
INTRODUCTION
Cancer is estimated to have claimed the lives of more than 8 million individuals in 2012 and remains a leading cause of death worldwide [1]. Increased knowledge concerning the etiology, pathobiology and treatment of cancer has meant that some patients now have a high chance of cure. However, despite promising advances in the treatment of some cancer-types, others continue to retain dismal survival rates, such as pancreatic cancer, which has usually metastasized at the time of diagnosis and is virtually impossible to cure [2].
Although many conventional chemotherapeutics extend lives, more potent and specific therapies are required to combat advanced disease. Recent advances in cancer research have been driven by a call to understand the molecular differences between normal and neoplastic cells in order to identify cancer-specific molecular targets. This need has generated the basis for the development of the next generation of chemotherapeutics, e.g., trastuzumab, erlotinib, etc., that have entered the clinics in the past decade and target key cell signaling pathways and effectors that are deregulated in cancers [3]. However, better therapies are still urgently needed to effectively treat more aggressive tumors, particularly those that have developed resistance and metastasized. One such strategy is to target cellular iron metabolism that is known to be markedly altered in cancer cells [4–7].
Considering that most cancers are characterized by the deregulation of multiple genes and pathways, therapeutics that can target multiple molecular signaling effectors present as promising strategies for the successful treatment of cancer. Herein, we review recent research on the targeting of iron for cancer therapy, with emphasis on the novel DpT class of thiosemicarbazone iron chelators. We will discuss recent advances in understanding how these potent compounds not only target cellular iron, but also multiple signaling pathways that are implicated in tumor progression and metastasis, and how they may be integrated into the current landscape of molecular targeted therapies for cancer treatment. These and continuing developments will help to facilitate the translation of these novel therapies as a potential multi-focal strategy for cancer treatment.
IRON AND CANCER
An excess of iron in the body is toxic due to its ability to engage in redox cycling and promote free radical formation [4]. Iron via the Fenton reaction can exacerbate the consequences of hydrogen peroxide production, leading to the generation of hydroxyl radicals, the most reactive chemical species in biological systems (Equation 1) [8, 9]. The superoxide ion can also participate in regenerating ferrous iron that is required for the Fenton reaction (Equation 2) [10].
(1)
(2)
These free radicals can instigate a series of chemical reactions, leading to DNA oxidation, mitochondrial damage and the peroxidation of membrane lipids, thereby initiating DNA damage, adversely affecting cell cycle progression and promoting cell apoptosis [8]. Therefore, it is crucial that iron levels are tightly regulated in the body via specialized proteins and processes responsible for transporting and storing iron in a soluble and non-toxic form [11]. Failure to maintain iron homeostasis in the body is associated with numerous disease states, including cancer, cardiovascular disease, diabetes mellitus, infections and neuro-degenerative disorders [5].
Iron metabolism
Iron from dietary sources is absorbed within the small intestine and is then transported from the duodenal enterocyte into the blood, where it binds to the iron-binding protein, transferrin [11–13]. Transferrin is present in physiological fluids, with each transferrin molecule able to bind two atoms of ferric iron with high affinity [14–16]. Transferrin plays a critical role in transporting bound iron from sites of absorption and heme degradation (i.e., the intestine and liver, respectively) to cells for critical functions such as erythropoiesis and storage in ferritin. The transferrin receptor 1 (TfR1) protein, which is present on the plasma membrane, is able to bind two transferrin molecules to facilitate iron uptake by cells [11, 14, 17].
The transferrin-TfR1 complex is then internalized into the endosome, where the ferric iron is released from transferrin due to the acidic environment and reduced to its ferrous form, at least in erythroid cells, by the ferrireductase, six-transmembrane epithelial antigen of the prostate 3 (STEAP3; Figure 1A) [18]. The iron can then be released across the endosomal membrane into the cytosol via the divalent metal transporter 1 (DMT1) [19] into the labile iron pool, a poorly characterized compartment where iron is proposed to complex with low molecular weight ligands, e.g., citrate, ATP, sugars, etc. [20, 21]. However, evidence for a low molecular weight pool of cellular iron in rapidly metabolizing cells was never substantiated [22]. In fact, more recently, cellular iron chaperones have been well characterized, i.e., human poly (rC)-binding proteins (i.e., PCBP1–4), which mediate the delivery of iron to ferritin and cytosolic non-heme iron-containing enzymes, i.e., dioxygenases [23–25].
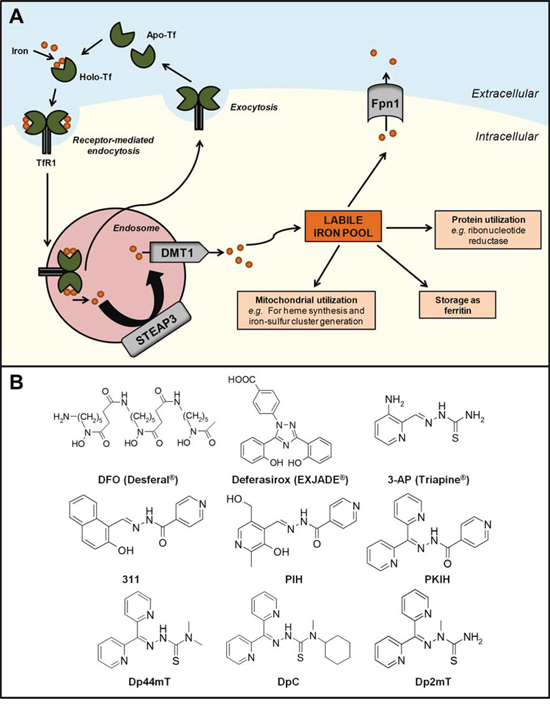
Figure 1: A. Cellular iron metabolism. Two atoms of ferric iron bind to transferrin (Tf) proteins present in the circulation to form the iron-saturated form of Tf (holo-Tf). Two molecules of holo-Tf can then bind to transferrin receptor 1 (TfR1) expressed at the cell membrane. The Tf-TfR1 complex undergoes endocytosis and is internalized into the endosome, inducing the release of Tf-bound ferric iron and reduction to its ferrous form by six-transmembrane epithelial antigen of the prostate 3 (STEAP3). Iron is then released into the cytosol via the divalent metal transporter 1 (DMT1) into the labile iron pool, where it can be: (i) incorporated into the active sites of proteins, such as ribonucleotide reductase; (ii) stored as cytoplasmic ferritin; (iii) utilized by the mitochondria; and (iv) exported out of the cell via the iron efflux pump, ferroportin 1 (Fpn1). The iron-deficient form of Tf (apo-Tf) and TfR1 molecules are subsequently recycled and returned to the cell surface. B. Chemical structures of key chelators discussed in this review. The structure of clinically available iron chelators, DFO and deferasirox, and anti-cancer thiosemicarbazone or PIH chelators, 3-AP and 311, respectively. The lead compounds of the first and second generation DpT thiosemicarbazones derived from PKIH analogues are shown, namely Dp44mT and DpC, respectively. Dp2mT is a structural analogue of Dp44mT that cannot bind iron and is used in studies as a negative control compound.
Cellular iron is utilized in multiple homeostatic processes. For instance, it can be: (i) incorporated into the active sites of proteins, e.g., ribonucleotide reductase (RR); (ii) stored in cytoplasmic ferritin; (iii) distributed to the mitochondria for heme synthesis, iron-sulfur cluster generation, or storage in mitochondrial ferritin; or (iv) exported out of the cell via the iron efflux pump, ferroportin 1 (Fpn1; Figure 1A) [5, 26]. Iron homeostasis is achieved, at least in part, by the iron-regulatory proteins (IRPs), which respond to intracellular iron concentrations and control protein expression at the post-transcriptional level, thereby regulating the uptake, usage and storage of iron [27, 28].
Altered iron metabolism in cancer cells
Deregulation of iron homeostasis in cancer cells compared to normal cells has been widely reported [5, 7]. These altered iron states in malignancies can occur through changes in iron uptake, efflux and storage, which may confer an increased iron acquisition phenotype by cancer cells to mediate their rapid proliferation [5, 29].
Iron uptake
Numerous studies have demonstrated that cancer cells have higher TfR1 levels compared to normal cells, and that higher TfR1 expression is correlated with more advanced cancers [5, 30–32]. This reflects the higher rate of iron uptake from transferrin seen in cancer cells [33]. Furthermore, antibodies against TfR1 have been shown to inhibit tumor growth [34]. The rate limiting enzyme required for DNA synthesis requires a continual supply of iron for its assembly and activity and is also up-regulated in cancer [35]. Hence, the rates of proliferation and DNA synthesis are increased in cancer cells relative to normal cells [35].
Iron uptake is also facilitated by the endosomal metalloreductase, STEAP3, which may reduce endosomal ferric iron bound to transferrin to its ferrous form in erythroid cells [18]. In fact, STEAP has been reported to be overexpressed in prostate, colorectal, lung, and pancreatic cancers, amongst others [36, 37], indicating another aspect of altered iron metabolism that may be linked with cancer.
Iron efflux
Studies have reported that the expression of the iron efflux pump, Fpn1, is decreased or absent in breast cancer, prostate cancer, hepatocellular carcinoma and leukemia, although these changes were not seen in brain or esophageal cancers [38–40]. Pinnix et al. [39] noted that reduction in Fpn1 levels occurred concomitantly with increased levels of the peptide hormone, hepcidin, which negatively regulates Fpn1 expression by promoting its internalization and degradation [41]. Moreover, decreased Fpn1 expression in breast cancer patients was correlated with poor prognosis and metastasis, independent of other risk factors, and was proposed as a prognostic marker for this disease [39], indicating potentially important roles for the Fpn1-hepcidin regulatory axis in oncogenic signaling.
Iron in cells can also be released via the action of the signaling molecule, nitric oxide (NO) [42, 43]. Interestingly, NO can directly bind iron and affect a variety of intracellular proteins and metabolic pathways [11, 44–47]. Furthermore, NO generated either intracellularly (via inducible nitric oxide synthase; iNOS) or extracellularly has been shown to result in marked release of iron from cells [48, 49]. In fact, in contrast to the mechanism of cellular iron release mediated by Fpn1 described above, NO-dependent iron release has been demonstrated to occur via the multi-drug resistance protein 1 (MRP1) [49, 50].
Iron storage
Studies in the literature also indicate that the regulation of iron storage is altered in tumors [5]. Assessment of ferritin expression levels in cancer cells and patient tissues have yielded conflicting conclusions, with some studies reporting up-regulation of ferritin [51], while others showing down-regulation of ferritin [5, 39]. Elevated serum ferritin levels have been observed in primary lung cancer [52], advanced stage C colorectal cancer [53], and neuroblastoma patients [54], which were all correlated with poorer survival rates or prognoses.
Interestingly, ferritin expression was reportedly decreased by the proto-oncogene, c-myc, which often has increased expression in human malignancies, allowing an increased availability of free iron for DNA synthesis and cell growth [55]. In addition, Kakhlon et al. [56] showed that HRAS-induced down-regulation of ferritin was sufficient to increase the labile iron pool and stimulate proliferation. Thus, aberrations in ferritin levels may facilitate tumor progression.
These alterations of iron metabolism in cancer demonstrate the potential for therapeutic manipulation by iron-binding ligands. In the past few decades, this has led to the discovery and development of iron chelators with anti-tumor activity [5].
IRON CHELATORS: FROM IRON OVERLOAD DISEASES TO CANCER TREATMENT
Clinically approved iron chelators
Iron chelation therapy was initially designed to prevent iron-mediated toxicity in iron overload diseases such as β-thalassemia [57]. In β-thalassemia, ineffective hemoglobin synthesis necessitates repeated blood transfusions to reverse the anemic state, and together with iron absorption from the gut, this leads to iron overload, resulting in potentially fatal tissue damage [58]. Sequestering iron from cells relieves the iron burden, thereby leading to the effective treatment of the iron overload associated with β-thalassemia therapy [58, 59].
Desferrioxamine (DFO), or Desferal®, is a hexadentate chelator isolated from the bacteria Streptomyces pilosus, and binds iron in a 1:1 ratio [60] (Figure 1B). It is currently the “gold standard” chelator used for the treatment of β-thalassemia [61]. However, due to its hydrophilicity and short plasma half-life, it requires administration by long subcutaneous infusion for periods of 8–12 hours [62]. This cumbersome and expensive method, along with pain and swelling at the injection site, has led to poor patient compliance [63], and the need to develop orally-active chelators.
The potential of iron chelators for the treatment of cancer first came to light in studies assessing the effect of DFO and other iron chelators on leukemia cells in culture and also in leukemia patients [64, 65]. This was subsequently followed by studies examining neuroblastoma tumor cells [66, 67] and then clinical trials in neuroblastoma patients, which showed promising results [68]. In these early studies, DFO administered in combination with other chemotherapeutic drugs, such as cyclophosphamide, etoposide and carboplatin, resulted in 50 out of 57 patients showing partial to complete positive responses [69]. However, the reported anti-tumor activity of DFO has been inconsistent, with a number of studies failing to demonstrate marked anti-tumor effects [70, 71]. This observation is likely due to the short half-life and poor membrane permeability of this compound that markedly limits its iron chelation efficacy and anti-tumor activity [4, 72, 73].
Many synthetic iron chelators have since been designed in attempts to increase efficacy, reduce toxicity and side effects, and improve oral activity [4]. More recently, the ligand 4-[3, 5-bis-(hydroxyphenyl)-1, 2, 4-triazol-1-yl]-benzoic acid (deferasirox) was successful in clinical trials in iron-loaded patients and released by Novartis as EXJADE® for the treatment of β-thalassemia as a relatively lipophilic, orally active alternative to DFO [74] (Figure 1B). Data from phase II and III trials showed that the efficacy of deferasirox was comparable to that of DFO in transfusion-dependent β-thalassemia patients [75, 76]. However, deferasirox appears to be better tolerated than DFO in both adults and children [76], and the number of serious adverse events has been relatively low [77].
In recent years, there has been growing interest in the potential of deferasirox as a potential anti-tumor agent, given that it is already clinically approved, well tolerated, and is orally administered [74]. Deferasirox has been reported to inhibit the growth of hepatoma, myeloid leukemia and mantle cell lymphoma cells [78–81]. It has also recently been demonstrated that deferasirox significantly inhibited the growth of DMS-53 lung tumor and esophageal (OE33, OE19, and OE21) tumor xenografts grown in mice, with no marked effects on biochemical and hematological indices [82, 83]. Interestingly, in a patient suffering chemotherapy-resistant acute monocytic leukemia, administration of deferasirox demonstrated a remarkable anti-leukemic effect resulting in complete remission [84]. Collectively, these studies suggest that deferasirox is a candidate chelator for chemotherapy, but further extensive studies are required to validate its clinical application.
Thiosemicarbazone chelators
In recent years, the development of more active chelators for cancer treatment has included a range of ligands [4, 6, 85, 86]. These compounds include: 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP; Triapine®; Figure 1B) [87], Tachypyridine [88], O-Trensox [89] and novel chelators derived from pyridoxal isonicotinoyl hydrazone (PIH; Figure 1B) [90, 91], di-2-pyridylketone isonicotinoyl hydrazone (PKIH; Figure 1B) [92] and hybrid ligands derived from aroylhydrazones and thiosemicarbazones [93]. To date, 3-AP (Triapine®) is the only other well characterized ligand [94] besides DFO that has reached phase I and II clinical trials specifically for the treatment of cancer [95, 96]. Administered to patients either alone or in combination with other chemotherapeutics, Triapine® could successfully reduce white blood cell counts by more than 50% in leukemia patients [97]. However, clinical trials using Triapine® have also reported a number of unwanted side effects including: anemia, thrombocytopenia, leucopenia, methemoglobinemia [98], neutropenia, hypoxia, hypotension, fatigue, nausea and vomiting [99]. The success of Triapine® in clinical trials has been further limited by the general lack of efficacy and failure of patients to respond, resulting in termination of studies before completion [100–102]. New classes of ligands have since been developed in attempts to overcome these negative effects and improve the potency of these anti-cancer agents.
