
INTRODUCTION
Prostate cancer (PC) is the leading cause of male cancer deaths in the western world and remains a major challenge to treat effectively [1, 2]. At presentation, PC growth is androgen-dependent hence the mainstay for treatment is hormone-ablation therapy using anti-androgens and/or androgen-deprivation therapies (ADT) [3, 4]. These act to repress the androgen receptor (AR), a member of the nuclear hormone receptor family of transcription factors that regulates expression of genes involved in prostate growth and transformation. By directly competing for androgen binding, anti-androgens such as bicalutamide, prevent activation of the AR and hence cause tumour regression [3, 5]. Unfortunately, the cancer invariably re-appears in an androgen-independent form, termed castrate-resistant PC (CRPC), that is largely fatal. Importantly, the AR signalling axis is active in this advanced stage of disease and thus remains a suitable therapeutic target [2, 6]. Indeed, the development of second generation anti-androgen and ADT therapies, such as enzalutamide [7], ARN-509 [8] and abiraterone [9] have shown promise in the treatment of CRPC. However, response rates of just 50% and the development of resistance has limited their success in the clinic [10–12].
Aberrant AR signalling is a hallmark of CRPC and is driven by numerous mechanisms including AR gene amplification [13], somatic receptor mutation [14, 15], expression of AR splice variants [16] and de-regulated co-factor expression [17, 18] that facilitate receptor activity in castrate conditions and contribute to treatment failure. Post-translational modification of the AR represents an additional level of receptor regulation with acetylation of key residues in the hinge region of the receptor playing a pivotal role in contact-independent growth and tumour development in vivo [19].
The acquisition of AR mutations during ADT, that either facilitate transcriptional activity of the receptor in the absence of androgens or switch anti-androgens to AR agonists, is a well characterised mechanism of hormone escape and has been reported to occur in upwards of 60% of CRPC patients [3, 14]. Importantly, the frequency of AR mutations in primary disease is low, but is elevated in advanced disease through therapy-specific selection of aberrantly functioning receptors [14, 15]. For example, chronic treatment with the anti-androgens bicalutamide and flutamide regularly drives selection of respective ARW741L/C and ARH874Y/ART877A mutations that utilise the agents as agonists to promote androgenic signalling and tumour cell growth [1]. More recently, the identification of an ARF876L mutation in patient samples refractory to enzalutamide and ARN-509 therapies has indicated that this is a phenomenon not limited to first-generation anti-androgens [20–22].
Modelling the function of CRPC-relevant AR mutants in their native context is challenging with most studies utilising non-PC cell lines, ectopically-expressed variant receptors and luciferase reporter-based transcriptional assays [15, 23, 24]. Outside of LNCaP cell studies, that express the ART877A mutant, there is a paucity of information on the functional dynamics and global transcriptomics of CRPC-associated AR mutants in a physiological setting that is likely to provide key biomarkers and additional treatment regimens for anti-androgen-resistant malignancies. Moreover, a major consideration for the development of next-generation AR-targeted therapies is whether they will be effective against pre-existing AR mutants in CRPC hence the development of key research tools to facilitate these studies is of high priority. To address this, we have developed a novel RNAi-rescue approach that utilises stable expression of specific AR mutants in LNCaP cells depleted of the endogenous receptor to facilitate more robust analyses of aberrant receptor signalling. Therefore, it is now possible to assess global transcriptional activity and sensitivity of CRPC-associated AR mutants to new receptor-targeting agents in a more relevant cellular context. Using the ARW741L variant as a paradigm, we demonstrate that this mutant activates several endogenous AR-target genes, including PSA and TMPRSS2, and promotes a hyper-proliferative phenotype in the presence of bicalutamide; a phenomenon that can be reversed by depletion of ARW741L. Global transcriptomics identified a sub-set of ARW741L-driven genes that are markedly up-regulated compared to the endogenous receptor, including SGK1, TIPARP and RASD1. Importantly, treatment with an SGK inhibitor down-regulated bicalutamide-driven receptor activity and cell growth, suggesting this could be a novel avenue of treatment for bicalutamide-resistant patients. In all, we have successfully applied a novel AR replacement strategy to physiologically model the ARW741L mutation in disease and highlighted key distinctions in receptor activity that could be therapeutically-exploited for improved CRPC treatment.
