
INTRODUCTION
Breast cancer is the most commonly diagnosed type of cancer and the second most fatal cancer of women in the world [1]. Over the last few decades, various targeted therapeutics have been introduced into the clinic, but there has been no corresponding improvement in patient survival, primarily because of the malignant behavior of breast cancer. Metastasis of breast cancer from primary location to distant sites is the main cause contributing to disease fatalities. However, factors controlling breast cancer cell migration remain to be elucidated. Therefore, it is urgent to identify key molecular regulators in metastasis to improve the prognosis assessment and treatment of breast cancer patients.
Friend leukemia virus integration 1 (FLI1) plays a critical role in normal development and hematopoiesis by functioning as both transcriptional activator and repressor [2–6]. The oncogenic role of FLI1 was first confirmed in mice erytholeukemia [7]. Now, it is clear that about 85% of Ewing sarcoma cases are characterized by the presence of the EWS/FLI1 fusion oncogene as a result of balanced chromosomal translocation t(11;22)(q24;q12). However, only recently has FLI1 been reported to show aberrant activation in patients with solid tumors [8]. Despite these advances, little is known about the role of FLI1 in metastasis, particularly in breast cancer.
In this communication, we examined the oncogenic activation of FLI1 and its correlation with clinicopathological features in patients with breast cancer. Notably, FLI1 activated the Rho GTPase pathway that is known to be associated with metastasis in breast cancer.
RESULTS
Upregulation of FLI1 in metastatic breast cancer cells
To identify factors that are associated with metastasis, we selected an aggressive breast cancer cell line MDA-MB231 and a non-aggressive cell line MCF-7. We aimed to identify factors that were differentially expressed between these two cell lines.
Using RT-PCR, we analyzed the expression of growth factors, oncogenes and factors that may be related to tumor metastasis. Fig. 1A shows the typical data for part of candidate genes analyzed. Interestingly, we found that FLI1 was one of the most notable genes that show distinct expression patterns. As a potential candidate target, FLI1 was highly expressed in the aggressive MDA-MB231 cells, but was almost undetectable in the non-aggressive MCF-7 cells (Fig. 1A, lane 3). Quantitative PCR also confirmed the upregulation of FLI1 in the aggressive MDA-MB231 cell line (Fig. 1B). In addition to FLI1, ETS-1 and MMP-1 were also significantly upregulated in the aggressive MDA-MB231 cells.
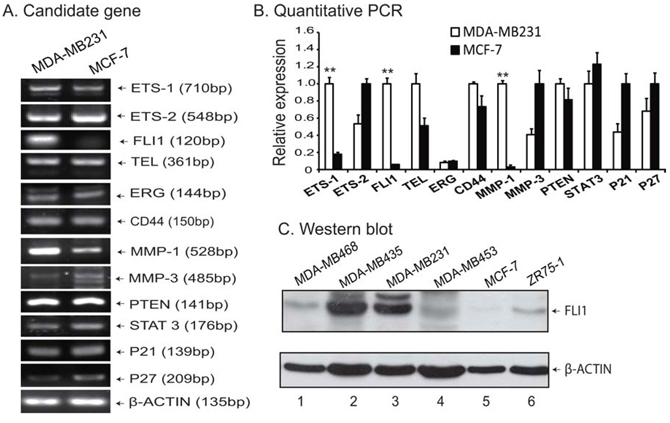
Figure 1: FLI1 expression is associated with metastasis of breast cancer cells. A. Representative RT-PCR data of genes analyzed. Differential expression of growth factors, oncogenes and target genes were compared between the aggressive MDA-MB231 and the non-aggressive MCF-7 cell lines. B. Confirmation of gene expression by quantitative PCR. Data shown are mean ± SEM from three independent experiments by normalization over the β-ACTIN control. **p < 0.01 as compared with MCF-7. C. Western blot of FLI1 in breast cancer cell lines.
We then used Western blot to compare the expression of FLI1 in five breast cancer cell lines that show varied metastatic abilities. As seen in Fig. 1C, FLI1 was highly expressed in cell lines with high ability of metastasis, including MDA-MB231 and MDA-MB453 (lanes 2–3). Three breast cancer cell lines (MDA-MB468, MDA-MB453, and ZR75-1) exhibited weak expression of FLI1, while the non-aggressive MCF-7 was almost negative for FLI1.
FLI1 activation correlates with breast cancer metastasis
To determine the clinical significance of FLI1, we examined its expression pattern in tumor samples collected from breast cancer patients. Using immunohistochemically staining with FLI1 specific antibody, we found that FLI1 were significantly upregulated in breast tumor tissues as compared with adjacent normal tissues (Fig. 2A).