Studies examining a range of PIH analogues demonstrated that 2-hydroxyl-1-naphthylaldehyde isonicotinoyl hydrazone (311; Figure 1B), possessed marked anti-proliferative activity and greater iron chelation efficacy compared to DFO [90, 91, 103]. This anti-proliferative effect was found to be due to the ability of 311 to deplete cellular iron pools required for RR activity and other processes, as well as to affect the expression of molecules regulating cell cycle progression [94, 103, 104].
Further structure-activity studies of aroylhydrazones, such as the PKIH analogues (Figure 1B) [92], and novel aroylhydrazone/thiosemicarbazone hybrid ligands [93], led to the identification of key structural characteristics important for iron chelating efficacy and potent anti-proliferative activity. From this analysis emerged the development of the di-2-pyridylketone thiosemicarbazone (DpT) series of iron chelators, of which di-2-pyridylketone-4, 4-dimethyl-3-thiosemicarbazone (Dp44mT; Figure 1B) showed far more potent chelating efficacy and anti-proliferative activity than both DFO and 311 [105]. This activity was demonstrated in vitro in SK-N-MC neuroepithelioma, SK-Mel-28 melanoma and MCF-7 breast cancer cells, as well as in vivo using cytotoxic drug-resistant, M109 mouse lung carcinoma tumors [105]. In these studies, the specifically synthesized DpT analogue, di-2-pyridylketone 2-methyl-3-thiosemicarbazone (Dp2mT; Figure 1B), which cannot bind metals, had no significant effect on proliferation [105]. This observation demonstrated that the potent activity of the DpT series was due to its ability to specifically interact with metal ions. Comparison of the activities of Dp44mT with Triapine® both in vitro and in vivo showed that Dp44mT displayed markedly and significantly greater anti-tumor activity across a range of 28 tumor cell lines, including etoposide- and vinblastine-resistant cells, indicating it could overcome resistance to other anti-tumor agents [106]. In mouse xenograft studies, Dp44mT was also more potent and effective at inhibiting tumor growth than Triapine® and was less toxic [106]. The potent anti-proliferative and anti-metastatic effects of Dp44mT have also been demonstrated in other studies in vivo using breast [107] and hepatocellular carcinoma cells [108]. Further, Dp44mT was shown to induce apoptosis via the mitochondrial pathway, where decreased Bcl-2 and increased Bax expression was observed with holo-cytochrome c release and caspase activation [105, 109, 110]. This indicated important roles for the mitochondria in mediating the molecular events that lead to chelator-induced cell death.
Attempts to further elucidate the potent anti-proliferative effects of Dp44mT revealed two important properties, namely: (1) that it had a high lipophilicity compared to DFO, enabling it to easily permeate cell membranes; and (2) that the iron complex had an appropriate redox potential to facilitate redox activity which could lead to cytotoxicity [111]. Moreover, Dp44mT was shown to enter the lysosome and become trapped due to its ionization properties and the acidic pH of this organelle [110]. Subsequently, Dp44mT binds iron and copper to form redox-active complexes that generate cytotoxic ROS, enabling permeabilization of the lysosomal membrane and induction of apoptosis [110]. More recently, it was demonstrated that Dp44mT is a substrate of P-glycoprotein (Pgp) that is also found on lysosomal membranes, and thus, mediating lysosomal trapping of Dp44mT and enhancing cytotoxicity in multidrug-resistant cells [112]. This finding revealed a novel mechanism by which Dp44mT could overcome multidrug-resistance involving the ‘hijacking’ of the lysosomal-Pgp transport system that normally mediates resistance [112].
The observation that high, non-optimal intravenous doses of Dp44mT resulted in cardiac fibrosis in nude mice [106] led to the development of the second generation of DpT analogues [5, 109, 113]. Of this series, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC; Figure 1B) is the lead compound that is currently undergoing clinical development through Oncochel Therapeutics LLC (San Francisco, CA). Significantly, DpC has demonstrated marked and selective anti-neoplastic activity when administered orally or intravenously, without inducing the cardiotoxicity of Dp44mT [109, 113]. Moreover, DpC was more effective than Dp44mT at inhibiting the growth of PANC-1 pancreatic tumor xenografts in vivo [109]. Elucidation of the molecular effects of Dp44mT and DpC is ongoing and critical for understanding their potent mechanism of action.
Recent advances in our knowledge of how iron chelators can modulate key cellular signaling pathways deregulated in cancer will be discussed in the next sections. In general, and where suitable comparisons can be drawn, these studies have demonstrated that iron chelators of different classes can modulate these signaling pathways in a similar manner, indicating that the underlying iron-binding activities of these compounds are important for their observed molecular effects. Differences in the properties of chelators, such as the ability of DpT chelators to generate cytotoxic ROS, may play additional roles in modulating these pathways, and are discussed in the relevant sections.
MODULATION OF CELLULAR SIGNALING PATHWAYS BY IRON CHELATORS
The cell cycle
It is now clear that the anti-proliferative effects of the novel chelators discussed above are due not only to their ability to bind iron, but also to their complex effects on multiple intracellular targets. Iron chelators are known to target multiple molecules which are critical in regulating progression of the cell cycle, including RR, cyclins, cyclin-dependent kinases (CDKs), retinoblastoma protein (Rb), p53, p21WAF1/CIP1, p27KIP1, N-myc downstream regulated gene 1 (NDRG1), and apoptotic proteins [104, 114].
Ribonucleotide reductase
RR is an enzyme responsible for catalyzing the synthesis of deoxyribonucleotides required for DNA replication, the rate limiting step of DNA synthesis, cell cycle progression and cellular repair [115]. Human RR consists of two homodimers, namely, R1, which contains the active site and binding site for allosteric modulators, and R2, which contains a di-nuclear iron center and stable tyrosyl radical per monomer crucial for enzymatic activity [116]. Furthermore, while the R2 subunit is necessary for normal DNA synthesis during the S/G2 phase of the cell cycle, a variant of the R2 subunit, the p53R2 subunit is known to supply dNTPs for DNA repair following DNA damage in a p53-dependent manner in G0/G1 phase cells [117]. Both the R2 and p53R2 subunits possess an iron-binding site that is critical for their enzymatic function and iron chelation is known to inhibit the activity of RR [118]. Considering increased RR activity has been associated with malignant transformation and cancer cell growth [119], the ability of iron chelators to inhibit its activity is likely to be important for their anti-cancer effects (Figure 2).
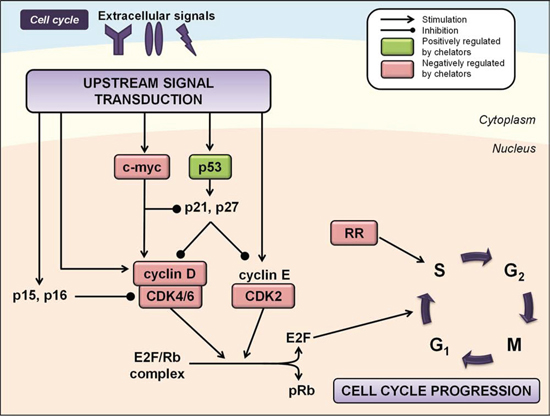
Figure 2: Iron chelation modulates multiple regulators of the cell cycle. Regulation of cell cycle progression is mediated by the integration of multiple cues from upstream signal transduction pathways. Iron chelation has been demonstrated to modulate the activities of ribonucleotide reductase (RR), cyclin/cyclin-dependent kinase (CDK) complexes, CDK inhibitors, and other cell cycle regulators including p53 and c-myc (see text for more detail). Molecules known to be negatively regulated by iron chelators are indicated by red boxes, while molecules known to be positively regulated by iron chelators are indicated by green boxes.
Cyclins and CDKs
Iron depletion by chelators has been shown to affect the expression of a number of cyclins and CDKs [104]. Iron chelation in SK-N-MC neuroepithelioma cells reduced expression of cyclins D1, D2 and D3, and to a lesser extent, cyclins A and B [104]. Inhibition of cyclin D1 expression by DFO and DpT chelators has been demonstrated in prostate and pancreatic cancer cells [120, 121], and by deferasirox in DMS-53 lung carcinoma cells [82] and mantle cell lymphoma cells [80]. This latter study also demonstrated deferasirox-mediated down-regulation of phosphorylated Rb (pRb), resulting in increased levels of the E2F/Rb complex and G1/S phase cell cycle arrest (Figure 2) [80]. Studies have also shown that iron chelators can decrease expression of CDK2 or CDK4, although this appears to occur in a cell-type-dependent manner and varies under different experimental conditions [94, 104, 122] (Figure 2).
p53
Iron chelation using DFO has been reported to up-regulate expression of the tumor suppressor, p53, at the post-transcriptional level [123]. Importantly, p53 has a critical role at the G1/S checkpoint and is activated in conditions of cell stress or DNA damage to either initiate repair mechanisms or induce apoptosis if damage is irreparable [124]. Incubation of cells with the chelators, DFO and 311, has also been shown to increase nuclear p53 protein and p53 DNA-binding activity (Figure 2) [125]. More recent studies have suggested that iron regulates p53 activity through direct interactions between heme and the p53 protein, which triggers nuclear export and cytosolic degradation of p53 [126]. Clearly, this latter effect is opposite to that observed after iron depletion and occurs when cells are iron replete.
CDK inhibitors
The CDK inhibitor, p21WAF1/CIP1, negatively regulates the cell cycle by binding to G1/S phase CDK-cyclin complexes, rendering them inactive [127]. This allows Rb to bind E2F1, thereby preventing the G1/S phase transition and causing cell cycle arrest. However, p21WAF1/CIP1 expression is required at low levels for the formation of cyclin D/CDK complexes [128], and it has also been shown to inhibit doxorubicin-induced apoptosis through repressing caspase-3 activation [129]. Iron chelation using DFO and 311 in MCF-7 breast adenocarcinoma cells has been shown to up-regulate p21 mRNA, while decreasing the expression of p21WAF1/CIP1 protein [130]. MCF-7 cells have abnormally high levels of p21WAF1/CIP1 expression, which probably provides these cells with a survival advantage by allowing suppression of apoptosis [131]. Therefore, post-transcriptional regulation of p21WAF1/CIP1 by DFO and 311 confers a mechanism by which these chelators inhibit the proliferation of cells which over-express p21WAF1/CIP1 [130]. Incubating other cell-types, namely DMS-53 lung carcinoma and SK-N-MC cells, with the iron chelators DFO, Dp44mT, or deferasirox, increased p21WAF1/CIP1 expression, which would likely contribute to the anti-tumor activities of these latter two compounds in vitro and in vivo [82].
c-Myc
Although not classical regulators of the cell cycle, Myc proteins have profound effects on enabling cells to enter the cell cycle and accelerating cell cycle progression [132]. Myc lies at the “crossroads” of multiple signal transduction pathways, including the mitogen-activated protein kinase (MAPK), transforming growth factor-β (TGF-β), Wnt and signal transducer and activator of transcription (STAT) pathways [133, 134]. In fact, induction of c-myc proto-oncogene expression is required for a robust cell cycle response to mitogenic signals [135]. This likely occurs through its ability to positively regulate cyclin D/CDK4/6 activity and repress transcription of p21WAF1/CIP1 [136, 137]. Iron chelation using DFO has been shown to decrease c-myc expression in leukemic blood cells [138]. Further, the chelators, DFO, Dp44mT and DpC, have been recently demonstrated to reduce c-myc levels in pancreatic and prostate cancer cells (Figure 2) [121]. The mechanism mediated by these agents could at least partially involve inhibition of the STAT3 pathway and subsequent expression of its downstream targets, including cyclin D1, c-myc, and Bcl-2 (discussed in further detail below) [121].
N-myc downstream regulated gene 1 (NDRG1)
NDRG1 (also known as differentiation related gene 1 or Cap-43) is a metastasis suppressor gene that encodes a protein with a range of significant cell functions including cell cycle regulation, differentiation, angiogenesis, maintenance of the myelin sheath of Schwann cells and mast cell maturation [139]. In fact, its pleiotropic nature is known to be cell-type-dependent [140]. While NDRG1 is widely expressed and prominent in normal cells and tissues [141], its expression is significantly lower in various cancers, including breast, colon and prostate tumors, particularly in patients with bone or lymph node metastases [142–144]. The expression of NDRG1 has also been demonstrated to have a significant inverse correlation with the Gleason grading and overall survival rate of prostate cancer patients [142], as well as the depth of tumor invasion in pancreatic adenocarcinoma patients [145]. These findings indicate an important role for NDRG1 in regulating tumor metastasis and subsequently render it an attractive target for the treatment of cancer and its spread. It is well established that intracellular iron levels regulate the expression of NDRG1, with iron-depletion markedly up-regulating NDRG1 at both the mRNA and protein levels in a variety of different cancer cell-types [146, 147]. This occurs via both hypoxia inducible factor 1α (HIF-1α)-dependent and -independent mechanisms [146]. Moreover, it was recently shown that translation of NDRG1 is enhanced under conditions of cellular iron depletion via eukaryotic initiation factor 3a (eIF3a) activity [148], which may correlate to the HIF-1α-independent mechanism previously identified [146].
NDRG1 is phosphorylated by serum and glucocorticoid-induced kinase-1 (SGK1) at Ser330 and Thr346, priming it as a substrate for glycogen synthase kinase 3 β (GSK3β), which can further phosphorylate NDRG1 at Ser342, Ser352 and Ser362 [149]. The ligands, DFO and Dp44mT, can increase expression of phosphorylated NDRG1 [109, 120]. However, the functional implications of NDRG1 phosphorylation have not been extensively explored in the literature and remain largely unclear. Recent studies by Murakami et al. [150] have suggested that in the human pancreatic cancer cell line, MIAPaCa-2, phosphorylation of NDRG1 at Ser330 and Thr346 is essential for its tumor-suppressive action on CXC chemokine expression and the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling pathway. Further, phosphorylated NDRG1 was found to be localized to centromeres and the cleavage furrow in dividing cells, implying roles for this protein in microtubule organization, the cell cycle and mitosis [151]. Of interest, recent studies demonstrated that NDRG1 is proteolytically cleaved at the N-terminus in the prostate cancer cell lines DU145, PC3 and LNCaP, but not in normal prostate epithelial cells [152]. This observation suggests that truncation of NDRG1 in cancer cells could abrogate its tumor growth and metastasis suppressive function.
Significant advances have been made in recent years in terms of dissecting the molecular mechanism by which NDRG1 exerts its tumor growth and metastasis suppressive activities (reviewed in [153, 154]). Importantly, NDRG1 alters the expression of a variety of transcription factors and genes involved in ribosome and protein synthesis and reduces the expression of cathepsin C, a molecule which has roles in invasion, in pancreatic cancer [147]. Additionally, whole genome gene array analysis revealed NDRG1 up-regulates thiamine triphosphatase (Thtpa) expression, which decreases levels of thiamine triphosphate that is required for cellular energy metabolism [147].
