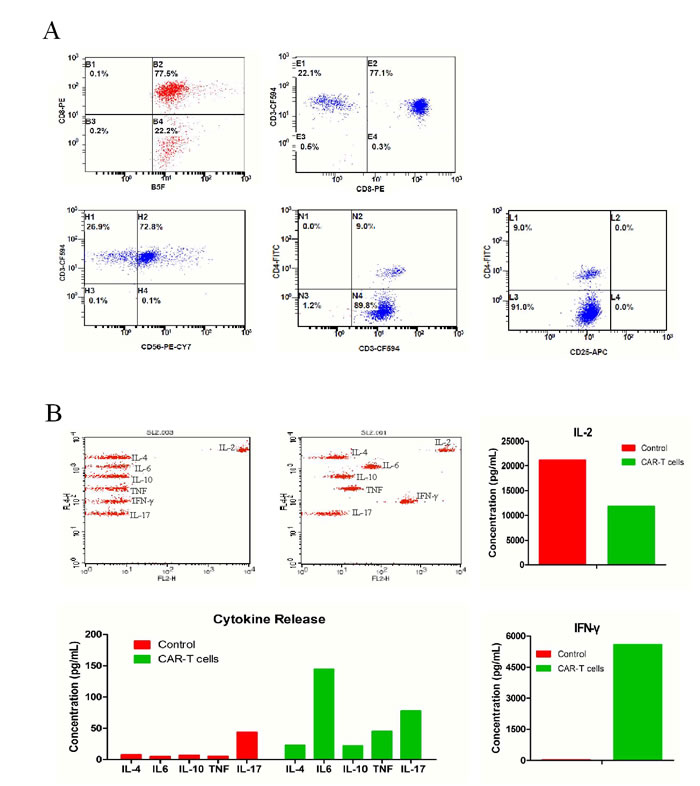
Figure 2: Phenotypes and cytokine release of avidin-CAR T cells. A. Expression of phenotypic markers including CD8, CD3, CD4, CD56, CD25 on avidin-CAR-T were determined by combinational flow cytometric analysis. B. Concentration of cytokines released by avidin-CAR T cells was determined by cytometric bead array (top middle panel); Cytokines concentration of non-activated avidin-CAR-T cells was also determined as control (top left panel).
In vitro determination of pre-target and re-target
Through labeling preparation and calculation, the moles biotin conjugated to per mole 4G1 are approximately equal to 7.2; and the moles dye conjugated to per mole biotin-4G1 are approximately equal to 3.5.
Results of western blotting (Figure 3A), flow cytometric analysis (Figure 3B), IFA and IHC (Figure 3C) determined that biotin-4G1 exclusively bound to EGFRvIII expressed by F98npEGFRvIII cells but not wild-type EGFR expressed by F98npEGFR cells (Figure 3A–3C). In vitro cell binding results showed that avidin-CAR-T cells targeted F98npEGFRvIII cells that were bound with biotin-4G1, whereas, few avidin-CAR-T cells could be observed on F98npEGFR cells pre-targeted with biotin-4G1 (Figure 4A).
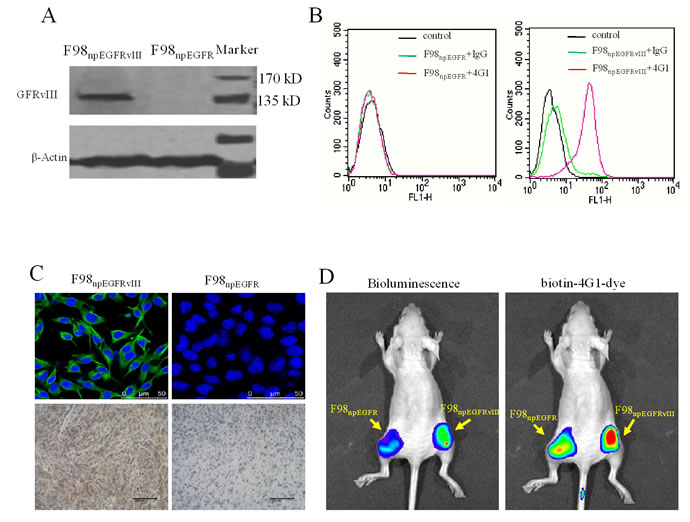
Figure 3: Biotinylated 4G1 exclusively recognizes with EGFRvIII. A. western-blotting, B. flow cytometry, C. IFA and IHC were undertaken for EGFRvIII detection. Biotin-4G1 was used as primary antibody. D. Biotin-4G1-dye as an optical molecular probe was intravenous injected and specifically uptaken by EGFRvIII+ xenograft tumor. Left panel: bioluminescent imaging after luciferin intraperitoneal injection; right panel: optical imaging at Ex/Em: 675/720 nm for biotin-4G1-dye detection.