RESULTS
Generation of an RNAi-rescue strategy for testing AR mutant activity
There is a paucity of physiologically-relevant information on the distinct functionality of CRPC-associated AR mutants and how they drive aggressive PC malignancy. Studies to date have primarily utilised reporter-based assays incorporating ectopically-expressed mutant receptors to assess activity and sensitivity to receptor-targeting agents in AR null cell lines [14]. Although useful to demonstrate that specific CRPC-associated receptor mutants are activated by distinct ligands and down-regulated by first- and second-generation anti-androgens, as demonstrated in Supplementary Figures S1A and S1B, the failure to assess global functionality of these aberrantly functioning receptors in this context is a major problem. Improved models for examining CRPC-relevant AR mutations are therefore required. To address this, we developed a more physiological read-out for AR mutant activity using an siRNA-mediated receptor replacement strategy in the androgenic LNCaP PC cell line. Using the bicalutamide-activated ARW741L as a paradigm for this study (Supplementary Figure S1), in part due to its relevance in current clinical practise, we generated an LNCaP derivative that stably expressed FLAG-tagged ARW741L, called LNCaP-ARW741L (Figure 1A). We next designed siRNA oligonucleotides (termed siART877A) to discriminately deplete endogenous ART877A (approximately 90% knockdown) by targeting the 3′-UTR of the AR transcript that is absent in ectopically-expressed ARW741L mRNA (see Supplementary Table S2 for sequences). These oligonucleotides down-regulated endogenous AR and PSA levels in LNCaP cells (Supplementary Figure S2A) and attenuated DHT-induced ART877A recruitment to cis-regulatory elements of the PSA gene (Supplementary Figure S2B). Importantly, siART877A failed to reduce levels of ectopically-expressed FLAG-AR in PC3 cells, while an oligonucleotide targeted to the coding region of the AR (siAR) down-regulated expression of this protein (Supplementary Figure S3A and S3B). In the context of the LNCaP-ARW741L cell line, as expected, siART877A reduced endogenous AR levels, but did not affect expression of the ARW741L variant (Figure 1A). Importantly, an siRNA targeted specifically to the linker region between the FLAG-tag and translation start site of the ARW741L transcript markedly depleted the ectopically-expressed protein, but failed to impact on endogenous ART877A.
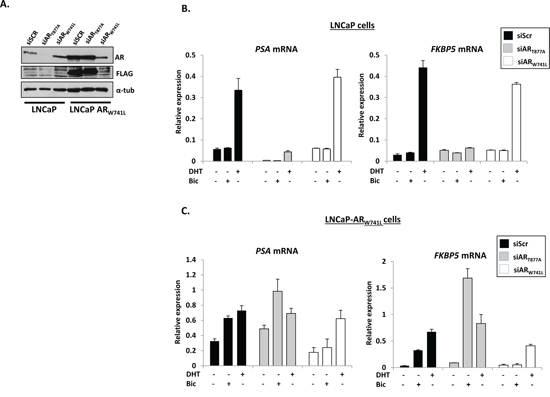
Figure 1: Stably-integrated ARW741L in LNCaP cells up-regulates endogenous PSA and FKBP5 in the presence of bicalutamide. A. Western analysis of parental and ARW741L-expressing LNCaP cells depleted of either endogenous (siART877A) or ectopic (siARW741L) receptors using AR, FLAG (to detect FLAG-tagged ARW741L) and α-tubulin antibodies. Scrambled siRNA (siScr) was used as a transfection control. Quantitative PCR analysis of PSA and FKBP5 expression in parental LNCaP cells B. and the LNCaP-ARW741L derivative C. depleted of either endogenous or ectopic receptors treated with 1 nM DHT or 10 nM bicalutamide for 24 hours. Data represents the mean of three independent experiments ± standard error.
To assess if expression of the bicalutamide-activated mutant impacts on the behaviour of the LNCaP derivative cell line, we firstly investigated the expression of known AR target genes PSA, FKBP5 (Figure 1B), KLK2 and TMPRSS2 (Supplementary Figure S4) in the presence and absence of 1 nM DHT, 10 nM bicalutamide (pro-proliferative dose; see Supplementary Figure S5) or vehicle control and compared to parental LNCaP cells. As expected, AR-target gene expression was up-regulated by DHT, but not bicalutamide, in LNCaP cells and this effect could be negated by depletion of endogenous ART877A (Figure 1B and Supplementary Figure S4A). In contrast, both DHT and bicalutamide enhanced transcription in LNCaP-ARW741L cells, and depletion of ART877A further increased bicalutamide-activated FKBP5 and TMPRSS2 expression (Figure 1C and Supplementary Figure S4B), suggesting a potential inhibitory role of ART877A when both receptors are co-expressed. The effect of bicalutamide on these genes was specific to the ARW741L variant as knockdown using the siARW741L oligonucleotide abolished anti-androgen-driven transcription, but still enabled endogenous ART877A to drive gene expression in the presence of DHT (Figure 1C and Supplementary Figure S4B).