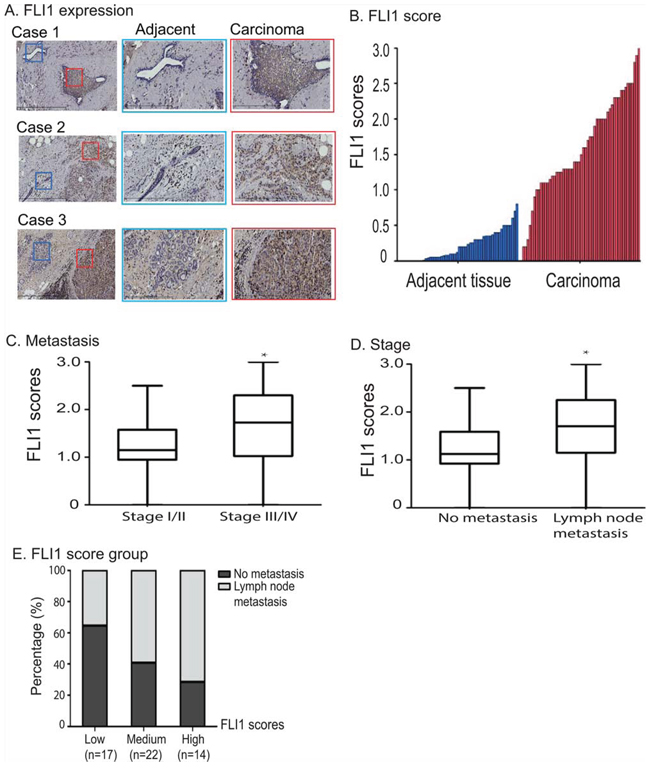
Figure 2: Overexpression of FLI1 in breast cancer tissues. A. Immunohistochemical staining of FLI1 in three representative breast cancer samples and their adjacent tissues. Blue square: adjacent normal tissue; red square: carcinoma. B. FLI1 is highly expressed in breast cancer tissues as compared with adjacent normal tissues. C. FLI1 expression scores in breast cancer patients in different clinical stages. D. High FLI1 expression in breast cancer patients with lymph node metastasis. E. Association between lymph node metastasis and FLI1 expression scores.
For comparison, we calculated the expression score for FLI1 and compared FLI1 score between breast cancer and adjacent tissues. There was a significantly higher FLI1 expression score in collected tumor tissues than that in their adjacent tissues (P < 0.01, Fig. 2B).
To better understand the role of FLI1 in tumor prognosis, the association of FLI1 expression with stage and lymph node metastasis was also investigated. The FLI1 scores were higher in stage III/IV patients (1.7 ± 0.5) than that in stages I/II patients (1.2 ± 0.3) (Fig. 2C, p < 0.05). Similarly, FLI1 scores were also higher in patients with lymph node metastasis (1.7 ± 0.5) than that in patients without metastasis (1.1 ± 0.4) (p < 0.05) (Fig. 2D).
We divided FLI1 expression scores into three groups (low, medium, and high) and examined the percentage rate of breast tumor metastasis between groups. There was a clear positive association between the FLI1 expression score and the percentage rate of lymph node metastasis (Fig. 2E).
Knockdown of FLI1 inhibits proliferation of breast cancer cells
To test whether FLI1 is functionally important in breast cancer, we used two small interference RNAs (siFLI1 1# and siFLI1 2#) to knockdown FLI1 in MDA-MB231 and MDA-MB453 cells. These two cell lines overexpressed FLI1 and exhibited high metastatic potential.
Two siRNAs showed distinct activities in these two cell lines, with siFLI1 1# exhibiting a better inhibition potency than siFLI1 2# (Fig. 3A-3B). Correspondingly, we found a dose-dependent inhibition of cell proliferation with FLI1 knockdown in these two breast cancer cells (Fig. 3C-3D).
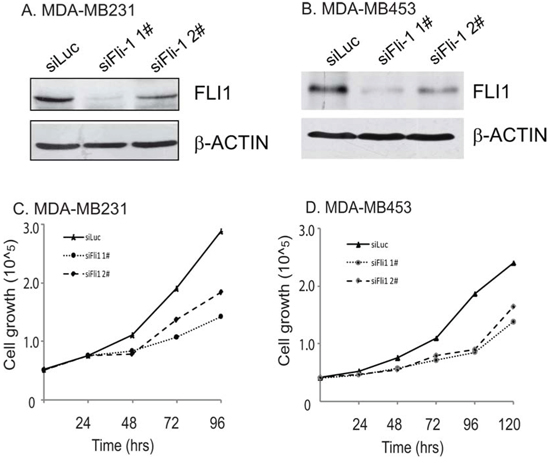
Figure 3: FLI1 knockdown decreases cell proliferation in two aggressive breast cancer cells. A-B. Knockdown of FLI1 by two siRNAs (siFLI1 1#, siFli1 2#) in MDA-MB231 (A) and MDA-MB453 (B) FLI1 expression was measured by Western blot. siLUC: control siRNA targeting the photinus pyralis luciferase gene. C-D. Inhibition of cell proliferation by FLI1 siRNAs in MDA-MB231 (C) and MDA-MB453 (D) cancer cells.