NDRG1 also up-regulates the CDK inhibitor, p21WAF1/CIP1, which may mediate its anti-tumor effects [155]. Further important roles for NDRG1 in the regulation of multiple key cell signaling pathways (e.g., phosphatidylinositol-3 kinase (PI3K)/AKT, MAPK and TGF-β pathways) which affect the epithelial-to-mesenchymal transition (EMT) and metastasis have also been identified [107, 109, 120, 156, 157]. These roles have been implicated in mediating, in part, the anti-tumor and anti-metastatic activities of the potent thiosemicarbazones, Dp44mT or DpC, due to the ability of these compounds to markedly up-regulate NDRG1 [107, 109, 120, 156, 157]. These findings will be discussed in further detail in the relevant upcoming sections.
PI3K/AKT/mTOR signaling
The PI3K/AKT pathway has emerged in recent years as a key regulator of mammalian cell proliferation, growth and survival [158] (Figure 3). Binding of insulin or insulin-like growth factors to a receptor tyrosine kinase (RTK), such as insulin growth factor (IGF) binding to insulin growth factor receptor (IGF-R) [159], results in direct interaction between the RTK and regulatory subunits of class I PI3Ks (e.g., p85α, p85β, p55γ) [160, 161]. This enables the PI3K p110α/β/δ catalytic subunits to exhibit their kinase activity and catalyze the phosphorylation of phosphatidylinositol-4, 5-biphosphate (PIP2) to phosphatyidylinositol-3, 4, 5-triphosphate (PIP3) [160, 161]. The generation of PIP3 in the membrane then acts as a second messenger to activate downstream signaling effectors, including the serine/threonine kinase AKT. Specifically, PIP3 provides a docking site to recruit 3-phosphoinositide-dependent kinases (PDKs), to which AKT subsequently binds to form the phosphorylated form of AKT (p-AKT) that is active [162]. PIP3 levels are tightly controlled in mammalian cells by the action of PIP3 phosphatases, the most well characterized of which is the tumor-suppressor, phosphatase and tensin homolog deleted on chromosome 10 (PTEN; Figure 3) [163].
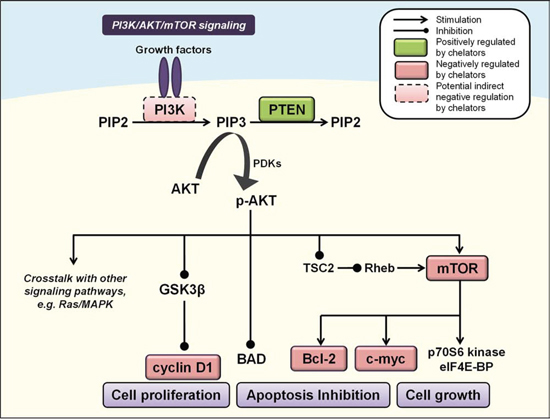
Figure 3: Iron chelators modulate PI3K/AKT signaling. Activated PI3K catalyzes the phosphorylation of PIP2 to PIP3, leading to phosphorylation and activation of AKT (p-AKT). PIP3 levels are controlled by the phosphatase PTEN to inhibit p-AKT, which affects multiple cascades that control cell growth, proliferation and apoptosis, including GSK3β, mTOR, cyclin D1, c-myc, BAD and Bcl-2 (see text for more detail). Molecules negatively regulated by iron chelators are indicated by red boxes, and those positively regulated are indicated by green boxes. Solid boxes indicate direct evidence for modulation by iron chelators, while dashed boxes indicate potential indirect modulation, e.g., through chelator-mediated up-regulation of NDRG1.
The activation of AKT occurs via phosphorylation of two residues, Ser473 and Thr308 [164], and leads to multiple signaling cascades with important consequences for cell survival, proliferation and growth [158]. PI3K/AKT signaling functions as an anti-apoptotic pathway by phosphorylating a pro-apoptotic member of the B-cell lymphoma 2 (Bcl-2) family of proteins, i.e., Bcl-2-associated death promoter (BAD; Figure 3), which forms a non-functional heterodimer with anti-apoptotic B-cell lymphoma-extra large (Bcl-XL) [165].
Activated AKT promotes cell proliferation by inhibiting GSK3β, a protein which increases cyclin D1 degradation and suppresses its expression [166, 167] (Figure 3). AKT can also regulate the activity of a number of transcription factors including cyclic-AMP response element binding protein (CREB) [168], E2F [169], NF-κB [170] and the forkhead transcription factors [171, 172], all of which can promote cell proliferation or survival.
Another major target of PI3K/AKT signaling is the serine/threonine kinase, mammalian target of rapamycin (mTOR). The activation of mTOR could occur through a number of mechanisms including direct interactions with p-AKT itself [173], or p-AKT-mediated inhibition of tuberous sclerosis 2 (TSC2), which then inhibits Rheb and leads to activation of mTOR [174–176] (Figure 3). Activation of mTOR has central roles in protein translation and cell growth, by activating p70S6 kinase and eukaryotic translation initiation factor 4E binding proteins (eIF4E-BP), which enhance mRNA recruitment to the ribosome for translation [177]. The mTOR pathway also regulates the Bcl-2 proteins involved in apoptosis and c-myc, thereby promoting cell survival, proliferation and oncogenesis [178, 179] (Figure 3).
PI3K/AKT signaling is inappropriately activated in many cancers by various mechanisms [180]. The first genetic mechanism identified that results in aberrant PI3K/AKT activation was the loss of the tumor suppressor, PTEN [180–182]. PTEN inactivation is seen in endometrial, central nervous system, prostate, colon, breast and liver tumors, amongst others [161]. Indeed, loss of PTEN expression has been associated with a higher Gleason grading in primary prostate tumors, with metastases displaying an accumulation of PTEN mutations [180].
Somatic activating mutations of the PI3K catalytic subunit, p110α, which is encoded by the PIK3CA gene, occur frequently in human cancers, including those of the breast, prostate, colon, and endometrium [161]. Moreover, some tumors (e.g., glioblastomas, colon and ovarian) also harbor activating mutations of the p85α regulatory subunit of PI3K, which is encoded by the PIK3R1 gene, leading to constitutive PI3K-AKT signaling [183]. Furthermore, constitutively activated p-AKT is found in 30–40% of solid tumors [184], and high levels of p-AKT in prostate cancer have been associated with a poorer prognosis for patients [185].
Hyperactivation of PI3K-AKT signaling in cancer has formed the underlying basis for targeted therapies against this cascade that are currently in clinical development, including dual PI3K-mTOR inhibitors, PI3K inhibitors, AKT inhibitors, and mTOR complex catalytic site inhibitors [186]. Preclinical data suggests that such agents may be effectively used to overcome acquired resistance to therapies that target RTKs, such as trastuzumab and erlotinib, and in combination with other targeted therapies, such as mitogen-activated protein kinase kinase (MEK) inhibitors [180].
Recent studies have elucidated how iron chelators target the PI3K/AKT/PTEN pathway in prostate cancer as part of their anti-tumor activity [120]. Incubation of prostate epithelial cells and the DU145 prostate cancer cell line with DFO or Dp44mT increased expression of tumor-suppressive PTEN and anti-metastatic NDRG1 [120]. Moreover, NDRG1 over-expression attenuated oncogenic p-AKT activity and significantly increased PTEN levels [120]. NDRG1-mediated inhibition of p-AKT was also observed in PTEN-null PC3 cells [107, 120], suggesting that this effect occurred independently of PTEN.
In MIAPaCa-2 pancreatic cancer cells, NDRG1 over-expression also increased PTEN levels and decreased p-AKT and p-mTOR levels [187]. Further, expression of the PI3K regulatory subunits phospho-p85α and phospho-p55γ was significantly reduced with NDRG1 over-expression, which prevents complex formation of the PI3K p110α/β/δ catalytic subunits [187]. Given that the thiosemicarbazones, Dp44mT and DpC, markedly up-regulate NDRG1 in MIAPaCa-2 and other pancreatic cancer cell lines [109], this suggests that NDRG1 could also mediate its demonstrated anti-tumor activity in pancreatic cancer through modulation of these critical molecules in the PI3K/AKT/PTEN cascade.
Interestingly, iron chelators have been reported to increase p-AKT levels [120, 188–190]. However, this may be a short-term stress-induced response that is not characterized by its classic signaling cascade, and may be induced by these compounds before their demonstrated anti-proliferative effects overwhelm this initial pro-survival response. In fact, this elevation in p-AKT levels was not found to correlate with downstream activation of mTOR or increased cyclin D1 expression in prostate cells [120], and was consistent with previous reports that DFO inhibited cyclin D1 expression [191].
Moreover, studies in K562 human myeloid leukemia cells have demonstrated that the orally active iron chelator, deferasirox, actually represses mTOR signaling [78]. This was observed to occur via enhanced expression of regulated in development and DNA damage response protein 1 (REDD1) and its downstream target, TSC2, which inhibits mTOR activity. Moreover, deferasirox repressed expression of S6 ribosomal protein and phosphorylated S6, a downstream target of mTOR [78]. Given the potential of clinically available deferasirox to be utilized as a chemotherapeutic agent [82, 83], this reveals an important mechanism by which deferasirox can modulate a key downstream mediator of oncogenic PI3K/AKT signaling, which can also integrate signals from other pathways (e.g., MAPK and TGF-β; see relevant sections below Figures 4A, 4B) [192, 193].
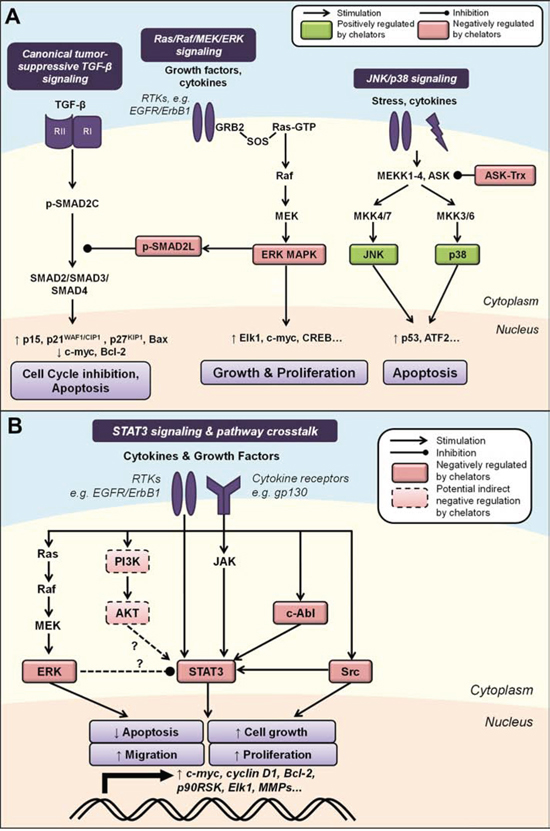
Figure 4: A. Iron chelation modulates mitogen-activated protein kinase (MAPK) signaling. Activated by growth factors and cytokines, the Ras/Raf/MEK/ERK cascade regulates cell growth and proliferation through multiple downstream effectors. ERK can also inhibit canonical TGF-β signaling by phosphorylating SMAD2 at the linker region (p-SMAD2L), preventing nuclear translocation of the SMAD2/3/4 complex and subsequent tumor-suppressive effects. The JNK and p38 pathways are two parallel MAPK cascades that are also classically activated by stress stimuli and may primarily mediate pro-apoptotic processes. Iron chelation is known to modulate the activities of ASK by dissociating its complex with thioredoxin (Trx), as well as JNK, p38, ERK and p-SMAD2L (see text for more detail). B. Modulation of STAT3 signaling by iron chelators and integrated pathway crosstalk between the MAPK and AKT pathways. STAT3 signaling is classically activated by cytokine receptor interactions, e.g., IL6/JAK, but can also be induced by growth factor receptor tyrosine kinases, e.g., EGFR, and non-receptor kinases, e.g., Src and c-Abl. Evidence also exists for potential STAT3 pathway crosstalk between the ERK and AKT pathways. Iron chelation has been demonstrated to inhibit STAT3 activity, which could be mediated by chelator-mediated effects on upstream regulators of STAT3 (see text for details). Molecules that are negatively regulated by iron chelators are indicated by red boxes, and those positively regulated are indicated by green boxes. Solid boxes indicate direct evidence for modulation by iron chelators, while dashed boxes indicate potential indirect modulation, e.g., through chelator-mediated up-regulation of NDRG1. Dashed arrows indicate interactions that remain to be fully characterized (see text for details).
Ras/Raf/MEK/ERK signaling
Mammalian cells possess four major MAPK protein kinase cascades [194]. These include the Ras/Raf/MEK/ERK pathway that is typically activated by growth factors and cytokines, and the c-Jun N-terminal kinase (JNK), p38 and ERK5 pathways that can be activated by environmental stress [194]. The Ras/Raf/MEK/ERK cascade regulates a range of important cellular processes including proliferation, survival, differentiation, migration and angiogenesis [195]. It is activated at the cell surface upon binding of ligands to RTKs such as epidermal growth factor receptor (EGFR; also known as ErbB1 or HER1), which was the first RTK to be discovered [196] (Figure 4A). Three additional family members have been subsequently identified, namely ErbB2/HER2, ErbB3/HER3 and ErbB4/HER4 [197]. The binding of a growth factor to its receptor (e.g., epidermal growth factor (EGF) binding to EGFR), induces an extracellular conformational change that facilitates the dimerization of two receptor monomers [198]. This enables the auto-phosphorylation of tyrosine residues in the cytoplasmic kinase domains of the receptors that creates a high-affinity binding site for the Src homology 2 (SH2) domain of growth factor receptor-bound protein 2 (GRB2), thereby recruiting it to the membrane [197]. Subsequently, the two Src homology 3 (SH3) domains of GRB2 interact with son of sevenless (SOS), which facilitates the activation of the intracellular transducer, Ras, by the exchange of GDP with GTP [199] (Figure 4A).
Ras was the first identified oncogene involved in neoplastic transformation, and is considered a key player in regulating cell growth, integrating growth factor signals with crucial effectors that relay these signals to the nucleus [200–202]. Activated RAS (RAS-GTP) subsequently recruits and activates one of its main effectors, the mitogen-activated protein kinase kinase kinase (MAPKKK, or MEKK), Raf [195]. Raf phosphorylates mitogen-activated protein kinase kinase (MAPKK, or MEK), which then phosphorylates the mitogen-activated protein kinases (MAPKs), otherwise known as extracellular signal-regulated kinases (ERKs; Figure 4A) [195]. Activated MAPKs can enter the nucleus to phosphorylate and regulate the activity of many transcription factors, including c-myc, Elk1, the O subclass member of the forkhead family of transcription factors (FOXO3), and CREB, to promote cell growth and proliferation (Figure 4A) [195]. The Ras/Raf/MEK/ERK pathway can also inhibit apoptosis by phosphorylating and inactivating pro-apoptotic Bim and BAD, thus promoting the anti-apoptotic roles of Bcl-2 and Bcl-xL [203–205]. Moreover, ERK has been implicated in the induction of cell motility and metastasis through its ability to enhance the activities of matrix metalloproteinases 2/9 (MMP-2/9) [206] and myosin light-chain kinase (MLCK) [207].
Over-expression of EGFR is observed in more than 50% of carcinomas, and ErbB2 over-expression is seen in 30% of breast cancers [208]. Mutations in Ras occur in more than 90% of pancreatic cancers, and are frequently found in non-small-cell lung carcinomas, liver cancers, melanomas and thyroid malignancies [209]. Identification of these oncogenes has led to the development of targeted therapeutics directed against critical molecules involved in the EGFR-Ras-MAPK pathway [194]. Indeed, most successful targeted therapies in the clinic are primarily directed against tyrosine kinases involved in this pathway, such as EGFR, ErbB2, BCR-ABL and KIT [210, 211].