Bicalutamide-activated ARW741L is recruited to AR-target genes and enhances cell proliferation
We next investigated recruitment of ARW741L to endogenous cis-regulatory elements of the AR-target genes PSA and TMPRSS2 in response to 1 nM DHT and 10 nM bicalutamide using a FLAG antibody. In cells expressing both AR variants (siSCR), DHT and bicalutamide activated a subtle increase in recruitment of FLAG-ARW741L to these loci (Figure 2A). Consistent with gene expression data (Figure 1), depletion of endogenous ART877A markedly elevated bicalutamide-activated ARW741L recruitment to the PSA enhancer and TMPRSS2 promoter and this effect was diminished upon ectopic ARW741L knockdown. Moreover, ARW741L is also recruited to the promoter of the KLK2 gene in response to bicalutamide and this can be attenuated with ectopic receptor knockdown (Supplementary Figure S6). Importantly, using a phospho-serine 5 RNA polymerase II antibody as a marker of transcriptional initiation, we found that DHT- and bicalutamide-activated ARW741L can facilitate the assembly of the transcriptional machinery to drive expression of endogenous PSA and TMPRSS2 genes (Figure 2B).
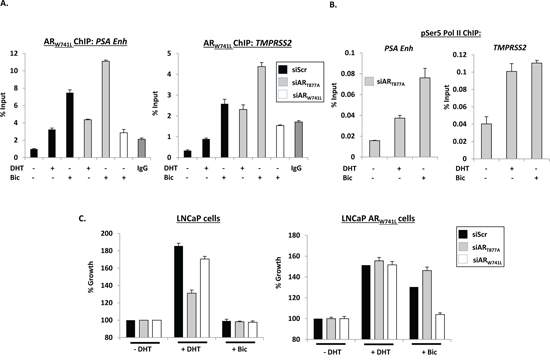
Figure 2: ARW741L is recruited to cis-regulatory elements of AR-target genes and drives a pro-proliferative phenotype in response to bicalutamide. A. LNCaP-ARW741L cells depleted of either endogenous or ectopic AR were treated with 1 nM DHT or 10 nM bicalutamide for 4 hours prior to chromatin immunoprecipitation (ChIP) analysis using a FLAG antibody to immunoprecipitate FLAG-ARW741L. Receptor recruitment to the PSA enhancer (PSA Enh) and TMPRSS2 promoter was assessed by quantitative PCR. B. LNCaP-ARW741L cells depleted of endogenous receptor and treated with 1 nM DHT or 10 nM bicalutamide for 4 hours were subject to ChIP analysis using a phospho-Serine 5 RNA polymerase II antibody (pSer5 Pol II) and enrichment at PSA and TMPRSS2 genes measured by quantitative PCR. C. LNCaP cells or the LNCaP-ARW741L derivative depleted of endogenous or ectopic AR were grown in the presence of 1 nM DHT or 10 nM bicalutamide for 96 hours prior to SRB staining. Percentage growth is relative to vehicle control for each siRNA. Data is the mean of triplicate experiments ± standard error.
We next examined the effect of ARW741L on proliferation of the LNCaP-ARW741L derivative cell line in response to 1 nM DHT and 10 nM bicalutamide, and compared to parental LNCaP cells. As shown in Figure 2C, growth of LNCaP cells was increased by DHT and this effect was reduced by siART877A, but not siARW741L. In contrast, growth of LNCaP-ARW741L was markedly increased in the presence of both DHT and bicalutamide and only depletion of ARW741L abolished bicalutamide-driven growth of these cells. To demonstrate that these findings were not an artefact of this specific clonal population of ARW741L-expressing cells, we tested an additional selected derivative (Clone 2) against the original (Clone 1; utilised in all previous experiments) and a control transduced cell line (LNCaP-LacZ) that does not overexpress ARW741L. Importantly, both Clone 1 and Clone 2 showed comparable growth stimulation in response to DHT and bicalutamide and was distinct from LNCaP-LacZ that only responded to DHT (Supplementary Figure S7).