Optical imaging evaluation of pre-target and re-target
As shown in Figure 3D, biotin-4G1-dye did not effectively bind to F98npEGFR tumor, confirming that biotin-4G1 specifically pre-targets to EGFRvIII+ tumor in an antigen-dependent manner in vivo. Additionally, the optical imaging of streptavidin-Cy7 uptake in EGFRvIII tumors demonstrated the stability of biotin-4G1 and indicated that avidin-CAR-T cells can efficiently target EGFRvIII+ tumors pre-targeted with biotin-4G1 (Figure 4B).
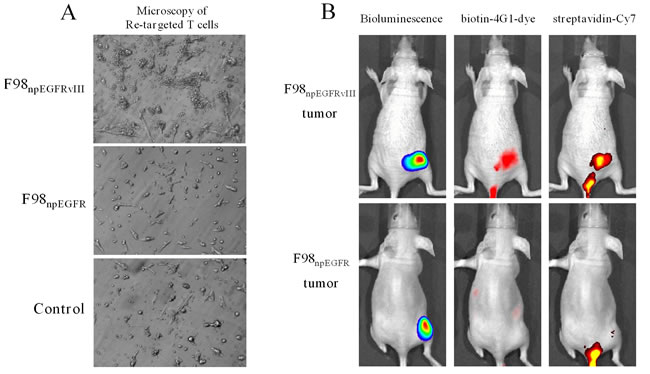
Figure 4: Avidin-CAR T cells re-target biotin-4G1. A. Microscopy observation of avidin-CAR T cells’ re-targeted to F98npEGFRvIII (upper) or F98npEGFR (middle) cells pre-targeted with biotin-4G1. F98npEGFRvIII pre-targeted with 4G1 was served as control (lower). B. Optical molecular imaging for pre-target and re-target evaluation. Left panel: bioluminescent imaging after luciferin intraperitoneal injection; middle panel: optical imaging at Ex/Em: 675/720 nm for biotin-4G1-dye detection; right panel: optical imaging at Ex/Em: 750/785 nm for Streptavidin-Cy7 detection.
Optimal time point for avidin-CAR-T cell transfer
Representative successive optical images of EGFRvIII+ tumor-bearing mice at different times after intravenous injection of 0.5 nmol biotin-4G1-dye are shown in Figure 5A. From the imaging results, all four tumors can’t be well visualized at 4 h after biotin-4G1-dye injection mainly due to prominent uptake of tracer by some normal organs. Start from 24 h, tumors can be clearly visible with good tumor-to-background contrast.
Quantification of tracer accumulation in tumors and major organs was realized by bio-distribution test. As shown in Figure 5B and Supplemental Table 3, the tumor uptakes were 6.68, 5.85, 2.41 and 2.99 %ID/g at 4, 24, 48 and 72 h post-injection, respectively (the weight and radiant efficiency of tumors and major organs were summarized in Supplemental Table 2 and Figure 1). Higher accumulation of biotin-4G1-dye in tumors at 4 and 24 h compared to other time points implies more avidin-CAR-T cells reach tumor at early time. However, accumulation of tracer in normal tissues is almost higher at 4 h than that of 24 h, therefore as shown in Figure 5C and Supplemental Table 4, the values of T/NT at 24 h are relatively higher, suggesting 24 h is supposed to be the optimal time point for avidin-CAR-T cell transfer both because of higher tracer accumulation in tumor and relative lower normal tissues uptake.
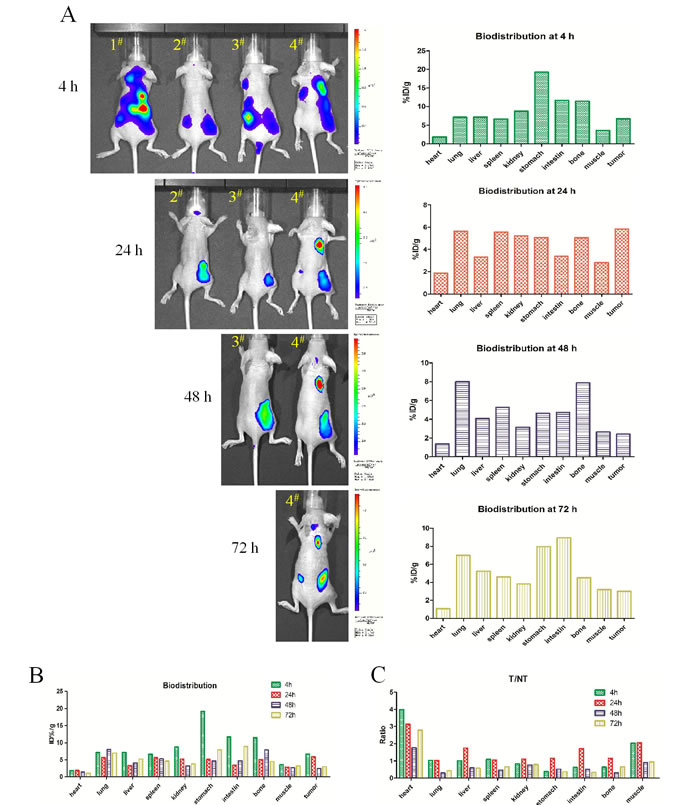
Figure 5: Optical imaging for determining time point of CAR-T cells transfer. A. Left panel: 4 mice injected with biotin-4G1-dye were undertaken optical imaging study at 4, 24, 48 and 72 h post-injection. Right panel: the corresponding bio-distribution analysis at those time point. B. Summarized bio-distribution analysis for all time points. C. The ratio of %ID/g between tumor and normal tissue (T/NT) analysis.