Recently, we demonstrated that DFO and Dp44mT could regulate the Ras/Raf/MEK/ERK cascade by decreasing phosphorylation of ERK1/2 in PC-3 and DU145 prostate cancer cells [120] (Figure 4A). Dp44mT could also inhibit ERK1/2 activity in hepatocellular carcinoma cells [108]. Given that ERK is activated by a range of mitogenic stimuli and represents a critical convergence point for multiple mitogenic signaling pathways [195], the ability of iron chelators to attenuate its activation could be an important mechanism by which they exert their anti-cancer effects. Subsequent inhibition of effectors downstream of ERK by these agents have also been demonstrated, including mTOR [78] and c-myc [121].
JNK/p38
The JNK and p38 pathways are two parallel MAPK pathways that are activated by various stress stimuli, including cytokines, ultraviolet radiation and oxidative stress [212]. These pathways participate in signaling cascades controlling cellular responses to such stimuli, and in contrast to ERKs, may primarily mediate anti-proliferative and pro-apoptotic processes [213] (Figure 4A). However, the complexity of stress-activated signaling networks means that this dichotomy is likely an oversimplification, with the effects of these MAPKs being context and cell-type dependent [214].
The JNK and p38 cascades can be activated by at least four MAPKKKs, including MEKK1-4, apoptosis signal-regulating kinase 1 (ASK1), TGF-β activated kinase 1 (TAK1) and mixed-lineage kinase (MLK) [214]. These MAPKKKs subsequently activate two distinct subgroups of MAPKKs, namely MKK4/7 and MKK3/6 that activate JNK and p38 MAP kinases, respectively [215] (Figure 4A). JNK and p38 regulate the activity of several downstream kinases and transcription factors, including p53, activating transcription factor 2 (ATF2), CREB and ETS domain-containing protein (Elk1), to affect cell transcription, proliferation and survival [215, 216]. While altered expression of the MAPK proteins, JNK and p38, are often observed in human tumor cell lines and specimens, there is little evidence that these are causally involved in tumorigenesis and tumor progression [217]. Interestingly, homozygous deletion or reduced expression of MKK4 was reported in 75% of ovarian serous carcinomas, which may contribute to development of this tumor type [218]. There is also evidence that MKK4 is mutated in pancreatic, breast, colon, lung and testis cancers, albeit at a low frequency of genetic inactivation of around 5% [219].
A role for iron depletion in mediating MAPK signaling was demonstrated in human lymphocytes, where DFO-induced apoptosis was mediated by activation of the JNK and p38 MAPKs [220] (Figure 4A). Activation of the p38 pathway was supported by phosphorylation of the upstream pathway proteins, MKK3 and MKK6, as well as the downstream targets, ATF2 and MAPK-activated protein kinase 2 (MAPKAPK2) [220].
More recent studies demonstrated that incubation of DMS-53 lung cancer cells with the chelators, DFO and Dp44mT, also increased JNK and p38 phosphorylation, resulting in increased phosphorylation of the downstream targets, p53 and ATF2 [221] (Figure 4A). Iron supplementation experiments indicated that the ability of DFO and Dp44mT to bind iron were crucial for their effects on JNK activation, and that the ability of Dp44mT to promote reactive oxygen species (ROS) generation also contributed to this effect [221]. DFO- and Dp44mT-induced JNK activation was found to be mediated by Trx oxidation following iron chelation, causing dissociation of the ASK1-Trx complex that normally inhibits ASK1 kinase activity and subsequent ASK1-dependent apoptosis [221, 222].
However, studies in other cell-types have reported some variable effects of chelators on MAPK signaling pathways. For instance, using immortalized human oral keratinocytes and the head and neck squamous cell carcinoma HN4 cell line, incubation with DFO resulted in increased levels of phosphorylated ERK1/2 and p38 MAPKs, but with no effect on JNK activation [223]. Moreover, it was shown that activation of ERK and p38 were key effectors mediating downstream apoptotic signaling through the caspase cascade and mitochondrial release of cytochrome c [223]. DFO also activated ERK, p38, and JNK in HL-60 human myeloid leukemia cells that was accompanied by a similar activation of the apoptotic cascade [224].
The observation that DFO increases phosphorylation of ERK in this latter investigation is in contrast to our previous study using prostate cancer cells that demonstrated suppressed phosphorylated ERK levels in response to DFO and Dp44mT [120]. This may indicate potential cell-type differences in the regulation of MAPK signaling and the complexities involved in cellular pathway regulation by iron depletion. However, the reported effects of iron chelation on activating p38 appear to be more consistent, and it has been suggested that p38 functions as a tumor suppressor since inhibition of p38 function plays an important role in Ras-induced transformation [225]. Moreover, p38 MAPK was found to be a major mediator of DFO-induced apoptosis over other MAPKs [224], suggesting that the ability of iron chelators to modulate this MAPK may be critical for their cytotoxic effects.
Canonical tumor-suppressive TGF-β signaling
The TGF-β pathway is activated when the cytokine TGF-β binds to the TGF-β receptor II (TGF-βRII) subunit expressed on cell membranes [226]. This recruits the TGF-β receptor I (TGF-βRI) subunit to the complex and causes phosphorylation of serine and threonine residues on TGF-βRI, which activates its kinase activity [226]. The receptor SMADs, SMAD2 and SMAD3, which are normally anchored by the cytoskeletal protein filamin A, are translocated to the active TGF-βRI/II complex where they are also phosphorylated at the C-termini at Ser465/467 (p-SMAD2C) and Ser423/425 [227, 228] (Figure 4A). This results in the formation of heteromeric complexes with cytosolic SMAD4 [229], which then translocates to the nucleus, binds to SMAD-binding elements (SBE) and thereby regulates TGF-β-responsive gene expression [229] (Figure 4A).
The canonical tumor-suppressive actions of TGF-β are mediated by its effects on the cell cycle, through the up-regulation of the CDK inhibitors, p15, p21WAF1/CIP1 and p27KIP1, as well as down-regulation of c-myc [230]. TGF-β also promotes apoptosis through up-regulation of pro-apoptotic Bax, increased activation of effector caspases, and down-regulation of anti-apoptotic Bcl-2 [231]. TGF-β has been shown to cause significant reduction in proliferation and induction of cell death in human prostatic epithelial cells [232] and some prostate cancer cell lines [233].
The level of TGF-β/SMAD signaling in cells has been reported to be negatively regulated by the Ras/Raf/MEK/ERK cascade, which is induced by EGF receptor signaling [234]. Specifically, Ras signaling inhibits the TGF-β-induced nuclear accumulation of the SMAD complex by phosphorylating SMAD2 at the linker region (p-SMAD2L), which is distinct from the TGF-β receptor phosphorylation site [234] (Figure 4A). In normal epithelial cells, TGF-β potently overcomes the proliferative effects of Ras-activating factors [235], suggesting Ras-mediated phosphorylation of SMAD2 serves to adjust the level of TGF-β/SMAD signaling according to the level of Ras activity in these cells. However, in cancer cells where Ras is hyper-activated by oncogenic mutations, this same mechanism may act to silence the tumor-suppressive functions of TGF-β/SMAD signaling and consequently promote tumor progression [234]. This suggests that in Ras-transformed cancer cells, inhibiting the phosphorylation of SMAD2 at the linker region may be a worthy therapeutic strategy to consider.
As previously mentioned, DFO and Dp44mT can attenuate phosphorylation of ERK1/2 in PC-3 and DU145 prostate cancer cells [120]. This study also demonstrated that these compounds could decrease levels of p-SMAD2L. Conversely, no changes were observed in the phosphorylation of SMAD2 at the C-terminal region (p-SMAD2C) that is regulated by TGF-β activation in these cells [120]. Thus, taken together, DFO and Dp44mT could suppress ERK1/2 to subsequently inhibit the oncogenic effects of SMAD2 phosphorylated at the linker region. This effect could modulate canonical tumor suppressive TGF-β signaling by overall promoting the effects of C-terminal-phosphorylated SMAD2 (Figure 4A).
STAT3 signaling
The STAT family consists of seven proteins that transduce extracellular signals to regulate genes involved in cell growth, survival and differentiation [236]. Of these, STAT3 is the most well characterized and has been linked to tumor progression [236]. STAT3 signaling is classically known to be activated by upstream cytokine receptor interactions, such as those resulting from interleukins and interferons activating the Janus tyrosine kinases (JAKs) [237]. However, STAT3 signaling can also be induced by growth factor RTKs (e.g., EGFR) and non-receptor kinases (e.g., Src, c-Abl) [238–240] (Figure 4B). Activation of STAT3 results in its phosphorylation at Tyr705 (p-STAT3), leading to dimer formation, nuclear translocation, binding to STAT3-specific DNA-binding elements and transcription of target genes [240].
The wide and varied cellular effects of STAT3 signaling include the promotion of cell cycle progression by up-regulating cyclin D1, c-myc and pim-1, and suppression of apoptosis through Bcl-2, myeloid leukemia cell differentiation protein 1 (Mcl-1) and cellular inhibitor of apoptosis 2 (c-IAP2) expression [236, 241] (Figure 4B). STAT3 has also been shown to repress p53 expression, with blockade of STAT3 in cancer cells leading to p53-mediated apoptosis [242]. Several lines of evidence also implicate STAT3 in tumor cell invasion, migration and immune evasion as well as the EMT by promoting Rho GTPase-regulated cell migration and regulating the expression of MMPs, E-cadherin, zinc-finger E-box-binding (ZEB) and vimentin, amongst others [236, 243–245].
Cancers of the prostate, pancreas and breast, as well as leukemias and melanomas, have all been reported to display constitutive activation of STAT3 [246, 247]. This has directed numerous recent efforts to develop new therapies targeting the STAT3 pathway, including direct inhibitors of STAT3, IL-6 receptor antagonists, antisense strategies and decoy phosphopeptides [248, 249].
Recently, we demonstrated that iron chelators regulate the STAT3 pathway in pancreatic and prostate cancer cells through inhibition of constitutive and IL-6-induced STAT3 activation [121] (Figure 4B). The chelators, DFO, Dp44mT and DpC, inhibited constitutive phosphorylation of STAT3, dimerization, binding of STAT3 to its target DNA sequence, and the expression of STAT3 targets (i.e., c-myc, cyclin D1 and Bcl-2) [121]. This may be, in part, mediated by the ability of these compounds to decrease activation of the upstream kinases, Src and c-Abl, that are known to promote STAT3 signaling [121] (Figure 4B). Further, the redox-active Dp44mT- and DpC-iron complexes also inhibited STAT3 activation and this could be prevented by the antioxidant, N-acetylcysteine (NAC), indicating that ROS generation by these compounds is involved in their STAT3-inhibitory properties [121]. Both Dp44mT and DpC could also inhibit IL-6-induced phosphorylation and nuclear translocation of STAT3, which has been implicated in tumor progression [121, 248]. These findings were reiterated in vivo in a PANC-1 tumor xenograft model where Dp44mT- or DpC-treated tumors displayed significantly lower staining for p-STAT3 and STAT3 compared to tumors from vehicle control-treated mice [121]. Similar findings were also observed in studies examining hepatocellular carcinoma cell lines, where Dp44mT suppressed p-STAT3 levels, possibly through up-regulation of the NDRG family member, NDRG2 [108].
Interestingly, serine phosphorylation of STAT3 by ERK has been reported to negatively modulate STAT3 tyrosine phosphorylation and activity [250, 251], while other studies implicate the MEK/ERK cascade in positively mediating STAT3 activity [252–254]. The AKT/PTEN pathway has also been reported to regulate STAT3 [255, 256], and it was recently suggested that STAT3 occupies a central role in integrating pathway crosstalk between the MAPK and AKT pathways [257]. Given these interactions, it is possible that the effects of iron chelators on the STAT3 pathway are, at least in part, influenced by their modulation of the ERK and AKT pathways (Figure 4B). Indeed, more detailed characterization of the complex crosstalk between these and potentially other pathways that mediate the anti-neoplastic activities of iron chelators is required.
The EMT
The EMT refers to the transient differentiation process whereby epithelial cells lose their polarity and cell-cell junctions, reorganize their cytoskeleton, and induce signaling changes that reprogram gene expression and cell shape [258]. While EMT plays important roles during embryogenesis and wound healing, its underlying processes are reactivated in the pathological processes of fibrogenesis and tumorigenesis [258]. Repression of the epithelial phenotype and activation of the mesenchymal phenotype involves changes in the gene expression of many molecules, including the master transcription factor regulators, Snail, Slug, Twist, and ZEB [258] (Figure 5). These proteins often regulate the expression of each other as well as other target genes, including the down-regulation of E-cadherin, occludin and claudins that maintain cell tight junctions, and the up-regulation of N-cadherin, MMPs, vimentin and fibronectin that are essential for promoting motility and invasiveness [259]. Two major signaling pathways that induce EMT via these effectors are the TGF-β and Wnt pathways, both of which have been demonstrated to be regulated by iron chelation [156, 260] (Figure 5).
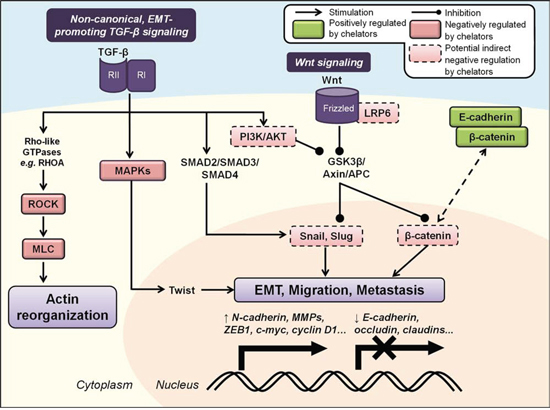
Figure 5: Iron chelators regulate the epithelial-to-mesenchymal transition (EMT) and molecular motors involved in cancer cell migration and metastasis. Non-canonical activation of TGF-β signaling promotes tumor progression, EMT and metastasis via SMAD, MAPK and PI3K/AKT pathways that up-regulate mesenchymal-associated transcription factors (e.g., Snail, Slug, Twist and ZEB) and repress E-cadherin expression. The Wnt signaling pathway also regulates EMT through its inhibition of GSK3β-mediated degradation of β-catenin, Snail and Slug. TGF-β can also activate Rho/ROCK/MLC signaling to drive dynamic actin reorganization required for metastasis. Iron chelators have been demonstrated to inhibit the EMT and metastasis by maintaining expression of E-cadherin and β-catenin at cell membranes, and may also mediate these effects through regulation of other upstream cascades (see text for more details). Molecules negatively regulated by iron chelators are indicated by red boxes, and those positively regulated are indicated by green boxes. Solid boxes indicate direct evidence for modulation by iron chelators, while dashed boxes indicate potential indirect modulation, e.g., through chelator-mediated up-regulation of NDRG1.
The role of TGF-β signaling in cancer is complex and paradoxical. In contrast to canonical tumor-suppressive TGF-β signaling in normal epithelial cells (described above), non-canonical TGF-β signaling is known to play important roles in promoting tumor progression and metastasis via induction of the EMT, stimulating cell proliferation and evasion of the immune system [259, 261]. This effect probably results from progressive mutations that collectively inactivate the core elements of the TGF-β signaling cascade and its downstream effectors, allowing tumor cells to escape the tumor-suppressive mechanisms of TGF-β and activate oncogenic signaling pathways [259]. In fact, TGF-β stimulation has been observed to induce cell proliferation via the Ras/Raf/MEK/ERK pathway, rather than suppress it via normal SMAD signaling, in TSU-Pr1 prostate cancer cells [262]. SMAD-dependent TGF-β signaling promotes EMT by activating the mesenchymal-associated transcription factors, Snail, Slug, Twist and ZEB1, amongst others [263–265]. Further, SMADs also up-regulate cytoskeleton-associated genes that mediate cell motility and invasion, including vimentin and fibronectin [266].