Interference of FLI1 results in Go/G1 cell cycle arrest
To study the potential mechanism of FLI1, we analyzed cell cycle in the FLI1-knockdwon MDA-MB231 and MDA-MB453 cells. The cells in G0/G1 and S-phase were measured by flow cytometry (Fig. 4A). After FLI1 knockdown, there was a dramatic decrease of cells in S-phase (28.6% vs 7.2% in MDA-MB231 cells and 24.9% vs 15.1% in MDA-MB453 cells). In parallel, there was an increment in the blockage of cells in the G0/G1 phase (61.5% vs 87.6% in MDA-MBA231 cells and 66.2% vs 78.1% in MDA-MB453 cells).
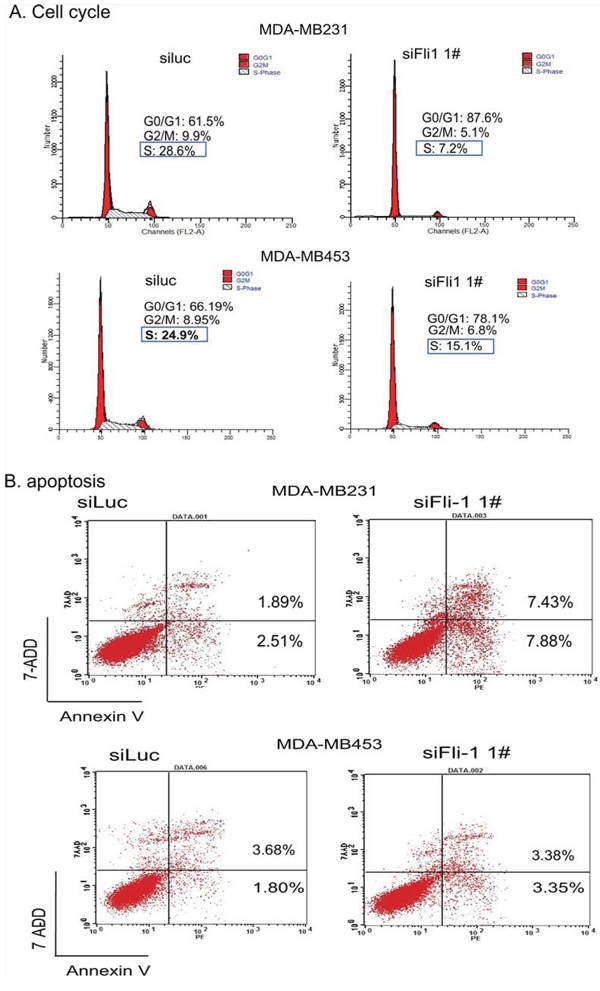
Figure 4: Analyses of cell cycle and apoptosis after interference of FLI1 expression. A. Cell cycle in MDA-MB231 cells treated with the control siRNA (siluc) and FLI1 siRNA (siFLI1 1#). B. Apoptosis in MDA-MB231 cells following the knockdown of FLI1.
Cell apoptosis was also analyzed after the knockdown of FLI1 in these two breast cancer cells (Fig. 4B). Using flow cytometry, we found that there was an increment in apoptosis cells in MDA-MB231 cells (top panel, 1.89% vs 7.43%), but this pattern was not notable in MDA-MB453 cells (bottom panel, 3.7% vs 3.4%).
FLI1 is critical to maintain breast cancer metastasis
The role of FLI1 in breast cancer metastasis was evaluated by cell migration, invasion, and adhesion assays. To determine the role of FLI1 in cell migration, we knocked down FLI1 with siRNAs in two highly metastatic human breast cancer cell lines (MDA-MB231 and MDA-MB453). Using the transwell assay, we showed that knockdown of FLI1 significantly decreased cell migration in both cell lines (Fig. 5A).
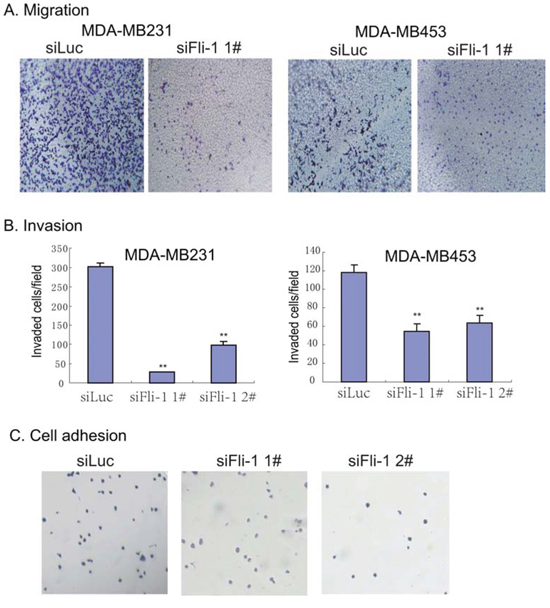
Figure 5: Knockdown of FLI1 inhibits metastasis of breast cancer cells. A. Reduced cell migration in MDA-MB231 and MDA-MB453 cells following the knockdown of FLI1. B. Cell invasion assay. Cells invaded through the collagen-coated membrane of the transwell were counted. All data shown are mean ± SEM from three independent experiments. *p < 0.05 as compared with control cells (siluc). C. Cell adhesion assay. Note the reduced ability of cell adhesion following the knockdown of FLI1.