Antitumor efficacy evaluation
To determine whether avidin-CAR-T cells are capable of imposing cytotoxicity against EGFRvIII positive cells, effector cells (avidin-CAR-T) were cultured with target cells (F98npEGFRvIII cells). The cytotoxicity assay results showed that the effector cells exerted significant cytotoxicity against F98npEGFRvIII cells, depending on the E:T ratio. The percentage of target cells (F98npEGFRvIII cells) that were killed increased with an increase in the E:T ratio and reached almost 60% when the E:T ratio was 50:1 (Figure 6).
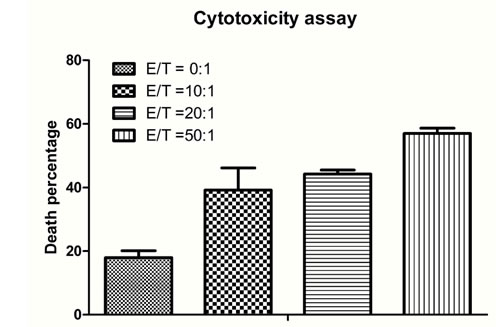
Figure 6: Cytotoxicity assay for therapy efficacy determination. Increasing numbers of avidin-CAR T cells (effector cells) were added to pre-targeted cells at effector-to-target ratios (E:T) of 0:1, 10:1, 20:1 and 50:1. The percentage of PI stained cells was analyzed by flow cytometric analysis. The p values of E:T at 0:1 and 20:1, 0:1 and 50:1, 20:1 and 50:1 were both lower than 0.001 (***); p values of 0:1 and 10:1, 10:1 and 50:1 were 0.072 (**) and 0.0127 (*), respectively; and p values of 10:1 and 20:1 was 0.2834 (no significant difference).
Through a succession of bioluminescent imaging for 5 weeks (Figure 7A), the antitumor efficacy of avidin-CAR-T cells was validated. After 2 weeks of slowly increasing (mean values were 5.78, 7.7, and 17.35 radiant counts at before therapy and the 1st, 2nd week respectively), the mean value of radiant counts of EGFRvIII+ tumors rose to near 100 at the 3rd week post-therapy and then sharply dropped to 36.5 at the 4th week post-therapy, which indicates that the therapy reduced the tumor-burden. In contrast, the radiant counts of EGFRvIII- tumors continuously increased (mean values were 14.77, 16.26, 44.10 and 58.61 at before therapy and the 1st, 3rd and 4th week respectively), excluding a slight decline to 7.99 at the 2nd week post-therapy (Figure 7B, Supplemental Figure 2).
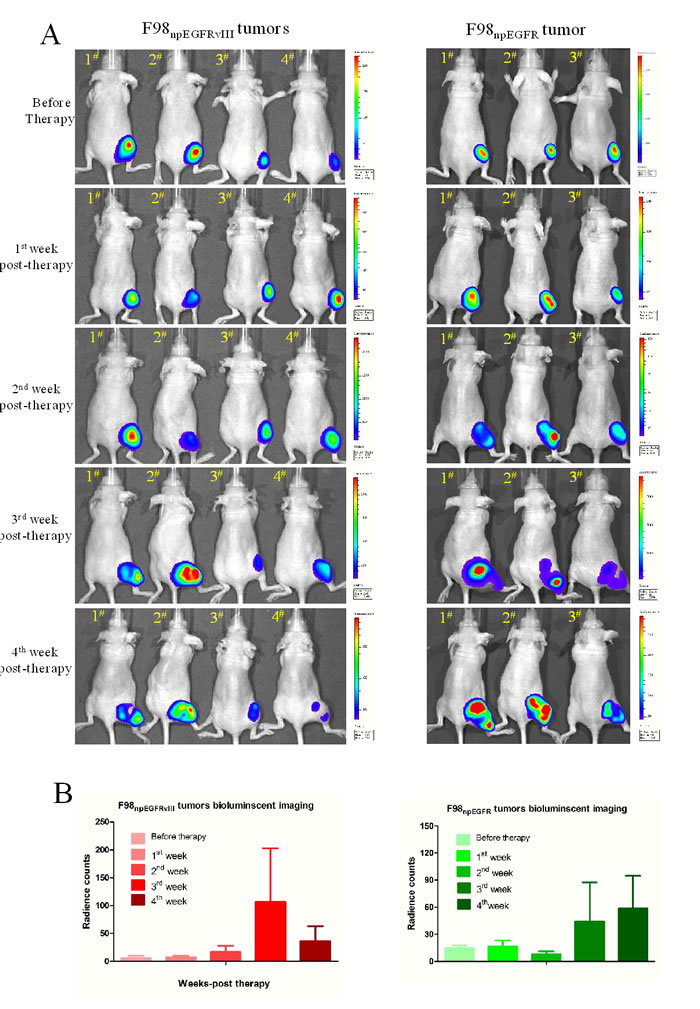
Figure 7: Bioluminescent imaging for therapy efficacy determination. A. Successive bioluminescent imaging to monitor avidin-CAR-T therapy efficacy. B. The radiant counts calculation according to bioluminescent imaging results.