ARW741L is inactivated by enzalutamide
Enzalutamide has shown great promise in the clinic [25], but the fact that not all patients respond to the drug may indicate the existence of a pre-determinant, such as an AR mutant, that compromises enzalutamide efficacy [20, 21]. Given that enzalutamide in many cases is given as a second-line therapy post-bicalutamide treatment, it is therefore important to establish if the ARW741L mutant is sensitive to enzalutamide in our more robust CRPC model system. To this end, we firstly assessed the effect of 10 μM enzalutamide on bicalutamide-activated ARW741L transcriptional activity in LNCaP-ARW741L cells. As expected, bicalutamide up-regulated PSA and KLK2 gene expression, and importantly, this was reduced to basal levels upon administration of enzalutamide (Figure 3A). Furthermore, ChIP analysis using a FLAG antibody demonstrated that enzalutamide markedly diminished bicalutamide-activated ARW741L recruitment to cis-regulatory elements of the PSA, TMPRSS2 and KLK2 genes (Figure 3B and Supplementary Figure S8).
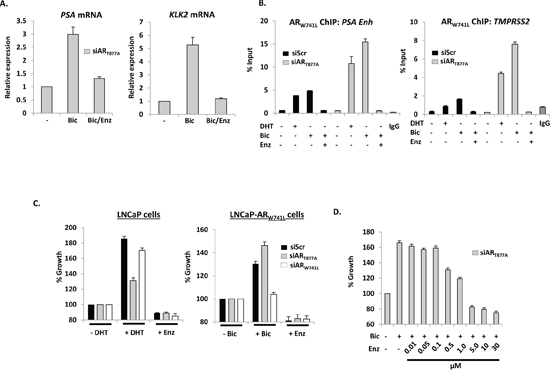
Figure 3: Bicalutamide-driven ARW741L activity and LNCaP-ARW741L cell growth is attenuated by enzalutamide. A. LNCaP-ARW741L cells depleted of endogenous AR were treated with 10 nM bicalutamide +/− 10 μM enzalutamide for 24 hours prior to quantitative analysis of AR target gene expression. B. LNCaP-ARW741L cells transfected with siScr or siART877A were treated for 4 hours with either 1 nM DHT or bicalutamide +/− 10 μM enzalutamide prior to ChIP and quantitative PCR analysis using primers specific to AR-target genes. C. LNCaP cells or the LNCaP-ARW741L derivative depleted of endogenous or ectopic AR were grown in the presence of 1 nM DHT, 10 nM bicalutamide or 10 μM enzalutamide for 96 hours prior to SRB staining. Percentage growth is relative to vehicle control for each siRNA. Data is the mean of triplicate experiments ± standard error. D. As for (C) except LNCaP-ARW741L cells depleted of endogenous AR were treated with 10 nM bicalutamide +/− increasing doses of enzalutamide to a maximum of 30 μM prior to SRB staining.
In proliferation assays, we showed that enzalutamide reduced growth of both parental and ARW741L-expressing LNCaP cells (Figure 3C and Supplementary Figure S9). Importantly, enzalutamide attenuated bicalutamide-driven proliferation of LNCaP-ARW741L cells (Figure 3D) indicating that this second-generation anti-androgen is likely to be effective in CRPC patients harbouring the ARW741L mutation.