The role of FLI1 in tumor cell invasion was examined by the collagen-coated transwell assay. In this assay, MDA-MB231 cells were allowed to invade through the collagen-coated trasnwell. As shown in Fig. 5B, we found that knocking-down of FLI1 significantly reduced the invasion of both MDA-MB231 and MDA-MB453 cells in the 3D collagen invasion assay.
To further understand the role of FLI1 in metastasis, we examined the capacity of cell adhesion in siFLI1 1#-treated cells. We found that MDA-MB231 cells treated with FLI1 siRNA were much less adhesive than cells treated with the control siRNA (Fig. 5C). These data suggest that the upregulated FLI1 may impair the cell adhesion phenotype.
FLI1 promotes metastasis through the Rho GTPase pathway
It is well established that Ras homologous (Rho) GTPases family proteins, like Rac1 and RhoA, play a key role in cell migration and tumor metastasis by regulating actin dynamics and cell-cell adhesion [9]. We thus investigated whether FLI1 may play a role in metastasis by activating the Rho GTPase family genes. For this, we purified GST-PBD and GST-RBD proteins (Fig. 6A) and used them to detect the active Rac1 and RhoA [10, 11]. In this binding assay, the p21-binding domain (PBD) of the Rac1 effector p21-activated kinases was used to affinity precipitate the active Rac1, and the RhoA binding domain (RBD) of the RhoA effector rhotekin was used to affinity precipitate the active RhoA.
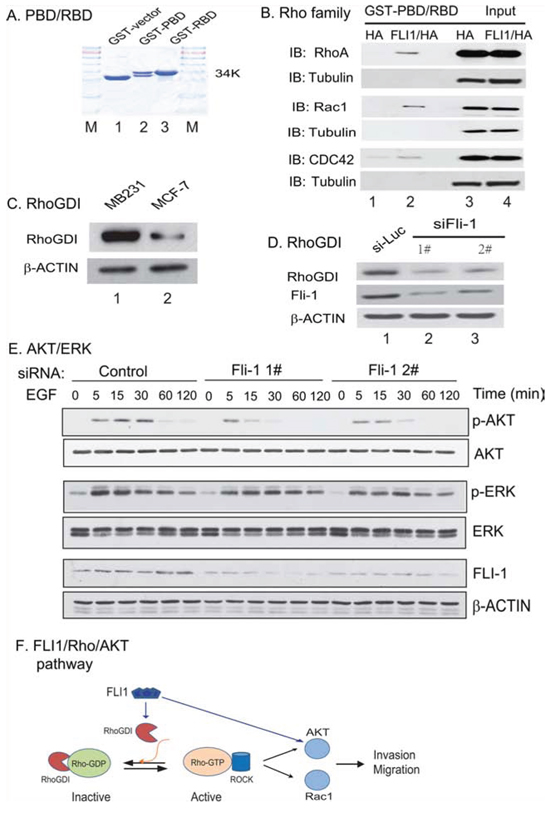
Figure 6: Activation of the Rho GTPase pathway. A. GST-PBD and GST-RBD fusion proteins used for quantitate the active RhoA and Rac1. B. Detection of the active Rho GTPases by the GST-PBD and GST-RBD pull-down. PBD: the p21-binding domain (PBD) of the Rac1 effector p21-activated kinases (PAK); RBD: the RhoA binding domain (RBD) of the RhoA effector rhotekin. Note the activation of RhoA, Rac1, and CDC42 in FLI1/HA-expressing breast cancer cells (lane 2) as compared with that in the HA control (lane 1). C. Upregulation of RhoGDI in the aggressive MDA-MB231 breast cancer cells. D. Knockdown of FLI1 reduces the Rho GDP-dissociation inhibitor (RhoGDI). E. Activation of the AKT/ERK cascade. Knockdown of FLI1 decreases the activity of the AKT pathway. F. The proposed model of the FLI1/Rho/AKT pathway.
To investigate the role of FLI1 in regulating Rho GTPase family protein function, we ectopically expressed FLI1 in MDA-MB231 cells. The control cells were transfected with the HA control vector. GST-pull down assay and coimmunoprecipitation assay were used to detect the active Rac1 and RhoA. Using this binding assay, we found that the ectopically expressed FLI1 was able to activate both Rac1 and RhoA GTPases, and enhanced the activity of CDC42 GTPase (Fig. 6B, lanes 1 vs 2).
Rho GDP-dissociation inhibitor (RhoGDI) is a critical regulator of Rho GTPase function [12]. Activation of Rho GTPase signaling pathways requires the regulated release of Rho GTPases from RhoGDI complexes, followed by their reuptake after membrane cycling. However, a clear picture of the role of RhoGDIs in breast cancer has not emerged. RhoGDIs can mediate both pro-tumorigenic and anti-tumorigenic signaling pathways [13]. We measured RhoGDI in our cell model using a Western antibody that recognizes both RhoGDI1 and RhoGDI2. We found that RhoGDI was overexpressed in the highly invasive MDA-231 cells as compared with the non-invasive MCF-7 cells (Fig. 6C). Similarly, FLI1 siRNA treatment dramatically downregulated RhoGDI (Fig. 6D, lanes 2–4), in parallel with the knockdown of FLI1 oncoprotein. These data suggest that FLI1 may regulate the Rho GTPase pathway by controlling the activity of the RhoGDI inhibitor.