DISCUSSION
Adoptive transfer of T cells with a specific CAR promotes cancer killing and has shown promise for the immunotherapy of human malignancies [25, 26]. Currently, a number of early phase clinical trials are underway that consist of using gene-modified peripheral blood lymphocytes, with CARs directed against a variety of tumor antigens [26-29]. In the current study, we used a recently reported strategy [20, 21] to treat EGFRvIII expressing glioma xenografts. We constructed avidin-CAR lentivirus plasmids and bioengineered T cells to express the CARs. To ensure the functional activity of the avidin-CAR-T cells, we analyzed their targeting, functional activity and cytotoxicity. CAR-T cells with high expression of avidin had phenotypes characteristic of cytotoxic T cells and they secreted significant amounts of IFN-γ. Avidin-CAR-T cells were then directed against the antigen EGFRvIII, as it is often expressed by malignant cancer cells and has been associated with a poor prognosis [7] and has been suggest to also be expressed by cancer stem cells [30]. We biotinylated 4G1 (biotin-4G1) and validated its capability to specifically bind EGFRvIII but not wild-type EGFR in vitro. After EGFRvIII on tumor cells was bound with biotin-4G1, avidin-CAR-T cells were either added to the culture for in vitro analysis or adoptively transferred into tumor bearing mice for in vivo analysis. The in vitro and in vivo analysis of avidin-CAR-T cell cytotoxicity indicated that the avidin-CAR-T cells were able to target and kill EGFRvIII expressing tumor cells.
Recent efforts to improve the antitumor efficacy of CAR-based therapies focus largely on the improvement of CAR design, including antigen receptor development [25, 28, 31, 32] or the introduction of costimulatory molecules [17, 33]. However, despite significant progress, some major limitations have not been solved and significant challenges still exist for the clinical application of CAR-T cells [34]. For instance, one limitation is the difficulty in visually observing the T cells in vivo, which is applicable to both the regular CAR and avidin-CAR strategies.
The avidin-CAR-T cells target TAAs and therefore one of the determining factors for this system’s therapeutic efficacy depends on the binding affinity and pharmacokinetics of biotinylated molecules and TAAs. Biotinylated polypeptides or antibodies should have different pharmacokinetics and binding affinities in vivo and in vitro, so it is necessary to develop a reliable method to evaluate the binding affinity and pharmacokinetics of different biotinylated molecules in vivo before and through CAR-T cells therapy process. For this reason, this study tried to use an optical molecular imaging approach to visualize the therapy. Through imaging study, we should achieve two goals: first, determine the specificity of pre-target and target; second, the appropriate time point for T cells adoptive transfer.
For confirming the specific binding of biotin-4G1, we labeled biotin-4G1 with near-infrared dye (biotin-4G1-dye) and injected it into mice bearing EGFRvIII positive or negative tumors. The uptake of biotin-4G1-dye by tumor cells in vivo was then analyzed. After biotin-4G1-dye injection, streptavidin-Cy7 was injected into experimental animals to confirm the binding of streptavidin to the target. Results of imaging study validated the high specificity of this biotin-avidin-T cells system.
On the other hand, one of the solutions to achieve more therapeutic benefits is to find an appropriate time point for transferring T cells. At that time point, more transferred T cells target tumor while less target normal tissue in order to reduce unnecessary cytotoxic effect to normal tissue. To determine this crucial time point, we performed optical imaging and bio-distribution study to evaluate accumulation of biotin-4G1-dye in tumors and normal tissues. From the results, we found accumulation of biotin-4G1-dye in tumors reached the peak at 4 and 24 h post-injection but rapidly declined at 48 and 72 h post-injection. So it seems that both 4 and 24 h are reasonable time for T cells transfer. However, in consideration of abundant non-specific uptake of biotin-4G1-dye in normal tissues and relative inferior T/NT values at 4 h, we finally determined 24 h as the optimal time point for T cells adoptive transfer.