ARW741L drives an androgenic signalling programme similar to ART877A
We next tested the utility of this model to provide much needed information on the global transcriptional targets of the CRPC-relevant ARW741L mutant. Using LNCaP-ARW741L cells depleted of endogenous ART877A, the expression profile of ARW741L in response to bicalutamide was compared to vehicle treated controls and a total of 869 genes were identified as being upregulated >1.5 fold (Figure 4A and 4B; siART877A). This threshold was chosen as it fell within fold change cut-offs that have been used in previous publications investigating AR signalling profiles [26–28]. As a control, LNCaP-LacZ cells were treated with 10 nM bicalutamide and the resultant transcriptome was compared to the gene list identified in LNCaP-ARW741L cells. Importantly, no genes were identified in the LNCaP-LacZ control line that exhibited >1.5 fold up-regulation in the presence of bicalutamide, suggesting that the identified set of bicalutamide-induced genes in the LNCaP-ARW741L cells were specific to the ectopically-expressed receptor (Figure 4A and 4B). To refine the list of core bicalutamide-induced genes further, we incorporated an additional control in which the expression profile of bicalutamide-treated LNCaP-ARW741L cells depleted of ARW741L was examined relative to siART877A vehicle and bicalutamide-stimulated experimental arms (Figure 4A and 4B; siARW741L). Genes that exhibited > 2 fold increase in expression following bicalutamide treatment in ARW741L-depleted cells were deemed to be bicalutamide-independent. We identified 38 genes matching to the 869 core data set that were subsequently eliminated (Figure 4A and 4B); resulting in a refined gene set of 831 genes whose expression were considered to be specifically driven by bicalutamide-activated ARW741L. The full list of bicalutamide-induced genes are listed in Supplementary Table S4 and include characterised AR-target genes such as PSA, KLK2, TMPRSS2, NKX3.1, KCNN2 and SPOCK1
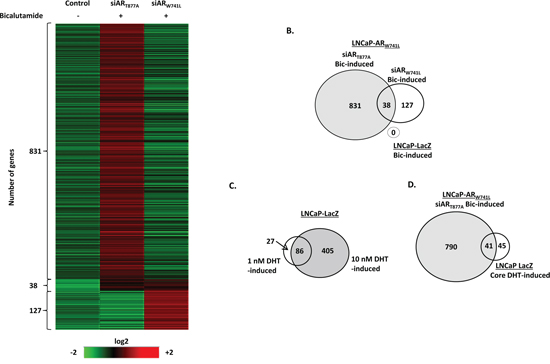
Figure 4: Bicalutamide activates an androgenic gene signature programme in LNCaP-ARW741L. LNCaP-ARW741L cells depleted of either endogenous or ectopic AR were treated with 10 nM bicalutamide for 24 hours prior to micro-array analysis. Resultant transcriptome was compared to control LNCaP-LacZ cells treated with 10 nM bicalutamide. Genes with a fold increase of >1.5 were considered to be ligand regulated and included in the analysis. Genes with a bicalutamide-induced fold increase of >2.0 in the ARW741L-depleted experimental arm were eliminated from the core list of bicalutamide-induced genes. Data is presented as a heat map A. and Venn diagram B, C. LNCaP-LacZ control cells were treated with either 1 or 10 nM DHT for 24 hours prior to micro-array analysis to identify a core of DHT-regulated genes between each condition. D. Overlap between bicalutamide-induced ARW741L transcriptome and the identified set of 86 core DHT-up-regulated genes in the LNCaP-LacZ cells.
We next compared the bicalutamide-driven transcriptome of LNCaP-ARW741L with the DHT-stimulated LNCaP-LacZ cell line derivative. In response to 1 and 10 nM DHT, microarray analysis revealed 113 and 491 up-regulated genes, respectively, relative to vehicle control (Figure 4C; Supplementary Tables S5 and S6). Comparison of both gene lists found 86 common genes (76% and 17% of 1 nM and 10 nM DHT up-regulated genes, respectively), highlighting a core set of androgen-regulated target genes that are activated in response to both 1 nM and 10 nM DHT (Figure 4C). As expected, a greater number of genes were activated in response to 10 nM DHT than 1 nM DHT, including FKBP5 (Supplementary Figure S10).
Direct comparison of the bicalutamide-induced LNCaP-ARW741L expression profile to the core set of androgen-regulated genes in LNCaP-LacZ, found that 41 of the 86 genes (48%) were common to both lists (Figure 4D). For example, PSA (KLK3) and KLK2, which were found to be the most DHT-stimulated in LNCaP-LacZ cells were also elevated in response to bicalutamide in LNCaP-ARW741L cells and is consistent with LNCaP-ARW741L gene expression data (Figure 1C). We next compared the bicalutamide-activated ARW741L transcriptome with two published androgenic gene signatures acquired from LNCaP cells [27, 29]. Of the respective 21 and 79 androgen-induced genes identified in the two studies, 15/21 (71%) and 52/79 (66%) matched directly to genes identified in the bicalutamide-activated LNCaP-ARW741L data-set (Supplementary Figure S11; see Supplementary Table S7), indicating robust commonality between the bicalutamide-induced ARW741L and DHT-stimulated ART877A transcriptional programmes.