In addition to activating the trimeric SMAD complex, TGF-β can also activate the MAPK, Rho/Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) and PI3K pathways, amongst others [261, 267] (Figure 5). Each of these pathways can also contribute to the EMT. For example, ERK MAPK signaling enhances repression of E-cadherin and activates Twist, N-cadherin and MMP expression [268, 269]. TGF-β-mediated activation of PI3K/AKT signaling induces activation of mammalian target of rapamycin complexes 1/2 (mTORC1/2) that aids the EMT, increasing expression of Snail and MMP-9, and repressing E-cadherin expression [270, 271]. Phosphorylation and inhibition of GSK3β that is regulated by AKT also stabilizes Snail and Slug expression [272, 273]. Moreover, TGF-β can also induce the activation of Rho-like GTPases, e.g., RhoA, that then activates the ROCK1 and LIM kinase (LIMK) pathways to drive dynamic actin reorganization, lamellipodia and filopodia formation required for the EMT [274, 275] (Figure 5).
Another key regulator of EMT is the Wnt signaling pathway that has canonical roles in development, tumorigenesis and metastatic progression [276]. It is activated at the plasma membrane by binding of Wnt ligands to its co-receptors, Frizzled and LDL receptor-related protein 5/6 (LRP5/6) [277]. In the absence of Wnt signaling, the destruction complex, which is composed of adenomatous polyposis coli (APC), axin and GSK3β, phosphorylates β-catenin that then promotes its proteasomal degradation [276] (Figure 5). However, in the presence of Wnt ligands, this complex disintegrates, stabilizing free β-catenin in the cytosol and allowing its nuclear translocation where it interacts with T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) transcription factors to promote transcription of pro-metastatic genes [276]. Wnt signaling also inhibits GSK3β-mediated degradation of the mesenchymal transcription factors, Snail and Slug [278, 279] (Figure 5).
Recent studies have identified important roles for cellular iron in regulating the EMT that occur via iron chelator-mediated up-regulation of the metastasis suppressor, NDRG1 [146, 147]. In DU145 prostate and HT29 colon cancer cells, the chelators DFO and Dp44mT attenuated TGF-β-induced loss of E-cadherin and β-catenin at the cell membrane (Figure 5), and also decreased TGF-β-induced up-regulation of vimentin [156]. This demonstrated that these iron chelators could inhibit TGF-β-induced EMT in these cells. Moreover, NDRG1 expression, which is markedly up-regulated by these compounds, also inhibited the TGF-β-induced EMT in DU145 and HT29 cells [156]. Specifically, NDRG1 knockdown in these cells assumed a spindle-shaped morphology, abolished expression of E-cadherin and β-catenin at the membrane, and increased nuclear expression of β-catenin [156]. NDRG1 over-expression in DU145 and HT29 cells attenuated the ability of TGF-β to induce changes to morphology and expression of EMT markers that are characteristic of the EMT, as well as the migratory and invasive potential of these cells [156]. This effect was found to involve the transcriptional E-cadherin repressors, Snail and Slug. Similar outcomes from NDRG1 inhibiting the EMT were also reported in oral squamous cell carcinoma cells through up-regulation of E-cadherin and down-regulation of N-cadherin, vimentin, Snail, Slug, MMP-2 and MMP-9 [280]. Further studies revealed that NDRG1 inhibits β-catenin phosphorylation and nuclear translocation, specifically by up-regulating FRAT1, which prevents GSK3β from binding the β-catenin destruction complex [281]. NDRG1 could also inhibit β-catenin nuclear translocation that is mediated by Wnt signaling via reducing nuclear localization of p21 activated kinase 4 (PAK4) [281]. Together, these NDRG1-mediated effects lead to increased levels of β-catenin that is expressed at the cell membrane, where it functions to promote cell adhesion, while inhibiting its localization in the cell nucleus, where it is oncogenic [281, 282].
Studies performed in PC3mm metastatic prostate and MCF7 breast cancer cells demonstrated that NDRG1 interacts with the Wnt receptor, LRP6, subsequently blocking the Wnt cascade to suppress the EMT and metastasis by reactivating GSK3β, promoting β-catenin degradation, and inhibiting downstream expression of ATF3 [107]. Moreover, these studies showed that Dp44mT could significantly enhance NDRG1 expression in human breast cancer cells and suppress bone metastasis in vivo [107]. This observation indicated that Dp44mT-mediated NDRG1 up-regulation could play a critical role in negatively regulating Wnt signaling during EMT and metastasis (Figure 5).
Multiple iron chelators, including DFO, deferasirox and a series of acyl hydrazones have been shown to destabilize β-catenin, subsequently inhibiting Wnt signaling to inhibit colorectal cancer cell growth [260]. These agents also decreased the expression of Wnt target genes, which was shown to occur via β-catenin degradation downstream of the destruction complex. Excess iron levels have also been implicated in colorectal tumorigenesis through enhancement of Wnt signaling in cells displaying APC loss or abnormal β-catenin expression [283]. This effect increased cellular proliferation, correlating with increased mRNA expression of its targets, c-myc and Naked cuticle 1 (Nkd1). Collectively, these studies identify important roles for iron in the regulation of multiple EMT mediators that could be exploited in cancer therapy using potent iron chelators.
Interestingly, DFO has also been reported to promote the EMT and enhance cell migration and invasion in HT29 cells, which was suggested to be mediated by DFO-induced HIF-1 expression [284]. However, this is in contrast with multiple reports previously described indicating that DFO exhibits inhibitory effects on tumor growth and the EMT [5, 156, 260], possibly reflecting differences in experimental conditions utilized in the study.
Migration and metastasis
In addition to regulating the EMT, directed cell invasion and migration of tumor cells is required for metastasis [285]. This process involves multiple events, including F-actin polymerization, its interaction with myosin II filaments, and the subsequent formation of contractile actomyosin structures such as stress fibers [286]. These are rearranged and extended within the cell in response to various signaling cues to promote cytoskeletal reorganization and cell motility [287]. The small Rho-GTPase family is involved in regulating stress fiber assembly, contraction and motility through effectors such as Rho and Rac [288]. These molecules can subsequently regulate multiple targets to regulate the actin cytoskeleton, including the p21 activated kinase (PAK), ROCK1/myosin light chain (MLC) and LIMK pathways that mediate the formation and contractility of stress fibers [289], of which phosphorylated MLC is a key mediator of actin filament polymerization, stress fiber assembly and cell migration [290].
We recently demonstrated that iron chelation could inhibit cell migration and metastasis by modulating the ROCK1/MLC2 cascade [157] (Figure 5). Specifically, DFO, Dp44mT and DpC decreased the expression of ROCK1 and phosphorylated MLC2 in DU145, HT29 and HCT116 cells [157]. Importantly, the observed effects of these iron chelators was likely mediated via the ability of these agents to up-regulate NDRG1, since suppression of endogenous NDRG1 expression at least partially attenuated the ability of the chelators to inhibit ROCK1 expression, MLC2 phosphorylation and migration [157]. These studies elucidated the inhibitory role of NDRG1 on the ROCK1/MLC2 pathway, with NDRG1 over-expression inhibiting F-actin polymerization and stress fiber formation through regulation of this cascade [157].
To further understand the role of NDRG1 in cellular migration, its effects on the oncogene, c-Src, were important to investigate as the downstream effectors of this molecule play critical roles in regulating this process [291, 292]. Importantly, NDRG1 inhibited Src-induced phosphorylation of p130Cas and prevented its complex formation with CrkII [293]. In addition, NDRG1 could decrease activation of c-Abl and subsequent CrkII phosphorylation [293]. Collectively, this resulted in the inhibition of Rac1 signaling and decreased levels of downstream phosphorylated PAK1, playing critical roles in cytoskeletal dynamics and the regulation of cell motility [294]. Given that the iron chelators, DFO, Dp44mT and DpC, have been demonstrated to inhibit Src and c-Abl activity in certain cancer cell-types [121] and that they can markedly up-regulate NDRG1, it is plausible that these compounds could exhibit their anti-metastatic effects via NDRG1-mediated modulation of these pathways.
ER stress and autophagy
Autophagy refers to a homeostatic, catabolic process that degrades cellular proteins and organelles to sustain cellular metabolism under conditions of stress [295]. It is initiated by the formation of crescent-shaped structures called phagophores that surround targeted cytoplasmic constituents [296] (Figure 6). Concomitantly, lipid-bound microtubule-associated protein 1A/1B-light chain 3 (LC3) is converted from its soluble form (LC3-I) to its lipid-bound form (LC3-II) and is expressed on the membrane of phagophores, subsequently leading to the formation of double-membrane vesicles, i.e., autophagosomes, which engulf the target cargo [296]. The autophagosome subsequently matures by fusing with a lysosome to form an autolysosome, exposing the cargo to lysosomal hydrolases that promote degradation of its contents and nutrient recycling [297] (Figure 6).
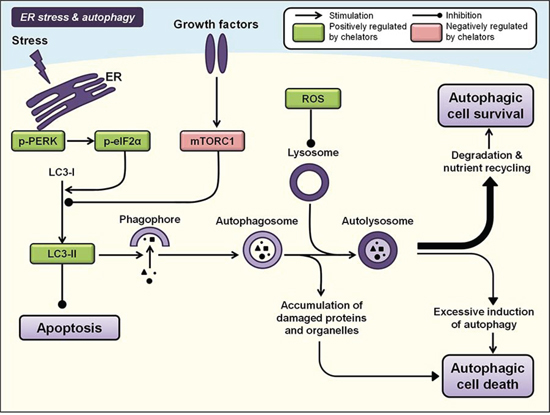
Figure 6: Iron chelation modulates ER stress and autophagy pathways. Autophagy involves the formation of double-membrane vesicles from phagophores, i.e., autophagosomes, which engulf cytoplasmic contents and organelles. Fusion of the autophagosome with a lysosome forms an autolysosome that results in the degradation of its contents, nutrient recycling and autophagic cell survival. However, prolonged autophagy in certain settings can also lead to non-apoptotic autophagic cell death. Two core regulators of autophagy include the PERK/eIF2α pathway that is activated by endoplasmic reticulum (ER) stress, and the PI3K/AKT/mTORC1 pathway. Iron chelators have been demonstrated to modulate both these pathways and subsequent autophagic processes, and also enhance expression of LC3-II, which is recruited to autophagosomal membranes during induction of autophagy (see text for more details). Molecules negatively regulated by iron chelators are indicated by red boxes, and those positively regulated are indicated by green boxes.
Complex signaling networks control mammalian autophagy, with the integration of stress signals through the PI3K/AKT/mTORC1 pathway acting as a core negative regulator of the autophagic machinery [297]. Activation of the PI3K/AKT signaling cascade inhibits TSC2 [174], activating the mTORC1 complex and inhibiting formation of a trimeric unc-51-like kinase (ULK) complex required for autophagosome formation from the phagophore [298]. Thus, mTORC1 activity inhibits autophagic initiation (Figure 6). Another key regulator of autophagy is adenosine monophosphate kinase (AMPK), which acts as a major stimulator of autophagy through its ability to activate TSC1/2 and suppress mTORC1 signaling [298–300], but also potentially through direct phosphorylation of ULK [301].
Activation of the endoplasmic reticulum (ER) stress pathway and unfolded protein response can also potently induce autophagy [299]. Specifically, the binding of accumulated unfolded proteins to the ER chaperone Grp78/Bip induces the release of three ER membrane-associated proteins that trigger distinct pathways, namely protein kinase-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1) [302]. Phosphorylation and activation of PERK directly induces phosphorylation of eukaryotic initiation factor 2α (eIF2α) that is critical for the transcription of key autophagy-associated genes during ER stress, mediating LC3 conversion and subsequent autophagosome formation [303] (Figure 6).
Autophagy serves dual roles in tumorigenesis and tumor progression [299]. Since defects in autophagy have been linked with susceptibility to genomic damage and tumorigenesis in mice [304], autophagy could suppress tumorigenesis by limiting cell growth and genomic instability [305]. However, autophagy can also maintain the survival of tumor cells, allowing adaptation to stress conditions induced by high metabolic demands, hypoxic tumor microenvironments or chemotherapeutics [299]. In human pancreatic adenocarcinoma cell lines and tumor specimens, high levels of basal autophagy allowed the maintenance of cellular energy production that fuelled tumor growth [306]. Moreover, inhibiting autophagy in these cells by genetic means or chloroquine treatment promoted tumor regression of xenografts in vivo and mice survival [306]. Indeed, this has formed the underlying basis for preclinical and ongoing clinical studies that are administering autophagy inhibitors, e.g., chloroquine and hydroxychloroquine, in combination with chemotherapy [299].
However, prolonged autophagy in certain cellular settings can overwhelm the capacity of the cell, leading to a non-apoptotic pro-death pathway known as autophagic cell death [307]. Thus, targeting autophagy for cancer treatment presents as a “double-edged sword”, with its role in tumor progression and treatment response remaining incompletely understood. Harnessing this pro-death cascade of the autophagic process could be a novel vantage point of attack against tumor cells already existing under conditions of high stress.
Recently, it was demonstrated that Dp44mT modulates the autophagic process to favor cell death in breast cancer cells by increasing autophagosome synthesis, but also inhibiting subsequent autolysosome formation by impairing lysosomal integrity [308]. This illustrates a unique mechanism by which the double-edged autophagic machinery can be exploited in cancer therapy. Specifically, Dp44mT induced autophagic initiation as demonstrated by increased LC3-II levels, and also increased expression of another autophagy marker, namely p62, due to suppressed autophagosome degradation [308]. This latter effect was found to be dependent on the ability of Dp44mT to generate ROS, which subsequently permeate and destabilize the lysosome, preventing autolysosome formation and autophagosome degradation [308]. Thus, rather than enabling completion of the autophagic process that typically promotes cancer cell survival through nutrient recycling, the accumulation of damaged proteins and organelles triggers cell death [308] (Figure 6). These studies also postulated that DFO inhibited autophagosome degradation, but did not increase autophagosome synthesis, and thus, led to LC3-II accumulation in these cells [308].
The chelators, DFO and Dp44mT, also induced autophagy in PANC-1 pancreatic cancer cells as demonstrated by increased LC3-II expression [309]. This occurred via induction of ER stress and activation of the PERK/eIF2α signaling cascade, as demonstrated by increased levels of p-PERK and p-eIF2α [309]. The ability of these compounds to sequester cellular iron was critical for its effects on autophagy induction, as incubation with the DFO:Fe complex or Dp2mT did not exhibit the same effects [309]. Incubation with iron- or copper-complexed Dp44mT also elevated LC3-II expression, which was attenuated by NAC, indicating that Dp44mT-mediated redox activity is important for its role in regulating LC3-II expression [309]. Interestingly, NDRG1 was found to suppress the stress-induced autophagic response, which could mediate the anti-tumor and anti-metastatic activities of NDRG1 by enhancing apoptosis and inhibiting metastasis [309]. This was shown to occur via inhibition of the PERK/eIF2α pathway involved in the unfolded protein response [309].