In future, we will directly label avidin-CAR-T cells or regular CAR-T cells and biotinylated molecules with appropriate isotopes for PET or SPECT imaging and we expect this strategy to make an important contribution to the clinical application of CAR-T cells. To our knowledge, this is the first study to employ molecular imaging in the CAR therapy research field to provide a real-time approach for evaluating the binding specificities and determining time point for T cells transfer in vivo. We strongly recommend widespread application of molecular imaging in future CAR therapy research.
In conclusion, we have confirmed the technical and therapeutic feasibility of the biotin-avidin system in adoptive immunotherapy of EGFRvIII positive gliomas and we developed optical molecular imaging approaches to evaluate it.
MATERIALS AND METHODS
Plasmids construction, cell lines and labeling kits
The retroviral plasmid for the expression of anti-EGFRvIII CARs, pMSGV1-huAb139scFv-hCD8.28BBZ, was kindly provided by Prof. Richard Morgan. pRSET-mSA-EGFP was a gift from Sheldon Park (Addgene plasmid #39862) [22]. pITA is a 2nd generation Lentiviral Plasmis. It is derived from pZSG and the promoter is switched from a UBC to an EF1 alpha promoter. pITA can be used for cDNA expresion with puro resistance. For construction of the third generation avidin-CAR lentiviral vector, pITA-avidin-CAR, the DNA fragment of streptavidin was derived by PCR from pRSET-mSA-EGFP. The streptavidin was then subcloned (5’- ccgCTCGAGATGGCTGAAGCTGGTATCACCG and 5’-ataagaatGCGGCCGCTAATGG TGGTGATGGTGATGGG) into the XhoI and NotI site of pMSGV1-huAb139scFv-hCD8.28BBZ to replace the previous plasmid huAb139scFv. The open reading frame (ORF) from streptavidin to CD3-ZETA was amplified by PCR (5’-ATGGCTGAAGCTGGTATCACCG and 5’-TTAGCGAGGGGGCAGGG) and inserted into a pGEM-T Easy Vector and the NotI (5’- TAAACCGTCCGCTGCTTCCaCGGCCGCATT and 5’- tGGAAGCAGCGGACGGTTTAACTTTGGTGAAGG) and BamHI (5’-CTGGTACAACCAGCTGGGtTCCACCTTCATCG and 5’-aCCCAGCTGGTTGTACCAGGTGCCGGTGAT) sites in the ORF were removed by site-directed Mutagenesis. The Streptavidin-hCD8.28BBZ ORF was then subcloned 5’- ataagaat GCGGCCGCATGGCTGAAGCTGGTATCACCG and 5’-cgcGGATCCTTAGCGAGGGGGCAGGG) into the NotI and BamHI site of the lentivector pITA (Figure 1A). For construction of the luciferase lentiviral vector pITA-Luci, the luciferase DNA fragment was derived by PCR and subcloned into the NotI and BamHI site of the lentiviral vector, pITA.
F98 npEGFR and F98npEGFRvIII rat glioblastoma cell lines were purchased from the American Type Culture Collection (Rockvill, MD, USA) and were bioengineered to express luciferase. Cells were cultured in high glucose DMEM supplemented with 10% fetal bovine serum and 0.2 mg/ml G-418 at 37°C with 5% CO2.
Kits for antibody biotinylation and near-infrared dye labeling were both purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA) and stored at -20°C.
Isolation, activation and lentiviral infection of human T cells
All peripheral blood samples were obtained from healthy volunteers under an institutional review board approved protocol. To isolate peripheral blood mononuclear Cells (PBMC), the red blood cells and platelets were removed with PBMC isolation kits according to the manufacturer’s instruction (AllCells, Alameda, CA, USA). For T cell separation and activation, the acquired cell pellet was re-suspended in media X-VIVO15 and cultured. After 5 h, the cells were transferred to another flask for culture in media X-VIVO15 supplemented with 50 IU of human recombinant interleukin-2, anti-CD3 and anti-CD28 monoclonal antibodies (Becton-Dickinson, Rutherford, NJ, USA) for T cell activation.
For lentiviral infection, T cells were transferred to a 6-well plate at 5×106 cells/well in 2 ml. After 12–24 h of activation, avidin-CAR lentiviral particles and 8 μg/ml polybrene was added to each well and the cells were incubated for 12–16 hours. Virus titres were adjusted to infect 90–99% of cells. Additionally, 8 μg/ml polybrene without lentiviral particles was added to another well as a control. Next, the medium containing lentiviral particles were removed and 2 ml fresh medium with 50 IU/ml of IL-2 was added to each well.