Exploiting ARW741L-driven SGK1 expression to inactivate CRPC cell growth
We next focused our attention on identifying ARW741L-driven genes that were significantly elevated in response to bicalutamide and distinct from our DHT-stimulated LNCaP-LacZ transcriptome to define biomarkers of this specific CRPC-associated AR mutation and potentially highlight avenues for therapeutic exploitation. Of the top 20 bicalutamide up-regulated genes in LNCaP-ARW741L cells (Figure 5A), several were well known AR target genes, including FKBP5 and NDRG1, and are elevated in response to 10 nM DHT treatment in our LNCaP-LacZ control line (Supplementary Table S6). Importantly, array data indicated that TIPARP (TCDD-inducible poly(ADP-ribose) polymerase), RASD1 (Ras dexamethasone-induced 1) and SGK1 (serum- and glucocorticoid-regulated kinase 1) were exclusively and markedly up-regulated by bicalutamide in the LNCaP-ARW741L cell line compared to the DHT-stimulated control LNCaP-LacZ derivative (data not shown). Robust bicalutamide-mediated up-regulation of TIPARP (57-fold), RASD1 (93-fold) and SGK1 (109.5-fold) was validated by QPCR, and demonstrated to be exclusively mediated by ARW741L as depletion of this mutant by siARW741L completely abrogated gene expression (Figure 5B). In contrast, treatment of LNCaP-LacZ cells with a dose-range of DHT (Figure 5B and Supplementary Figure S12) only modestly increased SGK1 (5-fold) and RASD1 (2-fold) expression and failed to elevate TIPARP transcript levels while, as expected, PSA was greatly up-regulated (Supplementary Figure S13). Moreover, analysis of an additional LNCaP derivative that ectopically expresses wild-type AR (LNCaP-wtAR) to levels comparable to that of ARW741L (data not shown), demonstrated modest enhancement of SGK1, TIPARP and RASD1 transcription in response to DHT stimulation with respective 4.5-, 8.6- and 5.9-fold induction, indicating that the robust up-regulation of the three genes in LNCaP-ARW741L cells is not due to the phenomenon of elevated cellular AR levels (Supplementary Figure S14).
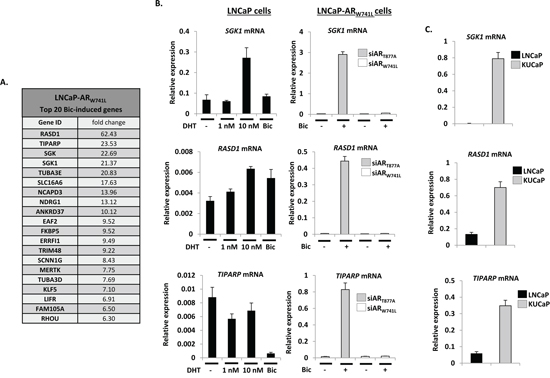
Figure 5: ARW741L markedly up-regulates genes distinct from endogenous ART877A. A. Top 20 bicalutamide-induced genes identified in siART877A-transfected LNCaP-ARW741L cells. B. LNCaP cells and LNCaP-ARW741L depleted of endogenous or ectopic receptors, were treated with either DHT (1 and 10 nM) or 10 nM bicalutamide for 24 hours prior to quantitative analysis of SGK1, RASD1 and TIPARP expression. C. Expression analysis of the same genes was compared between LNCaP cells and the ARW741C-expressing KUCaP xenograft model.
To investigate further the discriminate enhancement of TIPARP, RASD1 and SGK1 by ARW741L, we profiled expression of these genes in the KUCaP xenograft CRPC model. This xenograft was derived from a liver metastasis present in a bicalutamide-resistant CRPC patient and exclusively expresses the ARW741C mutation [30]. As shown in Figure 5C, expression of TIPARP, RASD1 and SGK1 were markedly elevated in KUCaP cells compared to the LNCaP line confirming that key distinctions exist between transcriptomes of ARW741L and ART877A that could be important in the pathobiology of disease in CRPC patients harbouring the bicalutamide-resistant mutation.
To address if the distinct gene-set of ARW741L could be exploited to provide key targets for CRPC therapy, we focussed on the potent up-regulation of SGK1 expression by this mutant receptor. Given that a previous study indicated that inactivation of SGK1 using the selective inhibitor GSK650394 reduces SGK1 expression and LNCaP cell growth [31], we hypothesised that the LNCaP-ARW741L derivative may be sensitive to this agent as they demonstrate markedly elevated SGK1 expression compared to our LNCaP control cells (Figure 5B). To this end, we assessed ARW741L-driven expression of SGK1 in the presence and absence of 10 μM GSK650395; a dose demonstrated to down-regulate SGK1 transcript levels in LNCaP cells [31]. As shown in Figure 6A, treatment of LNCaP-ARW741L cells with GSK650395 reduced expression of SGK1 by approximately 50% indicating that activity of the bicalutamide-activated receptor is potentiated by SGK1 and attenuation of the associated kinase activity down-regulates ARW741L-mediated transcription. Importantly, SGK1 inhibition reduced LNCaP-ARW741L proliferation by approximately 70% (Figure 6B) indicating SGK1 is a key down-stream effector of ARW741L-driven cell growth.