Given that Dp44mT markedly up-regulates NDRG1, the seemingly opposing effects of Dp44mT and NDRG1 on autophagy appear contradictory [309]. However, the initial Dp44mT-mediated activation of autophagic initiation is likely to be subsequently suppressed due to up-regulation of NDRG1 following longer periods of exposure to Dp44mT [309]. This is consistent with the notion that NDRG1 acts in response to cellular stress [148, 310], which could lead to suppression of stress-induced autophagy [309]. Further, in addition to the generation of ROS by Dp44mT that causes lysosomal membrane permeabilization that disturbs the autophagic process [308], it is important to note that Dp44mT affects multiple cellular targets to induce its anti-tumor activity, as described herein and elsewhere [5].
Autophagy has also been implicated in the anti-tumor mechanism of DFO and deferasirox in multiple myeloma cell lines [311]. Specifically, exposure of myeloma cell lines to DFO and deferasirox markedly induced LC3-II expression concomitant with induction of autophagy, and the autophagy inhibitor 3-methyladenine rescued these cells from chelator-induced cytotoxicity [311]. This was potentially mediated by the suppressive effect of these compounds on the mTORC1 substrate p70S6 kinase, which negatively regulates autophagy [312]. Moreover, DFO was shown to induce autophagy flux and decrease caspase activation, inhibiting TRAIL-mediated apoptosis, in colon cancer cells [313].
Of importance, the iron storage protein, ferritin, is reported to be degraded in the cell via autophagy [314, 315]. Multiple iron chelators can direct the pathway via which ferritin is degraded in cells [316]. In these studies, incubation of cells with poorly permeable DFO resulted in lysosome-mediated degradation of ferritin via autophagy [316]. The smaller, more lipophilic and membrane-permeable ligands, deferasirox and deferiprone, diverted ferritin degradation towards the proteasomal pathway [316]. This contrasted with previous findings [311], which may be explained by cell-type-dependent differences in the regulation of autophagic processing. Moreover, considering that autophagy is a dynamic process [317], and that these latter studies only visualized LC3-II-containing autophagosomes as a autophagic marker [311, 316], further studies utilizing late-stage autophagy inhibitors, such as bafilomycin A1 or choloroquine, in combination with iron chelators are necessary to determine whether the increased number of autophagosomes observed are due to enhanced autophagic initiation or suppression of lysosomal-mediated autophagic degradation [317]. Collectively, these recent studies clearly identify induction of autophagy as a potential mediator of cytotoxic iron chelators, requiring further elucidation of the precise mechanisms responsible to aid its clinical application (Figure 6).
CONCLUSIONS AND FUTURE DIRECTIONS
Compelling evidence exists to support the preclinical and clinical development of DpT thiosemicarbazones as anti-cancer agents. Moreover, these compounds demonstrate synergistic activities with existing chemotherapies including gemcitabine, cisplatin, doxorubicin and tamoxifen [113, 318]. Thus, these compounds could be effectively utilized in combination cancer therapies.
Major advances have been achieved in recent years in terms of gaining mechanistic insight into how the DpT ligands exert their multi-modal effects at the molecular level by targeting oncogenic signaling pathways. This is a critical step in the development of these compounds, providing valuable information related to their potential application as multi-targeted therapies based on the genetic profiles and stages of individual tumors. However, differences between cancer cell-types as well as complexities in signaling pathway crosstalk and feedback loops mean that the molecular mechanisms of DpT thiosemicarbazones still require careful elucidation. The studies described herein will no doubt provide a solid foundation for future studies and provide necessary justification for the planned clinical trials in humans.
ACKNOWLEDGMENTS
Des Richardson thanks the National Health and Medical Research Council of Australia (NHMRC) for a Senior Principal Research Fellowship [1062607] and project grants [1060482]. Danuta S. Kalinowski thanks the NHMRC for a project grant [1048972] and RD Wright Fellowship [1083057]. Zaklina Kovacevic acknowledges fellowship support from the NHMRC and the Cancer Institute New South Wales. Goldie Lui appreciates PhD scholarship support from the Australian Federal Government and University of Sydney. The authors kindly acknowledge critical comments on the manuscript prior to submission by Dr. Patric Jansson and Dr. Sumit Sahni (Molecular Pharmacology and Pathology Program).
CONFLICTS OF INTEREST
D.R.R. is a stakeholder in the companies, Oncochel Therapeutics LLC, USA, and Oncochel Therapeutics Pty Ltd, Australia, that are developing the thiosemicarbazone, DpC, for the treatment of cancer. D.R.R. also consults for Oncochel Therapeutics LLC and Pty Ltd.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013; 63:318–348.
3. Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010; 9:843–856.
4. Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005; 57:547–583.
5. Merlot AM, Kalinowski DS, Richardson DR. Novel chelators for cancer treatment: where are we now? Antioxid Redox Signal. 2013; 18:973–1006.
6. Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr Top Med Chem. 2004; 4:1623–1635.
7. Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011; 71:1511–1514.
8. Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004; 3:205–214.
9. Bergeron RJ, Wiegand J, McManis JS, Bussenius J, Smith RE, Weimar WR. Methoxylation of desazadesferrithiocin analogues: enhanced iron clearing efficiency. J Med Chem. 2003; 46:1470–1477.
10. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995; 82–83:969–974.
11. Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. 1997; 1331:1–40.
12. Crichton RR. (2009) Iron metabolism: from molecular mechanisms to clinical consequences. (Chichester, UK: John Wiley & Sons).
13. Lane DJ, Bae DH, Merlot AM, Sahni S, Richardson DR. Duodenal Cytochrome b (DCYTB) in Iron Metabolism: An Update on Function and Regulation. Nutrients. 2015; 7:2274–2296.
14. Morgan EH. Transferrin, biochemistry, physiology and clinical significance. Mol Aspects Med. 1981; 4:1–123.
15. Ponka P, Beaumont C, Richardson DR. Function and regulation of transferrin and ferritin. Semin Hematol. 1998; 35:35–54.
16. Crichton RR, Boelaert JR. (2001). Inorganic biochemistry of iron metabolism: From molecular mechanisms to clinical consequences. (New York: John Wiley and Sons).
17. Hanover JA, Dickson RB. (1985). Transferrin: Receptor-Mediated Endocytosis and Iron Delivery. In: Pastan I, Willingham M, eds. Endocytosis. (New York: Springer US), pp. 131–161.
18. Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005; 37:1264–1269.
19. Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998; 95:1148–1153.
20. Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic Biol Med. 2002; 33:1037–1046.
21. Jacobs A. Low molecular weight intracellular iron transport compounds. Blood. 1977; 50:433–439.
22. Richardson DR, Ponka P, Vyoral D. Distribution of iron in reticulocytes after inhibition of heme synthesis with succinylacetone: examination of the intermediates involved in iron metabolism. Blood. 1996; 87:3477–3488.
23. Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, Philpott CC. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc Natl Acad Sci U S A. 2014; 111:8031–8036.
24. Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, Stemmler TL, Philpott CC. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem. 2013; 288:17791–17802.
25. Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011; 14:647–657.
26. Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010; 107:10775–10782.
27. Eisenstein RS, Blemings KP. Iron regulatory proteins, iron responsive elements and iron homeostasis. J Nutr. 1998; 128:2295–2298.
28. Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci U S A. 1996; 93:8175–8182.
29. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013; 13:342–355.
30. Walker RA, Day SJ. Transferrin receptor expression in non-malignant and malignant human breast tissue. J Pathol. 1986; 148:217–224.
31. Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I, Zarkovic N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett. 2006; 238:188–196.
32. Soyer HP, Smolle J, Torne R, Kerl H. Transferrin receptor expression in normal skin and in various cutaneous tumors. J Cutan Path. 1987; 14:1–5.
33. Richardson DR, Baker E. The uptake of iron and transferrin by the human malignant melanoma cell. Biochim Biophys Acta. 1990; 1053:1–12.
34. Taetle R, Castagnola J, Mendelsohn J. Mechanisms of growth inhibition by anti-transferrin receptor monoclonal antibodies. Cancer Res. 1986; 46:1759–1763.
35. Takeda E, Weber G. Role of ribonucleotide reductase in expression in the neoplastic program. Life Sci. 1981; 28:1007–1014.
36. Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, Madraswala R, Zhou Y, Kuo J, Raitano AB, Jakobovits A, Saffran DC, Afar DE. STEAP: a prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci U S A. 1999; 96:14523–14528.
37. Isobe T, Baba E, Arita S, Komoda M, Tamura S, Shirakawa T, Ariyama H, Takaishi S, Kusaba H, Ueki T, Akashi K. Human STEAP3 maintains tumor growth under hypoferric condition. Exp Cell Res. 2011; 317:2582–2591.
38. Jiang XP, Elliott RL, Head JF. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010; 30:759–765.
39. Pinnix ZK, Miller LD, Wang W, D’Agostino R, Jr., Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010; 2: 43ra56.
40. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18, 000 cancer gene expression profiles. Neoplasia. 2007; 9:166–180.
41. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004; 306:2090–2093.
42. Hibbs JB, Jr., Taintor RR, Vavrin Z. Iron depletion: possible cause of tumor cell cytotoxicity induced by activated macrophages. Biochem Biophys Res Commun. 1984; 123:716–723.
43. Hibbs JB, Jr., Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun. 1988; 157:87–94.
44. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991; 43:109–142.
45. Richardson DR, Neumannova V, Nagy E, Ponka P. The effect of redox-related species of nitrogen monoxide on transferrin and iron uptake and cellular proliferation of erythroleukemia (K562) cells. Blood. 1995; 86:3211–3219.
46. Wardrop SL, Watts RN, Richardson DR. Nitrogen monoxide activates iron regulatory protein 1 RNA-binding activity by two possible mechanisms: effect on the [4Fe-4S] cluster and iron mobilization from cells. Biochemistry 2000; 39:2748–2758.
47. Richardson DR, Neumannova V, Ponka P. Nitrogen monoxide decreases iron uptake from transferrin but does not mobilise iron from prelabelled neoplastic cells. Biochim Biophys Acta. 1995; 1266:250–260.
48. Watts RN, Richardson DR. Nitrogen monoxide (NO) and glucose: unexpected links between energy metabolism and NO-mediated iron mobilization from cells. J Biol Chem. 2001; 276:4724–4732.
49. Lok HC, Suryo Rahmanto Y, Hawkins CL, Kalinowski DS, Morrow CS, Townsend AJ, Ponka P, Richardson DR. Nitric oxide storage and transport in cells are mediated by glutathione S-transferase P1-1 and multidrug resistance protein 1 via dinitrosyl iron complexes. J Biol Chem. 2012; 287:607–618.
50. Watts RN, Hawkins C, Ponka P, Richardson DR. Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proc Natl Acad Sci U S A. 2006; 103:7670–7675.
51. Vaughn CB, Weinstein R, Bond B, Rice R, Vaughn RW, McKendrick A, Ayad G, Rockwell MA, Rocchio R. Ferritin content in human cancerous and noncancerous colonic tissue. Cancer Invest. 1987; 5:7–10.
52. Milman N, Pedersen LM. The serum ferritin concentration is a significant prognostic indicator of survival in primary lung cancer. Oncol Rep. 2002; 9:193–198.
53. Lorenzi M, Lorenzi B, Vernillo R. Serum ferritin in colorectal cancer patients and its prognostic evaluation. Int J Biol Markers. 2006; 21:235–241.
54. Hann HW, Evans AE, Siegel SE, Wong KY, Sather H, Dalton A, Hammond D, Seeger RC. Prognostic importance of serum ferritin in patients with Stages III and IV neuroblastoma: the Childrens Cancer Study Group experience. Cancer Res. 1985; 45:2843–2848.
55. Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999; 283:676–679.
56. Kakhlon O, Gruenbaum Y, Cabantchik ZI. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem J. 2002; 363:431–436.
57. Ceci A, Felisi M, De Sanctis V, De Mattia D. Pharmacotherapy of iron overload in thalassaemic patients. Expert Opin Pharmacother. 2003; 4:1763–1774.
58. Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997; 89:739–761.
59. Richardson DR, Ponka P. Development of iron chelators to treat iron overload disease and their use as experimental tools to probe intracellular iron metabolism. Am J Hematol. 1998; 58:299–305.
60. Aouad F, Florence A, Zhang Y, Collins F, Henry C, Ward RJ, Crichton RR. Evaluation of new iron chelators and their therapeutic potential. Inorganica Chimica Acta. 2002; 339:470–480.
61. Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, Allen CJ, Farrell DE, Harris JW. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med. 1994; 331:567–573.
62. Dayani PN, Bishop MC, Black K, Zeltzer PM. Desferoxamine (DFO) - mediated iron chelation: rationale for a novel approach to therapy for brain cancer. J Neuro Oncol. 2004; 67:367–377.
63. Kontoghiorghes GJ. Comparative efficacy and toxicity of desferrioxamine, deferiprone and other iron and aluminium chelating drugs. Toxicol Lett. 1995; 80:1–18.
64. Kontoghiorghes GJ, Piga A, Hoffbrand AV. Cytotoxic and DNA-inhibitory effects of iron chelators on human leukaemic cell lines. Hematol Oncol. 1986; 4:195–204.
65. Estrov Z, Tawa A, Wang XH, Dube ID, Sulh H, Cohen A, Gelfand EW, Freedman MH. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood. 1987; 69:757–761.
66. Blatt J, Stitely S. Antineuroblastoma activity of desferoxamine in human cell lines. Cancer Res. 1987; 47:1749–1750.
67. Blatt J, Taylor SR, Stitely S. Mechanism of antineuroblastoma activity of deferoxamine in vitro. J Lab Clin Med. 1988; 112:433–436.
68. Donfrancesco A, Deb G, Dominici C, Pileggi D, Castello MA, Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990; 50:4929–4930.
69. Donfrancesco A, Debernardi B, Carli M, Mancini A, Nigro M, Desio L, Casale F, Bagnulo S, Helson L, Deb G. Deferoxamine followed by cyclophosphamide, etoposide, carboplatin, thiotepa, induction regimen in advanced neuroblastoma - preliminary results. Eur J Cancer. 1995; 31A:612–615.
70. Blatt J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994; 14:2109–2112.
71. Selig RA, White L, Gramacho C, Sterling-Levis K, Fraser IW, Naidoo D. Failure of iron chelators to reduce tumor growth in human neuroblastoma xenografts. Cancer Res. 1998; 58:473–478.
72. Aouad F, Florence A, Zhang Y, Collins F, Henry C, Ward RJ, Crichton RR. Evaluation of new iron chelators and their therapeutic potential. Inorg Chim Acta. 2002; 339:470–480.
73. Richardson D, Ponka P, Baker E. The effect of the iron(III) chelator, desferrioxamine, on iron and transferrin uptake by the human malignant melanoma cell. Cancer Res. 1994; 54:685–689.
74. Cappellini MD, Taher A. Deferasirox (Exjade) for the treatment of iron overload. Acta Haematol. 2009; 122:165–173.
75. Piga A, Galanello R, Forni GL, Cappellini MD, Origa R, Zappu A, Donato G, Bordone E, Lavagetto A, Zanaboni L, Sechaud R, Hewson N, Ford JM, Opitz H, Alberti D. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica. 2006; 91:873–880.
76. Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, Aydinok Y, Kattamis A, Kilinc Y, Porter J, Capra M, Galanello R, Fattoum S, Drelichman G, Magnano C, Verissimo M, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood. 2006; 107:3455–3462.
77. List AF, Baer MR, Steensma DP, Raza A, Esposito J, Martinez-Lopez N, Paley C, Feigert J, Besa E. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. J Clin Oncol. 2012; 30:2134–2139.
78. Ohyashiki JH, Kobayashi C, Hamamura R, Okabe S, Tauchi T, Ohyashiki K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. 2009; 100:970–977.