Activated T cell phenotype analysis and cytokine secretion assays
The expression of avidin-CAR on T cells was detected by B5F (Biotin-5-fluorescein conjugate, Sigma, St. Louis, MO, USA) or by flow cytometry using mouse-avidin monoclonal antibodies (Santa Cruz, Dallas, TX, USA) as the primary antibody and rabbit anti-mouse IgG conjugated to FITC (Santa Cruz) as the secondary antibody (Becton-Dickinson). The following monoclonal antibodies were used for T cell phenotype determination: mouse anti-Human CD3-CF594, mouse anti-human CD4-FITC, mouse anti-human CD8-PE, mouse anti-human CD25-APC and mouse anti-human CD56-PE-CY7 (Becton-Dickinson). The presence of IL-2, IL-4, IL-6, IL-10, interferon-γ (IFN-γ), tumor necrosis factor (TNF) and IL-17 of activated avidin-CAR-T cells in culture supernatant were analyzed using Human Th1/Th2 Cytokine Kit II (Becton-Dickinson) according to manufacturer’s instructions. The cytokines concentration of non-activated avidin-CAR-T cells was also determined as control. Values represent the mean of triplicate wells.
Biotinylation and dye labeling of the anti-EGFRvIII antibody 4G1
A novel mouse anti-EGFRvIII monoclonal antibody, 4G1, was prepared according to our unpublished study. To prepare biotin-4G1, 1.33×10−4 mmol of Sulfo-NHS-Biotin solution (Thermo Fisher) was added to 1 mg of 4G1 dissolved in phosphate-buffered saline (20:1 molar ratio of Sulfo-NHS-Biotin and antibody) and the reaction was incubated at room temperature for 30–60 minutes. The reaction mix was then purified with a Zeba Spin Desalting Column (Thermo Fisher) to remove excess biotin reagent, the flow through of which was the purified biotinylated antibody sample. To measure the level of biotin incorporation, 100 µl of biotin-4G1 was added to a cuvette containing HABA/Avidin. The absorbance was measured at 500 nm and the HABA assay calculation was performed according to the manufacturer’s instructions.
To prepare the optical imaging probe, we labeled biotin-4G1 with a near infrared dye, Dylight 680 NHS (Ex/Em: 682/715 nm, Thermo Fisher). Briefly, 140 µg Dylight 680 NHS was added to 2 mg biotin-4G1 solution (molar ratio =10:1) and the reaction was incubated at room temperature for 2 h. The reaction mix was purified with a PD-10 column (Amersham, Piscataway, NJ, USA) to remove excess dye. The collected flow-through solution contained the purified labeled biotin-4G1 (biotin-4G1-dye). The degree of labeling was calculated by analyzing the sample using spectrophotometer at an absorbance of 280nm and 684 nm according to the manufacturer’s instructions.
F98npEGFR and F98npEGFRvIII xenograft model of glioma
All animal experiments were performed in accordance with the Guidelines of Peking University Health Science Center Animal Care and Use Committee. 2×106 tumor cells in 100 µl were injected subcutaneously into the right or both legs of female BALB/c nude mice.
Evaluation of biotin-4G1’s pre-target to EGFRvIII
In vitro tests were performed to verify the binding specificity of biotin-4G1. EGFRvIII expression by F98npEGFR and F98npEGFRvIII cells was analyzed by western blotting and flow cytometry using biotin-4G1 and Dylight 800-rabbit anti-mouse IgG (EarthOx, San Francisco, CA, USA) as the primary and secondary antibodies, respectively. Mouse IgG was used as an isotype primary antibody. For the indirect immunofluorescent assay (IFA), cells grown on LabTek chamber slides were incubated with biotin-4G1 antibodies at 4°C overnight and then incubated with FITC-rabbit anti-mouse IgG at room temperature for 2 h. DAPI was used for nuclear staining. Fluorescence signals were detected using a confocal microscope (TCS SP5; Leica, Germany). For immunohistochemistry (IHC), paraffin sections of F98npEGFR and F98npEGFRvIII xenograft tumors were made. Biotin-4G1 and HRP-rabbit anti-mouse IgG were used as primary and secondary antibodies, respectively. The sections were incubated in diaminobenzidine–hydrogen peroxide solution (Boster, DAB staining kit, Wuhan, China) and examined microscopically.
For in vivo molecular imaging, 0.5 nmol of biotin-4G1-dye was intravenously injected into mice when the tumor size reached between 50 and 100 mm3. Mice were then anesthetized by inhalation of 2% isoflurane and were injected with 2 mg D-luciferin (SynChem, Inc, IL, USA) dissolved in PBS. Bioluminescent and optical imaging at Ex/Em: 675/720 nm were then performed respectively under an IVIS small animal imaging system (Xenogen, Alameda, CA) at successive time points (4, 24, 48 and 72 h). Besides for biotin-4G1 binding effect evaluation, the optimal time point for in vivo avidin-CAR T cell transfer was determined according to imaging results.