During cell migration, there is a close crosstalk between the Rho GTPase signal pathway and AKT/ERK cascades [14–16]. To examine this interaction in our model, we used EGF to stimulate cells that were treated with siFLI1 1# and siFLI1 #2. We found that the level of active phospho-AKT (p-AKT) was correlated with FLI1 down-regulation (Fig. 6D). However, no such crosstalk was observed for the ERK cascade.
DISCUSSION
Metastasis remains the primary cause of cancer-related death in patients with breast cancer. A comprehensive understanding of genetic determinants that promote metastatic dissemination is critical for the development and implementation of novel diagnostic and treatment strategies. In this study, we for the first time identify FLI1 as a critical molecular factor associated with breast cancer metastasis.
FLI1, an Ets family member of transcription factors, was originally identified as a proto-oncogene for retroviral integration of Friend virus-induced erythroleukemias [17]. The EWS/FLI1 oncoprotein is formed by t(11;22)(q24;q12) rearrangement, in which FLI1 becomes juxtaposed 3’ to the EWS gene [18]. EWS/FLI1 is detected in 95% of Ewing’s sarcomas [19]. As a hallmark of Ewing’s sarcoma, detection of this genetic alteration has provided a powerful tool in confirming the diagnosis of this childhood disease.
Oncogenic activation of FLI1 leads to tumorigenesis, such as Ewing sarcoma and ovarian cancer. However, little is known about the role of FLI1 in other solid tumors, particularly in breast cancer. In this study, we demonstrate that FLI1 is overexpressed in cancer tissues as compared with the adjacent tissues. The FLI1 expression score is closely correlated with clinicopathologic features, including advanced stages and lymph node metastasis. Consequently, knockdown of FLI1 attenuates the metastatic potential in highly aggressive breast cancer cell lines. Our findings thus support a distinct paradigm for the involvement of FLI1 in the progression of breast cancer.
It is well known that FLI1 drives Ewing sarcoma through a mechanism of EWS-FLI1 fusion oncoprotein [20, 21]. By directly inducing or repressing gene enhancers, EWS-FLI1 establishes an oncogenic regulatory landscape in Ewing sarcoma governing both tumor survival and differentiation [22]. However, this EWS-FLI1 fusion mechanism is not active in breast cancer. Thus, the oncogenic role of FLI1 in breast cancer metastasis, as observed in the present study, must be mediated through a distinct mechanism.
Rho GTPase comprise a major branch of the Ras superfamily of small GTPase, with Rac1, CdC42 and RhoA being best characterized as the important regulators of actin cytoskeleton organization, cell migration and cell progression. G proteins and their downstream signaling targets influence aberrant cell growth and survival, largely through the activation of AKT/mTOR, MAPKs, and Hippo signaling pathways [23]. Rho GTPases, including Rac1, CDC42 and RhoA, switch the cycle between inactive (GDP-bound) and active (GTP-bound) states [24]. During the complex process in tumor metastasis, tumor cell migration and invasion are two critical steps and responsible for the entry of tumor cells into blood vessels and lymph nodes, even the secondary tissues [25, 26]. In addition, cell adhesion, an important step in initiating tumor cell migration, is also tightly regulated by Rho GTPases [27]. The Rac signaling pathway is hyperactived in human breast cancer [28–30]. Constitutively activated Rac1 promotes focal adhesions in breast cancer cells. The activated Rac signal mediates breast cancer cell motility, invasion and breast cancer metastasis [30, 31].
We were thus interested in examining if FLI1 affects the Rho GTPases pathway. Functional studies revealed that the forced expression of FLI1 led to the activation of all three Rho GTPases: CDC42, Rac1 and Rho-A (Fig. 6B). Correspondingly, knockdown of FLI1 decreased cell migration, invasion, and cell attachment (Fig. 5). Taken together, these data suggest that the activation of this signal pathway by FLI1 may be associated with the metastatic potential of the breast cancer cells tested in this study.
The activities of Rho GTPases are regulated by RhoGDI [12]. The expression of RhoGDIs is altered in a variety of cancers. However, its specific role in various cancers remains controversial. RhoGDIs can mediate both pro-tumorigenic and anti-tumorigenic signaling pathways [13]. While early studies establish the role of RhoGDIs in maintaining Rho GTPase in inactive form [32], recent evidence has come to suggest that it may also act as a positive regulator in cancer progression. For example, RhoGDI was identified as an up-regulated protein in metastatic colorectal cancer [33], essential for cell proliferation, migration and distant metastasis. Similarly, Zhang et al reported that RhoGDI2 was highly expressed in tumor, but not in benign breast cell lines. Knockdown of RhoGDI2 in MDA-MB-231 cells resulted in decreased motility and invasion [34]. Ota et al also demonstrate that RhoGDIs positively regulate Rho GTPase activity by a distinct mechanism through direct interaction with GTPase activating protein (GAP) [35].