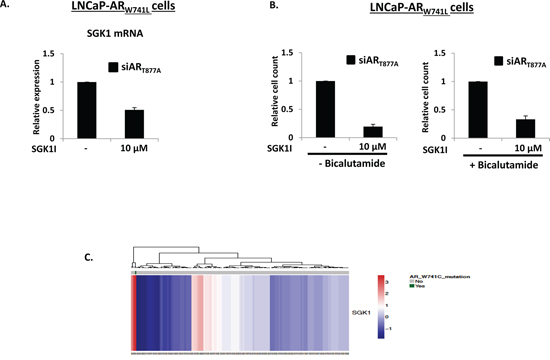
Figure 6: SGK1 inhibition reduces ARW741L activity and attenuates LNCaP-ARW741L proliferation. A. LNCaP-ARW741L cells depleted of ART877A were treated with 10 μM SGK inhibitor GSK650395 for 24 hours prior to quantitative analysis of SGK1 expression. B. As above, but cells were treated with GSK650395 for 96 hours in the presence and absence of 10 nM bicalutamide prior to proliferation analysis. C. In silico SGK1, TIPARP and RASD1 expression profiling in 150 primary prostate cancer specimens (28); ARW741C-expressing patient sample is indicated in green.
Finally, we conducted a systematic in silico analysis of a comprehensive PC cohort containing 150 primary tumour samples [32] and identified one patient biopsy expressing the ARW741C mutation. Consistent with our findings from the LNCaP-ARW741L derivative and KUCaP xenograft, SGK1 gene expression was found to be significantly up-regulated in this sample compared to all other samples suggesting that this may be a bona fide biomarker for aberrant ARW741L function (Figure 6C and Supplementary Figure S15) and manipulating activity of this enzyme may provide an additional means of treatment for CRPC patients resistant to bicalutamide.
DISCUSSION
The selection of AR mutations during androgen-depravation therapy is a well-defined mechanism of therapy resistance that, to date, has been reported to occur in upwards of 60% advanced CRPC patients [14, 15]. Although unclear, this figure may increase further due to two key developments: (i) the utility of more sensitive approaches for detecting mutations in both diagnostic and basic research [32], and (ii) ease of access to disseminated disease through the study of circulating tumour cells that offers a non-invasive means for AR sequencing in CRPC [33, 34]. Importantly, the presence of mutant AR in CRPC poses particular clinical challenges as many of the identified mutations promote promiscuous receptor activity that enable androgenic signalling by non-conventional ligands, including bicalutamide, flutamide [14], and more recently, enzalutamide/ARN-509 [10, 21]; hence limiting available treatment options in advanced disease. From a biological perspective, our understanding of the global functioning of these aberrant receptors is remarkably limited. Outside of studies in the PC cell lines LNCaP, that harbour the ART877A mutation, and CWR22RV1, that express ARH874Y and also numerous alternatively spliced AR isoforms, there is a reliance upon transient expression of mutant receptors in non-androgenic cell lines to study, in most cases, the transcriptional dynamics of these proteins [23, 24]. Although useful, these experiments offer little or no insight into global transcriptomics of AR mutants and lack physiological context. Given the prevalence of AR mutations in advanced CRPC, defining their activity in more robust and disease-relevant models is imperative to help improve our understanding of these receptors and to potentially exploit their distinct activities for the development of new PC treatments.