Evaluation of avidin-CAR T cells’ re-target
For in vitro evaluation of avidin-CAR-T cells binding to biotin-4G1, 20 µg of of biotin-4G1 was added to F98npEGFRvIII cells at a concentration of 1×105 cells in a 6-well plate, which were then incubated at 4°C for 1 h; 20 µg of 4G1 was also added to tumor cells as a control. After the cells were washed three times to remove excess biotin-4G1, 5×106 avidin-CAR T cells were added to each well and the cells were incubated for 1 h. The cells were then washed three times to remove non-binding T cells. Plates were analyzed under a phase contrast microscope to determine avidin-CAR T cells’ re-target.
For in vivo molecular imaging, 0.5 nmol of biotin-4G1-dye was intravenously injected into mice. 0.5 nmol Strepavidin-Cy7 (Amyjet Scientific Inc. Wuhan, CHN) dissolved in PBS was then intravenously injected into mice at the optimal time, as determined by the pre-target imaging results (mentioned above). After 0.5 h, mice were subjected to bioluminescent imaging after 2 mg of luciferin injection and optical imaging with excitation and emission absorbance of 675/720 nm for Dylight 680 detection and 750/785 nm for Cy7 detection.
Determination of optimal time for T cells adoptive therapy
Four F98npEGFRvIII tumor-bearing mice were intravenously injected with 10 nmol of biotin-4G-dye and used for optical imaging at 4, 24, 48 and 72 h post-injection. After each time point imaging study, one mouse was sacrificed to bio-distribution analysis. Briefly, mice were killed and dissected, tumors and major organs were collected and wet-weighed. The fluorescence radiant efficiency in these tissues was measured using IVIS imaging system. The results are presented as percentage injected dose per gram of tissue (%ID/g). And the ratios of the levels of tissue %ID/g between the tumors and normal tissues (T/NT ratios) were calculated.
Therapeutic efficacy evaluation
A modified and simplified cytotoxicity assay was undertaken for in vitro evaluation. Briefly, 20 µg of biotin-4G1 was added to F98npEGFR or F98npEGFRvIII cells at density of 1×105 cells in 6-well plates, which were then incubated at room temperature for 1 h. The cells were then washed three times and avidin-CAR T cells (effector cells) were added to the pre-targeted cells at effector-to-target ratios (E:T) of 0:1, 10:1, 20:1 and 50:1. The cells were incubated at room temperature for 4 h and then washed three times. The cells were then trypsinized, suspended in phosphate-buffered saline and stained with propidium iodide for 3 min before flow cytometric analysis. Tumor cells stained with propidium iodide were regarded as non-viable cells.
For in vivo antitumor efficacy evaluation, F98npEGFR or F98npEGFRvIII tumor-bearing mice were intravenously injected with 100 ng of biotin-4G when the total radiant efficiency of each tumor reached 20 counts. At the optimal time point post injection of biotin-4G (as determined by in vivo 4G1 imaging), 1×107 activated avidin-CAR T cells were intraperitoneally injected. Bioluminescent imaging was performed every week to evaluate the antitumor activity of the avidin-CAR T cells.
Statistical analysis
Data are expressed as mean ± SD. Statistical evaluation was performed using 2-tailed Student’s t tests. P values less than 0.05 were considered significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number: 81272790).
Conflicts of Interest
There is no conflict of interest.
References
1. Yamazaki H, Fukui Y, Ueyama Y, Tamaoki N, Kawamoto T, Taniguchi S and Shibuya M. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Molecular and cellular biology. 1988; 8:1816-1820.
2. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS and Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences of the United States of America. 1992; 89:2965-2969.
3. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK and Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:7727-7731.
4. Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS and Cavenee WK. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. The Journal of biological chemistry. 1997; 272:2927-2935.
5. Tang CK, Gong XQ, Moscatello DK, Wong AJ and Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer research. 2000; 60:3081-3087.
6. Lammering G, Hewit TH, Valerie K, Contessa JN, Amorino GP, Dent P and Schmidt-Ullrich RK. EGFRvIII-mediated radioresistance through a strong cytoprotective response. Oncogene. 2003; 22:5545-5553.
7. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y and Ushio Y. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer research. 2003; 63:6962-6970.
8. Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, Friedman HS, Bigner DD and Sampson JH. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004; 10:3216-3224.