In this study, we found that RhoGDIs were upregulated in the aggressive MDA-MB-231 breast cancer cells as compared with that in the non-aggressive MCF-7 breast cancer cells (Fig. 6C). Knockdown of FLI1 using shRNAs dramatically downregulated RhoGDIs (Fig. 6D), in parallel with the reduced migration and invasion of MDA-MB-231 breast cancer cells. Our data thus suggest that FLI1 may regulate the Rho GTPase pathway in breast cancer, at least partially by controlling the activity of the RhoGDIs.
The Rho GTPases are also in a close crosstalk with the RAS-MAPK-ERK and PI3K-AKT-mTOR signaling pathways, which are the established factors controlling cell survival, differentiation, proliferation, metabolism, and motility [14–16, 36]. In normal cells, sustained activation of ERK1/ERK2 is necessary for G1- to S-phase progression and is associated with induction of positive regulators of the cell cycle [37]. Cellular processes regulated by AKT include cell proliferation and survival and tissue invasion. All these processes represent hallmarks of cancer [38]. Although both AKT and ERK pathways play an important role in malignant proliferation of breast cancer [39], our FLI1-transfection data only validate the role of p-AKT in MDA-MB231 and MDA-MB453 cells (Fig. 6D). p-AKT was significantly inhibited when FLI1 was interfered, thus supporting the concept that FLI1 may activate the AKT pathway in breast cancer cells.
In conclusion, this study for the first time demonstrates FLI1 as a target gene that is associated with breast cancer metastasis (Fig. 6E). In this FLI1/Rho/AKT pathway, overexpression of FLI1 in breast cancer activates the Rho GTPase family genes, either directly or by modulating the RhoGDI inhibitor. Through the pathway crosstalk, the AKT is activated to promote metastasis. Together, they induce a malignant phenotype that is associated with the metastasis of breast cancer. This study thus identifies FLI1 as an attractive target for therapeutic intervention in breast cancer.
MATERIALS AND METHODS
Breast cancer samples
Formalin-fixed and paraffin-embedded tissues of breast cancer and control samples were obtained from the First Hospital of Jilin University between 2005 and June 2009. Primitive neuroectodermal tumor (PNET) was chosen as the positive control. Clinical data related to disease stage, histological grade and follow-up data were available for these patients. The pathological diagnosis was made in accordance with the histological classification of tumors developed by the World Health Organization. Tumor stage was defined according to American Joint Committee on Cancer/International Union Against Cancer classification system. Tumors were histologically graded according to the Elston and Ellis method. The study was approved by the Research Ethics Board of the First Hospital of Jilin University [40]. Informed consent was obtained from each breast cancer patient and normal subject. The clinical characteristics of the patients and the control subjects were shown in Table S1.
Immunohistochemical staining
Tissue slides were deparaffinized with xylene and rehydrated through a gradual decline of alcohol (100–80%), and then incubated in 3% hydrogen peroxide for 15 minutes to block endogenous peroxidase activity. Antigen retrieval was carried out by immersing the slides in 10 mM sodium citrate buffer (pH 6.0) and maintained at a sub-boiling temperature for 15 minutes. The slides were rinsed in phosphate-buffered saline and incubated with 10% normal goat serum to block non-specific staining for 30 minutes at 37°C. Primary anti-FLI1 polyclonal antibodies (Neomarker) were diluted in 1:100, and incubated with the sections at 4°C overnight. After washing with PBS, the secondary antibodies (biotinylated goat anti-rabbit immunoglobulin) and streptavidin peroxidase complex reagent were applied. Subsequently, the visualization signal was processed according to the Polink-2 HRP DAB Detection kit. Finally, the slides were counterstained with hematoxylin for 15 min and dehydrated in ascending concentrations of alcohol (80–100%). After xylol treatment, slides were covered.
Two investigators were asked to evaluate each stained section independently without knowing any clinical information. The proportion of positive cells varied from 10 to 100%. Accordingly, the intensity of FLI1 staining was scored as 0 (negative), 1 (weak), 2 (moderate), and 3 (intense). The immunoreactivity score for each case was calculated as the percentage of positive cells per field multiplied by the intensity of staining.
Knockdown of FLI1 by RNA interference
FLI1-specific siRNAs were purchased from Invitrogen (CA, USA), siFLI1 1#: 5′-GGGAAAGUUCAC UGUUGGCCUAUAA-3′ and siFLI1 #2: 5′-AGGAGU GGAUCAAUCAGCCAGUGAG-3′. The control siRNA (siluc) against photinus pyralis luciferase gene (Invitrogen, CA) was 5′-GGAUUUCGAGUCGUCUUAAUGU AUA-3′. RNAiMAX transfection reagent was used for transient transfection following manufacturer’s protocol (Invitrogen, CA).
Cell proliferation assay
Breast cancer cell lines (MDA-MB468, MDA-MB435, MDA-MB231, MDA-MB43, MCF-7, ZR75-1) were purchased from ATCC and were cultured in DMEM containing 1% penicillin and streptomycin, supplemented with 10% fetal bovine serum (FBS). For cell proliferation assay, cells were seeded in 96-well plates with density 10% per well. The number of cell proliferation was measured by Trypan-blue exclusion assay from day 1 to day 4.