79. Lescoat G, Chantrel-Groussard K, Pasdeloup N, Nick H, Brissot P, Gaboriau F. Antiproliferative and apoptotic effects in rat and human hepatoma cell cultures of the orally active iron chelator ICL670 compared to CP20: a possible relationship with polyamine metabolism. Cell Prolif. 2007; 40:755–767.
80. Vazana-Barad L, Granot G, Mor-Tzuntz R, Levi I, Dreyling M, Nathan I, Shpilberg O. Mechanism of the antitumoral activity of deferasirox, an iron chelation agent, on mantle cell lymphoma. Leuk Lymphoma. 2013; 54:851–859.
81. Chantrel-Groussard K, Gaboriau F, Pasdeloup N, Havouis R, Nick H, Pierre JL, Brissot P, Lescoat G. The new orally active iron chelator ICL670A exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur J Pharmacol. 2006; 541:129–137.
82. Lui GYL, Obeidy P, Ford SJ, Tselepis C, Sharp DM, Jansson PJ, Kalinowski DS, Kovacevic Z, Lovejoy DB, Richardson DR. The iron chelator, deferasirox, as a novel strategy for cancer treatment: oral activity against human lung tumor xenografts and molecular mechanism of action. Mol Pharmacol. 2013; 83:179–190.
83. Ford SJ, Obeidy P, Lovejoy DB, Bedford M, Nichols L, Chadwick C, Tucker O, Lui GY, Kalinowski DS, Jansson PJ, Iqbal TH, Alderson D, Richardson DR, Tselepis C. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br J Pharmacol. 2013; 168:1316–1328.
84. Fukushima T, Kawabata H, Nakamura T, Iwao H, Nakajima A, Miki M, Sakai T, Sawaki T, Fujita Y, Tanaka M, Masaki Y, Hirose Y, Umehara H. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011; 31:1741–1744.
85. Yu Y, Wong J, Lovejoy DB, Kalinowski DS, Richardson DR. Chelators at the cancer coalface: Desferrioxamine to Triapine and beyond. Clin Cancer Res. 2006; 12:6876–6883.
86. Richardson DR. Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol. 2002; 42:267–281.
87. Cory JG, Cory AH, Rappa G, Lorico A, Liu MC, Lin TS, Sartorelli AC. Inhibitors of ribonucleotide reductase. Comparative effects of amino- and hydroxy-substituted pyridine-2-carboxaldehyde thiosemicarbazones. Biochem Pharmacol. 1994; 48:335–344.
88. Torti SV, Torti FM, Whitman SP, Brechbiel MW, Park G, Planalp RP. Tumor cell cytotoxicity of a novel metal chelator. Blood. 1998; 92:1384–1389.
89. Rakba N, Loyer P, Gilot D, Delcros JG, Glaise D, Baret P, Pierre JL, Brissot P, Lescoat G. Antiproliferative and apoptotic effects of O-Trensox, a new synthetic iron chelator, on differentiated human hepatoma cell lines. Carcinogenesis. 2000; 21:943–951.
90. Richardson DR, Tran EH, Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995; 86:4295–4306.
91. Darnell G, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents III: the effect of the ligands on molecular targets involved in proliferation. Blood. 1999; 94:781–792.
92. Becker EM, Lovejoy DB, Greer JM, Watts R, Richardson DR. Identification of the di-pyridyl ketone isonicotinoyl hydrazone (PKIH) analogues as potent iron chelators and anti-tumour agents. Br J Pharmacol. 2003; 138:819–830.
93. Lovejoy DB, Richardson DR. Novel “hybrid” iron chelators derived from aroylhydrazones and thiosemicarbazones demonstrate selective antiproliferative activity against tumor cells. Blood. 2002; 100:666–676.
94. Chaston TB, Lovejoy DB, Watts RN, Richardson DR. Examination of the antiproliferative activity of iron chelators: multiple cellular targets and the different mechanism of action of Triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin Cancer Res. 2003; 9:402–414.
95. Yen Y, Margolin K, Doroshow J, Fishman M, Johnson B, Clairmont C, Sullivan D, Sznol M. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother Pharmacol. 2004; 54:331–342.
96. Yee KWL, Cortes J, Ferrajoli A, Garcia-Manero G, Verstovsek S, Wierda W, Thomas D, Faderl S, King I, O’Brien SM, Jeha S, Andreeff M, Cahill A, Sznol M, Giles FJ. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leukemia Res. 2006; 30:813–822.
97. Gojo I, Tidwell ML, Greer J, Takebe N, Seiter K, Pochron MF, Johnson B, Sznol M, Karp JE. Phase I and pharmacokinetic study of Triapine (R), a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leukemia Res. 2007; 31:1165–1173.
98. Wadler S, Makower D, Clairmont C, Lambert P, Fehn K, Sznol M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J Clin Oncol. 2004; 22:1553–1563.
99. Knox JJ, Hotte SJ, Kollmannsberger C, Winquist E, Fisher B, Eisenhauer EA. Phase II study of Triapine in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Invest New Drugs. 2007; 25:471–477.
100. Ma B, Goh BC, Tan EH, Lam KC, Soo R, Leong SS, Wang LZ, Mo F, Chan AT, Zee B, Mok T. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest New Drugs. 2008; 26:169–173.
101. Mackenzie MJ, Saltman D, Hirte H, Low J, Johnson C, Pond G, Moore MJ. A Phase II study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) and gemcitabine in advanced pancreatic carcinoma. A trial of the Princess Margaret hospital Phase II consortium. Invest New Drugs. 2007; 25:553–558.
102. Traynor AM, Lee JW, Bayer GK, Tate JM, Thomas SP, Mazurczak M, Graham DL, Kolesar JM, Schiller JH. A phase II trial of Triapine (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Invest New Drugs. 2010; 28:91–97.
103. Richardson DR, Milnes K. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II: the mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood. 1997; 89:3025–3038.
104. Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: the mechanisms involved in inhibiting cell-cycle progression. Blood. 2001; 98:842–850.
105. Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. 2004; 104:1450–1458.
106. Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006; 103:14901–14906.
107. Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, Fukuda K, Li Y, Watabe K. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropicallysuppresses metastasis. EMBO Mol Med. 2012; 4:93–108.
108. Wang J, Yin D, Xie C, Zheng T, Liang Y, Hong X, Lu Z, Song X, Song R, Yang H, Sun B, Bhatta N, Meng X, Pan S, Jiang H, Liu L. The iron chelator Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway. Oncotarget. 2014; 5:8478–8491.
109. Kovacevic Z, Chikhani S, Lovejoy DB, Richardson DR. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: a new strategy for the treatment of pancreatic cancer. Mol Pharmacol. 2011; 80:598–609.
110. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011; 71:5871–5880.
111. Richardson DR, Sharpe PC, Lovejoy DB, Senaratne D, Kalinowski DS, Islam M, Bernhardt PV. Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J Med Chem. 2006; 49:6510–6521.
112. Jansson PJ, Yamagishi T, Arvind A, Seebacher N, Gutierrez E, Stacy A, Maleki S, Sharp D, Sahni S, Richardson DR. Di-2-pyridylketone 4, 4-Dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug-resistance by a novel mechanism involving the hijacking of lysosomal P-Glycoprotein (Pgp). J Biol Chem. 2015; 290:9588–9603.
113. Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, Bernhardt PV, Richardson DR. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J Med Chem. 2012; 55:7230–7244.
114. Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007; 6:1982–1994.
115. Thelander L, Reichard P. Reduction of ribonucleotides. Annu Rev Biochem. 1979; 48:133–158.
116. Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998; 67:71–98.
117. Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000; 404:42–49.
118. Nyholm S, Mann GJ, Johansson AG, Bergeron RJ, Graslund A, Thelander L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J Biol Chem. 1993; 268:26200–26205.
119. Tsimberidou A-M, Alvarado Y, Giles FJ. Evolving role of ribonucleoside reductase inhibitors in hematologic malignancies. Expert Rev Anticancer Ther. 2002; 2:437–448.
120. Dixon KM, Lui GY, Kovacevic Z, Zhang D, Yao M, Chen Z, Dong Q, Assinder SJ, Richardson DR. Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br J Cancer. 2013; 108:409–419.
121. Lui GYL, Kovacevic Z, S VM, Kalinowski DS, Merlot AM, Sahni S, Richardson DR. Novel thiosemicarbazones regulate the signal transducer and activator of transcription 3 (STAT3) pathway: inhibition of constitutive and interleukin 6-induced activation by iron depletion. Mol Pharmacol. 2015; 87:543–560.
122. Kulp KS, Green SL, Vulliet PR. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin D CDK4 protein levels in asynchronous MDA-MB-453 human breast cancer cells. Exp Cell Res. 1996; 229:60–68.
123. Fukuchi K, Tomoyasu S, Watanabe H, Kaetsu S, Tsuruoka N, Gomi K. Iron deprivation results in an increase in p53 expression. Bio Chem Hoppe Seyler. 1995; 376:627–630.
124. Kastan MB, Canman CE, Leonard CJ. p53, cell-cycle control and apoptosis - implications for cancer. Cancer Metast Rev. 1995; 14:3–15.
125. Liang SX, Richardson DR. The effect of potent iron chelators on the regulation of p53: examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis. 2003; 24:1601–1614.
126. Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, Hu H, Huang Y, Hui L, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014; 7:180–193.
127. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. (2008) Molecular Biology of the Cell. (New York: Garland Science).
128. Weiss RH. p21(Waf1/Cip1) as a therapeutic target in breast and other cancers. Cancer Cell. 2003; 4:425–429.
129. Tang JJ, Shen C, Lu YJ. Requirement for pre-existing of p21 to prevent doxorubicin-induced apoptosis through inhibition of caspase-3 activation. Mol Cell Biochem. 2006; 291:139–144.
130. Fu D, Richardson DR. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21(CIP1/WAF1) by iron depletion. Blood. 2007; 110:752–761.
131. Dupont JL, Karas M, LeRoith D. The cyclin-dependent kinase inhibitor p21(CIP/WAF) is a positive regulator of insulin-like growth factor I-induced cell proliferation in MCF-7 human breast cancer cells. J Biol Chem. 2003; 278:37256–37264.
132. Tansey WP. Mammalian MYC Proteins and Cancer. New J Sci. 2014; 2014:757534.
133. Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med. 1999; 189:63–73.
134. Dang CV. MYC on the path to cancer. Cell. 2012; 149:22–35.
135. de Alboran IM, O’Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001; 14:45–55.
136. Mateyak MK, Obaya AJ, Sedivy JM. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999; 19:4672–4683.
137. Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, Tyner AL. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A. 2001; 98:4510–4515.
138. Kyriakou D, Eliopoulos AG, Papadakis A, Alexandrakis M, Eliopoulos GD. Decreased expression of c-myc oncoprotein by peripheral blood mononuclear cells in thalassaemia patients receiving desferrioxamine. Eur J Haematol. 1998; 60:21–27.
139. Kovacevic Z, Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006; 27:2355–2366.
140. Ellen TP, Ke Q, Zhang P, Costa M. NDRG1, a growth and cancer related gene: regulation of gene expression and function in normal and disease states. Carcinogenesis. 2008; 29:2–8.
141. Lachat P, Shaw P, Gebhard S, van Belzen N, Chaubert P, Bosman FT. Expression of NDRG1, a differentiation-related gene, in human tissues. Histochem Cell Biol. 2002; 118:399–408.
142. Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, Saito K, Commes T, Hayashi S, Watabe M, Watabe K. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003; 63:1731–1736.
143. Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Piquemal D, Commes T, Watabe M, Gross SC, Wang Y, Ran S, Watabe K. Role of the putative tumor metastasis suppressor gene Drg-1 in breast cancer progression. Oncogene. 2004; 23:5675–5681.
144. Guang RJ, Ford JL, Fu YN, Li YZ, Shaw LM, Pardee AB. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000; 60:749–755.
145. Maruyama Y, Ono M, Kawahara A, Yokoyama T, Basaki Y, Kage M, Aoyagi S, Kinoshita H, Kuwano M. Tumor growth suppression in pancreatic cancer by a putative metastasis suppressor gene Cap43/NDRG1/Drg-1 through modulation of angiogenesis. Cancer Res. 2006; 66:6233–6242.
146. Le NTV, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood. 2004; 104:2967–2975.
147. Kovacevic Z, Fu D, Richardson DR. The iron-regulated metastasis suppressor, Ndrg-1: identification of novel molecular targets. Biochim Biophys Acta. 2008; 1783:1981–1992.
148. Lane DJ, Saletta F, Suryo Rahmanto Y, Kovacevic Z, Richardson DR. N-myc downstream regulated 1 (NDRG1) is regulated by eukaryotic initiation factor 3a (eIF3a) during cellular stress caused by iron depletion. PLoS One. 2013; 8:e57273.
149. Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Lang F, Wulff P, Kuhl D, Cohen P. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J. 2004; 384:477–488.
150. Murakami Y, Hosoi F, Izumi H, Maruyama Y, Ureshino H, Watari K, Kohno K, Kuwano M, Ono M. Identification of sites subjected to serine/threonine phosphorylation by SGK1 affecting N-myc downstream-regulated gene 1 (NDRG1)/Cap43-dependent suppression of angiogenic CXC chemokine expression in human pancreatic cancer cells. Biochem Biophys Res Commun. 2010; 396:376–381.
151. McCaig C, Potter L, Abramczyk O, Murray JT. Phosphorylation of NDRG1 is temporally and spatially controlled during the cell cycle. Biochem Biophys Res Commun. 2011; 411:227–234.
152. Ghalayini MK, Dong Q, Richardson DR, Assinder SJ. Proteolytic cleavage and truncation of NDRG1 in human prostate cancer cells, but not normal prostate epithelial cells. Biosci Rep. 2013; 33. art:e00042.doi:00010.01042/BSR20130042.
153. Sun J, Zhang D, Bae DH, Sahni S, Jansson P, Zheng Y, Zhao Q, Yue F, Zheng M, Kovacevic Z, Richardson DR. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis. 2013; 34:1943–1954.
154. Fang BA, Kovacevic Z, Park KC, Kalinowski DS, Jansson PJ, Lane DJ, Sahni S, Richardson DR. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim Biophys Acta. 2014; 1845:1–19.
155. Kovacevic Z, Sivagurunathan S, Mangs H, Chikhani S, Zhang D, Richardson DR. The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis. 2011; 32:732–740.
156. Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z, Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. 2012; 287:17016–17028.
157. Sun J, Zhang D, Zheng Y, Zhao Q, Zheng M, Kovacevic Z, Richardson DR. Targeting the metastasis suppressor, NDRG1, using novel iron chelators: regulation of stress fiber-mediated tumor cell migration via modulation of the ROCK1/pMLC2 signaling pathway. Mol Pharmacol. 2013; 83:454–469.
158. Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008; 27:6473–6488.
159. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JFM. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008; 8:187–198.
160. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129:1261–1274.
161. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009; 4:127–150.
162. Cheng GZ, Park S, Shu SK, He LL, Kong W, Zhang WZ, Yuan ZQ, Wang LH, Cheng JQ. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr Cancer Drug Targets. 2008; 8:2–6.
163. Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999; 96:4240–4245.
164. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996; 15:6541–6551.
165. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997; 91:231–241.
166. Lee HK, Kwak HY, Hur J, Kim IA, Yang JS, Park MW, Yu J, Jeong S. beta-catenin regulates multiple steps of RNA metabolism as revealed by the RNA aptamer in colon cancer cells. Cancer Res. 2007; 67:9315–9321.
167. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998; 12:3499–3511.