9. Liu X, Liu K, Qin J, Hao L, Li X, Liu Y, Zhang X, Liu X, Li P, Han S, Mao Z and Shen L. C/EBPbeta promotes angiogenesis through secretion of IL-6, which is inhibited by genistein, in EGFRvIII-positive glioblastoma. International journal of cancer Journal international du cancer. 2015; 136:2524-2534.
10. Jutten B, Dubois L, Li Y, Aerts H, Wouters BG, Lambin P, Theys J and Lammering G. Binding of cetuximab to the EGFRvIII deletion mutant and its biological consequences in malignant glioma cells. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2009; 92:393-398.
11. Jiang H, Wang H, Tan Z, Hu S, Wang H, Shi B, Yang L, Li P, Gu J, Wang H and Li Z. Growth suppression of human hepatocellular carcinoma xenografts by a monoclonal antibody CH12 directed to epidermal growth factor receptor variant III. The Journal of biological chemistry. 2011; 286:5913-5920.
12. Gan HK, Burgess AW, Clayton AH and Scott AM. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer research. 2012; 72:2924-2930.
13. Zalutsky MR, Boskovitz A, Kuan CT, Pegram CN, Ayriss J, Wikstrand CJ, Buckley AF, Lipp ES, Herndon JE, 2nd, McLendon RE and Bigner DD. Radioimmunotargeting of malignant glioma by monoclonal antibody D2C7 reactive against both wild-type and variant III mutant epidermal growth factor receptors. Nuclear medicine and biology. 2012; 39:23-34.
14. Porter DL, Levine BL, Kalos M, Bagg A and June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011; 365:725-733.
15. Cheadle EJ, Sheard V, Hombach AA, Chmielewski M, Riet T, Berrevoets C, Schooten E, Lamers C, Abken H, Debets R and Gilham DE. Chimeric antigen receptors for T-cell based therapy. Methods in molecular biology (Clifton, NJ). 2012; 907:645-666.
16. Sadelain M, Brentjens R and Riviere I. The basic principles of chimeric antigen receptor design. Cancer discovery. 2013; 3:388-398.
17. Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA and Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. Journal of immunology (Baltimore, Md : 1950). 2009; 183:5563-5574.
18. Hombach AA and Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. International journal of cancer Journal international du cancer. 2011; 129:2935-2944.
19. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK and Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. The Journal of clinical investigation. 2011; 121:1822-1826.
20. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N and Powell DJ, Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer research. 2012; 72:1844-1852.
21. Urbanska K and Powell DJ. Development of a novel universal immune receptor for antigen targeting: To Infinity and beyond. Oncoimmunology. 2012; 1:777-779.
22. Lim KH, Huang H, Pralle A and Park S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnology and bioengineering. 2013; 110:57-67.
23. Lu PH and Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. Journal of immunology (Baltimore, Md : 1950). 1994; 153:1687-1696.
24. Baecher-Allan C, Viglietta V and Hafler DA. Human CD4+CD25+ regulatory T cells. Seminars in immunology. 2004; 16:89-98.
25. Gonzalez S, Naranjo A, Serrano LM, Chang WC, Wright CL and Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. The journal of gene medicine. 2004; 6:704-711.
26. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011; 118:6050-6056.
27. Kandalaft LE, Powell DJ, Jr. and Coukos G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. Journal of translational medicine. 2012; 10:157.
28. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA and Rosenberg SA. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human gene therapy. 2012; 23:1043-1053.
29. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, Wang LL and Han WD. Treatment of CD33-directed Chimeric Antigen Receptor-modified T Cells in One Patient With Relapsed and Refractory Acute Myeloid Leukemia. Molecular therapy : the journal of the American Society of Gene Therapy. 2015; 23:184-191.
30. Liu XJ, Wu WT, Wu WH, Yin F, Ma SH, Qin JZ, Liu XX, Liu YN, Zhang XY, Li P, Han S, Liu KY, Zhang JM, He QH and Shen L. A minority subpopulation of CD133(+) /EGFRvIII(+) /EGFR(-) cells acquires stemness and contributes to gefitinib resistance. CNS neuroscience & therapeutics. 2013; 19:494-502.
31. Klebanoff CA, Yamamoto TN and Restifo NP. Immunotherapy: Treatment of aggressive lymphomas with anti-CD19 CAR T cells. Nature reviews Clinical oncology. 2014; 11:685-686.
32. Sautto GA, Wisskirchen K, Clementi N, Castelli M, Diotti RA, Graf J, Clementi M, Burioni R, Protzer U and Mancini N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut. 2015.
33. Zhong XS, Matsushita M, Plotkin J, Riviere I and Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Molecular therapy : the journal of the American Society of Gene Therapy. 2010; 18:413-420.
34. Lipowska-Bhalla G, Gilham DE, Hawkins RE and Rothwell DG. Targeted immunotherapy of cancer with CAR T cells: achievements and challenges. Cancer immunology, immunotherapy : CII. 2012; 61:953-962.