Cell migration and invasion assay
Cell migration and invasion assays were carried out using Transwell (Corning, MA) membrane filter inserted in 24-well tissue culture plates (6.5-mm diameter, 8-μm pore size) as previously described [41]. For migration assay, 4 × 104 cells suspended in serum-free medium were seeded on the upper chamber of transwell filters. Serum-containing medium was added to the lower chamber and incubated for 16 h at 37°C. The non-migrating cells were removed by wiping the upper side of the filter, and the migrated cells on the bottom side of the filter were fixed with 4% formaldehyde, stained with crystal purple and counted under a microscopy.
A similar protocol was used for the invasion assay, except that cells were seeded in Transwell chambers coated with 0.5 μg/μl type I collagen (Invitrogen, CA, #A1048301) [41]. Each assay represents the average of three independent experiments.
Western immunoblotting
Cells were lysed with immunoprecipitation assay buffer (1% Nonidet P-40, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, plus protease inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride). Proteins were separated by SDS-PAGE and analyzed by Western blotting. Antibodies to FLI1 and β-ACTIN were obtained from Santa Cruz Biotechnology (Santa Cruz Biotechnology, CA).
RhoA GTPase pathway assays
The Rac1 activity assay was performed using the method as previously described [10]. The active Rac1 was quantitated by the affinity binding to a GST-PBD fusion protein, consisting of glutathione S-transferase (GST) and the p21-binding domain (PBD) of the Rac1 effector p21-activated kinases (PAK). Briefly, after washing with phosphate-buffered saline (PBS), cells were lysed immediately with PBD lysis buffer (50 mM Tris-HCl, pH 7.2, 100 mM NaCl, 5 mM MgCl2, 10% glycerol, 1% NP-40, 1 mM DTT and complete protease inhibitor Cocktail). Cell lysates were clarified by centrifugation at 16, 000 g at 4°C for 10 min. Equal volumes of lysates were incubated with GST-PBD fusion proteins on glutathione S-transferase beads to pull down active Rac1 proteins. After incubation at 4°C for 2 hours, the beads were washed three times with cold PBD lysis buffer. Rac1 protein was eluted with sample buffer and subjected to SDS-PAGE. Western blot was performed using anti-Rac1 antibody. Protein bands were visualized with an enhanced chemiluminescence reagent (Amersham Biosciences, PA).
The RhoA activity assay was performed as previously described [11]. The activated RhoA was determined by the binding to a GST-RBD fusion protein, consisting of glutathione S-transferase (GST)-RhoA binding domain (RBD) of the RhoA effector rhotekin. Briefly, the same protocol as in the above section was performed except using Rho-binding domain fused to GST (GST-RBD) and RBD lysis buffer (50 mM Tris-HCl, pH 7.2, 500 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 1 mM DTT and Cocktail).
The activities of the AKT and ERK cascades were measured by Western blot as previously described [41].
Statistical analysis
Comparisons between groups were analyzed by t-test. We assessed score comparisons between groups by one-way ANOVA test. P value of less than 0.01 was considered significant. Statistical calculations were performed using SPSS 13.0.
ACKNOWLEDGMENTS
This work was supported by California Institute for Regenerative Medicine (CIRM) grant (RT2-01942), Jilin International Collaboration Grant (#20120720), the National Natural Science Foundation of China grant (#81272294, #31430021) to J.F.H; the National Natural Science Foundation of China grant (#81071920, #81372835) and Jilin Science and Technique Program grant (11GG003) to W.L.; and the grant of Key Project of Chinese Ministry of Education (#311015) to C.J.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA: A Cancer Journal for Clinicians. 2014; 64:9–29.
2. Liu F, Walmsley M, Rodaway A, Patient R. Fli1 Acts at the Top of the Transcriptional Network Driving Blood and Endothelial Development. Current Biology. 18:1234–1240.
3. Mélet F, Motro B, Rossi DJ, Zhang L, Bernstein A. Generation of a novel FLI1 protein by gene targeting leads to a defect in thymus development and a delay in Friend virus-induced erythroleukemia. Molecular and Cellular Biology. 1996; 16:2708–2718.
4. Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Molecular and Cellular Biology. 2000; 20:5643–5652.
5. Lakhanpal GK, Vecchiarelli-Federico LM, Li Y-J, Cui J-W, Bailey ML, Spaner DE, Dumont DJ, Barber DL, Ben-David Y. The inositol phosphatase SHIP-1 is negatively regulated by FLI1 and its loss accelerates leukemogenesis. Blood. 2010; 116:428–36.
6. Truong AH, Ben-David Y. The role of FLI1 in normal cell function and malignant transformation. Oncogene. 2000; 19:6482–6489.
7. Li YJ, Zhao X, Vecchiarelli-Federico LM, Li Y, Datti A, Cheng Y, Ben-David Y. Drug-mediated inhibition of FLI1 for the treatment of leukemia. Blood Cancer J. 2012.