168. Du KY, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998; 273:32377–32379.
169. Brennan P, Babbage JW, Burgering BMT, Groner B, Reif K, Cantrell DA. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997; 7:679–689.
170. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappa B by the Akt PKB kinase. Curr Biol. 1999; 9:601–604.
171. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999; 96:857–868.
172. Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000; 20:8969–8982.
173. Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000; 60:3504–3513.
174. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002; 4:648–657.
175. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell. 2002; 10:151–162.
176. Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009; 37:217–222.
177. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 18:1926–1945.
178. West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene. 1998; 17:769–780.
179. Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005; 16:525–537.
180. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9:550–562.
181. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997; 275:1943–1947.
182. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997; 15:356–362.
183. Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001; 61:7426–7429.
184. Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006; 18:77–82.
185. Wegiel B, Bjartell A, Culig Z, Persson JL. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer. 2008; 122:1521–1529.
186. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014; 46:372–383.
187. Kovacevic Z, Chikhani S, Lui GY, Sivagurunathan S, Richardson DR. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid Redox Signal. 2013; 18:874–887.
188. Alvarez-Tejado M, Naranjo-Suarez S, Jimenez C, Carrera AC, Landazuri MO, del Peso L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem. 2001; 276:22368–22374.
189. Box AH, Yuen C, Ponjevic D, Fick GH, Demetrick DJ. Signaling and apoptosis differences between severe hypoxia and desferoxamine treatment of human epithelial cells. Biochem Cell Biol. 2008; 86:425–436.
190. Dongiovanni P, Valenti L, Fracanzani AL, Gatti S, Cairo G, Fargion S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am J Pathol. 2008; 172:738–747.
191. Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007; 109:4045–4054.
192. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011; 36:320–328.
193. Zhang L, Zhou F, ten Dijke P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. 2013; 38:612–620.
194. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26:3291–3310.
195. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003; 17:1263–1293.
196. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004; 4:361–370.
197. Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005; 12:S17–S27.
198. Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002; 110:669–672.
199. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773:1263–1284.
200. Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007; 1773:1150–1160.
201. Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A. 1982; 79:4848–4852.
202. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003; 3:459–465.
203. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996; 87:619–628.
204. Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004; 101:15313–15317.
205. Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003; 278:18811–18816.
206. Welch DR, Sakamaki T, Pioquinto R, Leonard TO, Goldberg SF, Hon Q, Erikson RL, Rieber M, Rieber MS, Hicks DJ, Bonventre JV, Alessandrini A. Transfection of constitutively active mitogen-activated protein/extracellular signal-regulated kinase kinase confers tumorigenic and metastatic potentials to NIH3T3 cells. Cancer Res. 2000; 60:1552–1556.
207. Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997; 137:481–492.
208. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3:11–22.
209. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007; 26:3279–3290.
210. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007; 357:39–51.
211. Izar B, Rotow J, Gainor J, Clark J, Chabner B. Pharmacokinetics, clinical indications, and resistance mechanisms in molecular targeted therapies in cancer. Pharmacol Rev. 2013; 65:1351–1395.
212. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001; 22:153–183.
213. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995; 270:1326–1331.
214. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001; 410:37–40.
215. Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013; 4:342–359.
216. Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004; 3:156–161.
217. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009; 9:537–549.
218. Nakayama K, Nakayama N, Davidson B, Katabuchi H, Kurman RJ, Velculescu VE, Shih Ie M, Wang TL. Homozygous deletion of MKK4 in ovarian serous carcinoma. Cancer Biol Ther. 2006; 5:630–634.
219. Cunningham SC, Gallmeier E, Hucl T, Dezentje DA, Calhoun ES, Falco G, Abdelmohsen K, Gorospe M, Kern SE. Targeted deletion of MKK4 in cancer cells: a detrimental phenotype manifests as decreased experimental metastasis and suggests a counterweight to the evolution of tumor-suppressor loss. Cancer Res. 2006; 66:5560–5564.
220. Kim BM, Chung HW. Desferrioxamine (DFX) induces apoptosis through the p38-caspase8-Bid-Bax pathway in PHA-stimulated human lymphocytes. Toxicol Appl Pharmacol. 2008; 228:24–31.
221. Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. 2011; 286:15413–15427.
222. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998; 17:2596–2606.
223. Lee SK, Jang HJ, Lee HJ, Lee J, Jeon BH, Jun CD, Lee SK, Kim EC. p38 and ERK MAP kinase mediates iron chelator-induced apoptosis and -suppressed differentiation of immortalized and malignant human oral keratinocytes. Life Sci. 2006; 79:1419–1427.
224. Kim BS, Yoon KH, Oh HM, Choi EY, Kim SW, Han WC, Kim EA, Choi SC, Kim TH, Yun KJ, Kim EC, Lyou JH, Nah YH, Chung HT, Cha YN, Jun CD. Involvement of p38 MAP kinase during iron chelator-mediated apoptotic cell death. Cell Immunol. 2002; 220:96–106.
225. Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J Biol Chem. 2002; 277:31808–31817.
226. Rahimi RA, Leof EB. TGF-beta signaling: A tale of two responses. J Cell Biochem. 2007; 102:593–608.
227. Abdollah S, MaciasSilva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. T beta RI phosphorylation of Smad2 on Ser(465) and Ser(467) is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997; 272:27678–27685.
228. Souchelnytskyi S, Tamaki K, Engstrom U, Wernstedt C, tenDijke P, Heldin CH. Phosphorylation of Ser(465) and Ser(467) in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-beta signaling. J Biol Chem. 1997; 272:28107–28115.
229. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005; 19:2783–2810.
230. Massague J, Blain SW, Lo RS. TGF beta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103:295–309.
231. Guo YP, Kyprianou N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res. 1999; 59:1366–1371.
232. Sutkowski DM, Fong CJ, Sensibar JA, Rademaker AW, Sherwood ER, Kozlowski JM, Chung L. Interaction of epidermal growth factor and transforming growth factor-beta in human prostatic epithelial cells in culture. Prostate. 1992; 21:133–143.
233. Wilding G. Response of prostate cancer cells to peptide growth factors - transforming growth factor-beta. Cancer Surv. 1991; 11:147–163.
234. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGF beta/Smad signaling by oncogenic Ras. Genes Dev. 1999; 13:804–816.
235. Alexandrow MG, Moses HL. Transforming growth-factor-beta and cell-cycle regulation. Cancer Res. 1995; 55:1452–1457.
236. Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013; 2013:421821.
237. Ihle JN. Cytokine receptor signalling. Nature. 1995; 377:591–594.
238. Zhong Z, Wen Z, Darnell JE Jr,. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994; 264:95–98.
239. Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995; 269:81–83.
240. Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000; 19:6613–6626.
241. Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, Hirano T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity. 1999; 11:709–719.
242. Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, Briggs J, Karras J, Cress WD, Pardoll D, Jove R, Chen J, Yu H. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005; 25:7432–7440.
243. Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004; 23:3550–3560.
244. Debidda M, Wang L, Zang H, Poli V, Zheng Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J Biol Chem. 2005; 280:17275–17285.
245. Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC, Su WY, Wang JL, Cui Y, Wang ZH, Fang JY. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem. 2012; 287:5819–5832.
246. Scholz A, Heinze S, Detjen KM, Peters M, Welzel M, Hauff P, Schirner M, Wiedenmann B, Rosewicz S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology. 2003; 125:891–905.
247. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000; 19:2474–2488.
248. Sansone P, Bromberg J. Targeting the interleukin-6/Jak/Stat pathway in human malignancies. J Clin Oncol. 2012; 30:1005–1014.
249. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014; 1845:136–154.
250. Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997; 17:6508–6516.
251. Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene. 1998; 17:3157–3167.
252. Nakagami H, Morishita R, Yamamoto K, Taniyama Y, Aoki M, Matsumoto K, Nakamura T, Kaneda Y, Horiuchi M, Ogihara T. Mitogenic and antiapoptotic actions of hepatocyte growth factor through ERK, STAT3, and AKT in endothelial cells. Hypertension. 2001; 37:581–586.
253. Lim CP, Cao X. Regulation of Stat3 activation by MEK kinase 1. J Biol Chem. 2001; 276:21004–21011.
254. Higashi N, Kunimoto H, Kaneko S, Sasaki T, Ishii M, Kojima H, Nakajima K. Cytoplasmic c-Fos induced by the YXXQ-derived STAT3 signal requires the co-operative MEK/ERK signal for its nuclear translocation. Genes Cells. 2004; 9:233–242.
255. Sun S, Steinberg BM. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol. 2002; 83:1651–1658.
256. Kortylewski M, Feld F, Kruger KD, Bahrenberg G, Roth RA, Joost HG, Heinrich PC, Behrmann I, Barthel A. Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)-dependent mechanism. J Biol Chem. 2003; 278:5242–5249.
257. Gong C, Zhang Y, Shankaran H, Resat H. Integrated analysis reveals that STAT3 is central to the crosstalk between HER/ErbB receptor signaling pathways in human mammary epithelial cells. Mol Biosyst. 2015; 11:146–158.
258. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15:178–196.
259. Katz LH, Li Y, Chen JS, Munoz NM, Majumdar A, Chen J, Mishra L. Targeting TGF-beta signaling in cancer. Expert Opin Ther Targets. 2013; 17:743–760.
260. Song S, Christova T, Perusini S, Alizadeh S, Bao RY, Miller BW, Hurren R, Jitkova Y, Gronda M, Isaac M, Joseph B, Subramaniam R, Aman A, Chau A, Hogge DE, Weir SJ, et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 2011; 71:7628–7639.
261. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009; 19:128–139.
262. Park BJ, Park JI, Byun DS, Park JH, Chi SG. Mitogenic conversion of transforming growth factor-beta 1 effect by oncogenic Ha-Ras-induced activation of the mitogen-activated protein kinase signaling pathway in human prostate cancer. Cancer Res. 2000; 60:3031–3038.
263. Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009; 11:943–950.
264. Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell. 2007; 18:3533–3544.
265. Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003; 11:915–926.
266. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994; 127:2021–2036.
267. Lamouille S, Derynck R. Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs. 2011; 193:8–22.
268. Weiss MB, Abel EV, Mayberry MM, Basile KJ, Berger AC, Aplin AE. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012; 72:6382–6392.
269. Uttamsingh S, Bao X, Nguyen KT, Bhanot M, Gong J, Chan JL, Liu F, Chu TT, Wang LH. Synergistic effect between EGF and TGF-beta1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2008; 27:2626–2634.
270. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007; 178:437–451.
271. Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci. 2012; 125:1259–1273.
272. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005; 168:29–33.
273. Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT, Wang SP, Nesvizhskii AI, Chen YJ, Hong TM, Yang PC. GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene. 2014; 33:3172–3182.
274. Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001; 12:27–36.
275. Vardouli L, Moustakas A, Stournaras C. LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem. 2005; 280:11448–11457.
276. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004; 20:781–810.
277. Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010; 106:1798–1806.
278. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006; 8:1398–1406.
279. Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A. 2012; 109:16654–16659.
280. Lee JC, Chung LC, Chen YJ, Feng TH, Juang HH. N-myc downstream-regulated gene 1 downregulates cell proliferation, invasiveness, and tumorigenesis in human oral squamous cell carcinoma. Cancer Lett. 2014; 355:242–252.
281. Jin R, Liu W, Menezes S, Yue F, Zheng M, Kovacevic Z, Richardson DR. The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of beta-catenin through mechanisms involving FRAT1 and PAK4. J Cell Sci. 2014; 127:3116–3130.
282. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012; 586:1959–1970.
283. Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, Iqbal T, Tselepis C. A role for iron in Wnt signalling. Oncogene. 2008; 27:966–975.
284. Zhang W, Wu Y, Yan Q, Ma F, Shi X, Zhao Y, Peng Y, Wang J, Jiang B. Deferoxamine enhances cell migration and invasion through promotion of HIF-1alpha expression and epithelial-mesenchymal transition in colorectal cancer. Oncol Rep. 2014; 31:111–116.
285. Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012; 24:277–283.
286. Tojkander S, Gateva G, Lappalainen P. Actin stress fibers—assembly, dynamics and biological roles. J Cell Sci. 2012; 125:1855–1864.
287. Shutova M, Yang C, Vasiliev JM, Svitkina T. Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS One. 2012; 7:e40814.
288. Schwartz M. Rho signalling at a glance. J Cell Sci. 2004; 117:5457–5458.
289. Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004; 116:167–179.
290. Katoh K, Kano Y, Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface. 2011; 8:305–311.
291. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003; 22:337–358.
292. Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004; 117:989–998.
293. Liu W, Yue F, Zheng M, Merlot AM, Bae DH, Huang M, Lane DJ, Jansson P, Lui GY, Richardson V, Sahni S, Kalinowski D, Kovacevic Z, Richardson D. The proto-oncogene c-Src and its downstream signaling pathways are inhibited by the metastasis suppressor, NDRG1. Oncotarget. 2015; 6:8851–8874.
294. Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009; 28:51–63.
295. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005; 120:159–162.
296. Hamacher-Brady A. Autophagy regulation and integration with cell signaling. Antioxid Redox Signal. 2012; 17:756–765.
297. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010; 22:124–131.
298. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009; 20:1992–2003.
299. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011; 10:1533–1541.
300. Krishan S, Jansson PJ, Gutierrez E, Lane DJ, Richardson DES, Sahni S. Iron metabolism and autophagy: a poorly explored relationship that has important consequences for health and disease. Nagoya J Med Sci. 2015; 77:1–6.
301. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011; 331:456–461.
302. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007; 18:716–731.
303. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007; 14:230–239.
304. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003; 112:1809–1820.
305. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009; 137:1062–1075.
306. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011; 25:717–729.
307. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005; 6:505–510.
308. Gutierrez E, Richardson DR, Jansson PJ. The anti-cancer agent, Dp44mT, overcomes pro-survival autophagy by two mechanisms: persistent induction of autophagosome synthesis and impairment of lysosomal integrity. J Biol Chem. 2014; 289:33568–33589.
309. Sahni S, Bae DH, Lane DJ, Kovacevic Z, Kalinowski DS, Jansson PJ, Richardson DR. The metastasis suppressor, N-myc downstream-regulated gene 1 (NDRG1), inhibits stress-induced autophagy in cancer cells. J Biol Chem. 2014; 289:9692–9709.
310. Segawa T, Nau ME, Xu LL, Chilukuri RN, Makarem M, Zhang W, Petrovics G, Sesterhenn IA, McLeod DG, Moul JW, Vahey M, Srivastava S. Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene. 2002; 21:8749–8758.
311. Pullarkat V, Meng Z, Donohue C, Yamamoto VN, Tomassetti S, Bhatia R, Krishnan A, Forman SJ, Synold TW. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk Res. 2014; 38:988–996.
312. Liang C. Negative regulation of autophagy. Cell Death Differ. 2010; 17:1807–1815.
313. Moon JH, Jeong JK, Park SY. Deferoxamine inhibits TRAIL-mediated apoptosis via regulation of autophagy in human colon cancer cells. Oncol Rep. 2015; 33:1171–1176.
314. Hultcrantz R, Glaumann H. Intracellular fate of ferritin in HeLa cells following microinjection. Exp Cell Res. 1987; 171:203–212.
315. Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol. 2006; 291:C445–455.
316. De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine the route of ferritin degradation. Blood. 2009; 114:4546–4551.
317. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010; 140:313–326.
318. Potuckova E, Jansova H, Machacek M, Vavrova A, Haskova P, Tichotova L, Richardson V, Kalinowski DS, Richardson DR, Simunek T. Quantitative analysis of the anti-proliferative activity of combinations of selected iron-chelating agents and clinically used anti-neoplastic drugs. PLoS One. 2014; 9:e88754.