8. Mhawech-Fauceglia P, Herrmann FR, Bshara W, Odunsi K, Terracciano L, Sauter G, Cheney RT, Groth J. Friend leukaemia integration-1 expression in malignant and benign tumours: a multiple tumour tissue microarray analysis using polyclonal antibody. Journal of Clinical Pathology. 2007; 60:694–700.
9. Fukata M, Kaibuchi K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nature reviews Molecular cell biology. 2001; 2:887–897.
10. Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999; 274:13198–13204.
11. Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008; 283:35622–35629.
12. Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. The Biochemical journal. 2005; 390:1–9.
13. Harding MA, Theodorescu D. RhoGDI signaling provides targets for cancer therapy. European journal of cancer. 2010; 46:1252–1259.
14. Chew TW, Liu XJ, Liu L, Spitsbergen JM, Gong Z, Low BC. Crosstalk of Ras and Rho: activation of RhoA abates Kras-induced liver tumorigenesis in transgenic zebrafish models. Oncogene. 2014; 33:2717–2727.
15. Robinson JD, Pitcher JA. G protein-coupled receptor kinase 2 (GRK2) is a Rho-activated scaffold protein for the ERK MAP kinase cascade. Cellular signalling. 2013; 25:2831–2839.
16. Chen J, Xia H, Zhang X, Karthik S, Pratap SV, Ooi LL, Hong W, Hui KM. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. Journal of hepatology. 2015; 62:S0168–8278.
17. Ben-David Y, Giddens EB, Bernstein A. Identification and mapping of a common proviral integration site FLI1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc Natl Acad Sci U S A. 1990; 87:1332–1336.
18. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992; 359:162–165.
19. Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. The New England journal of medicine. 1994; 331:294–299.
20. Paronetto MP. Ewing sarcoma protein: a key player in human cancer. International journal of cell biology. 2013; 2013:642853.
21. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R, Denny CT. The Ewing’s sarcoma EWS/FLI1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI1. Molecular and Cellular Biology. 1993; 13:7393–7398.
22. Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, Formey A, Patel A, Gymrek M, Thapar V, Deshpande V, Ting DT, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer cell. 2014; 26:668–681.
23. O’Hayre M, Degese MS, Gutkind JS. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Current Opinion in Cell Biology. 2014; 27:126–135.
24. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002; 420:629–635.
25. De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. The Journal of pathology. 2003; 200:429–447.
26. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nature medicine. 2006; 12:895–904.
27. Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nature reviews Molecular cell biology. 2010; 11:633–643.
28. Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast cancer research and treatment. 2004; 84:21–32.
29. Yang H-Y, Kim J, Lee K-Y, Jang Y-S. Rac/ROS-related protein kinase C and phosphatidylinositol-3-kinase signaling are involved in a negative regulating cascade in B cell activation by antibody-mediated cross-linking of MHC class II molecules. Molecular Immunology. 2010; 47:706–712.
30. Yang C, Liu Y, Lemmon MA, Kazanietz MG. Essential Role for Rac in Heregulin β1 Mitogenic Signaling: a Mechanism That Involves Epidermal Growth Factor Receptor and Is Independent of ErbB4. Molecular and Cellular Biology. 2006; 26:831–842.
31. Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997; 390:632–636.
32. Stevens EV, Banet N, Onesto C, Plachco A, Alan JK, Nikolaishvili-Feinberg N, Midkiff BR, Kuan PF, Liu J, Miller CR, Vigil D, Graves LM, Der CJ. RhoGDI2 antagonizes ovarian carcinoma growth, invasion and metastasis. Small GTPases. 2011; 2:202–210.
33. Wang H, Wang B, Liao Q, An H, Li W, Jin X, Cui S, Zhao L. Overexpression of RhoGDI, a novel predictor of distant metastasis, promotes cell proliferation and migration in hepatocellular carcinoma. FEBS letters. 2014; 588:503–508.
34. Zhang Y, Zhang B. D4-GDI, a Rho GTPase regulator, promotes breast cancer cell invasiveness. Cancer Res. 2006; 66:5592–5598.
35. Ota T, Maeda M, Okamoto M, Tatsuka M. Positive regulation of Rho GTPase activity by RhoGDIs as a result of their direct interaction with GAPs. BMC systems biology. 2015; 9:3.
36. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR Pathways: Cross-talk and Compensation. Trends in biochemical sciences. 2011; 36:320–328.
37. Meloche S, Pouyssegur J. The ERK1//2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007; 26:3227–3239.
38. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Advances in cancer research. 2005; 94:29–86.
39. McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Advances in enzyme regulation. 2006; 46:249–279.
40. Kang L, Sun J, Wen X, Cui J, Wang G, Hoffman AR, Hu JF, Li W. Aberrant allele-switch imprinting of a novel IGF1R intragenic antisense non-coding RNA in breast cancers. European journal of cancer. 2015; 51:260–270.
41. Zhang S, Zhong B, Chen M, Yang G, Li Y, Wang H, Wang G, Li W, Cui J, Hoffman AR, Hu J. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. Int J Cancer. 2014; 134:1583–1594.