To this end, we developed a novel RNAi-rescue system to enable the study of AR mutants in the physiological background of LNCaP cells that have been depleted of the endogenous receptor. Using the bicalutamide-activated ARW741L as a proof of concept mutation, that is also clinically-relevant, we generated an LNCaP cell line derivative that stably-expressed this mutant receptor (LNCaP-ARW741L) and developed key siRNAs to deplete either endogenous or ectopic ART877A and ARW741L, respectively. Using this model, we demonstrated that in the presence of bicalutamide, ARW741L associates with cis-regulatory elements of several AR target genes, including PSA and TMPRSS2, and facilitates their expression. Significantly, these effects were attenuated upon depletion of the ectopic receptor indicating that the bicalutamide-driven functionality of ARW741L previously characterised in AR null cell line studies (Supplementary Figure S1 and [35]) has been phenocopied in LNCaP cells with promotion of chromatin-binding and endogenous target gene expression by the anti-androgen akin to the DHT-activated endogenous ART877A isoform. Interestingly, data from both chromatin immunoprecipitation and candidate gene expression analysis (PSA, TMPRSS2, KLK2) experiments demonstrated an inhibitory effect of ART877A on bicalutamide-activated ARW741L when both receptor isoforms were expressed (siScr control; Figures 1 and 2). We hypothesise that dimerization between the two distinct proteins may occur in the presence of bicalutamide and impact on their activity; homodimers of ARW741L will be transcriptionally potent, while ART877A-ARW741L heterodimers are likely to be functionally compromised. By depleting endogenous ART877A from LNCaP cells, the equilibrium is pushed toward the generation of active ARW741L dimers to promote more robust AR-target gene binding and transcription.
To further support the utility of this RNAi-rescue system to model distinct CRPC-relevant AR mutations, we demonstrated that the ARW741L mutant promoted growth of the LNCaP derivative line in the presence of bicalutamide. The pro-proliferative effect of bicalutamide was solely driven by the activity of ARW741L confirming that the ectopic receptor replaces the activity of ART877A and illustrates the ability for this model to recapitulate conditions of a bicalutamide-resistant CRPC disease state.
From these promising indications, we next tested the impact of the second-generation anti-androgen enzalutamide on mutant activity and growth of the LNCaP-ARW741L derivative. This is a particularly important experiment when one considers that new AR-targeting compounds will be applied to advanced, ADT-resistant CRPC that are likely to harbour pre-existing AR mutations [3]; hence defining efficacy of agents toward CRPC-relevant receptor mutations in a physiological model system is critical for optimal pre-clinical drug testing. In both the presence and absence of bicalutamide, ARW741L-chromatin binding, endogenous target gene expression and LNCaP-ARW741L growth was down-regulated by enzalutamide indicating that this mutation is sensitive to the second-generation anti-androgen. From this study, it is therefore possible to predict that patients refractory to bicalutamide through the selection of specific AR mutations are likely to demonstrate a clinical response to enzalutamide. However, given that up to 50% of patients do not respond to enzalutamide [7, 25], it is likely that other pre-determinants in advanced disease may compromise AR-targeting agent efficacy; the selection of other distinct mutations during first-line ADT could represent one such mechanism of new treatment failure. Importantly, our novel rescue system would enable the robust modelling of other CRPC-relevant receptor mutants that could facilitate patient stratification towards those predicted to receive benefit from sequential anti-AR targeted therapy and those that would not.
In keeping with the utility of this model as an important pre-clinical tool to identify distinctions in the functionality of disease-associated AR mutations, we hypothesised that global transcriptomic profiling of ARW741L would provide key gene targets that could be exploited for novel CRPC treatment strategies. Examination of the bicalutamide-activated ARW741L target gene-set against a control transduced LNCaP derivative (LNCaP-LacZ) identified a series of bicalutamide-induced genes; many of which were known AR-target genes including PSA, KLK2 and TMPRSS2. Furthermore, although the number of bicalutamide-activated genes were in excess of those controlled by DHT in our control cells and those reported in Hieronymous et al., [29] and Nelson et al., [27] there was considerable overlap in the bicalutamide- and androgen-activated transcriptomes supporting the concept that ARW741L maintains a common androgenic expression programme. Importantly, a number of robustly up-regulated bicalutamide-dependent genes were identified by micro-array in LNCaP-ARW741L cells that were not greatly enhanced by DHT in the LNCaP-LacZ line, including TIPARP, RASD1 and SGK1. Depletion of ectopic receptor in LNCaP-ARW741L cells attenuated this bicalutamide-driven response indicating that these genes are potentially discriminate targets of ARW741L. This dramatic and selective activity of the ARW741L mutant is intriguing and may be a consequence of subtle allosteric re-organisation of the receptor that permits hyper-activation of the receptor at distinct genomic loci. By repositioning the C-terminal activation function-2 (AF-2) domain of the receptor, the leucine residue may enable more robust interaction with selective co-regulators to drive acetylation and methylation of the AR to enhance inherent transcriptional activity [19, 36].
In contrast to our data, however, SGK1 has been identified to be robustly up-regulated by endogenous ART877A in LNCaP cells and the gene product found to positively reinforce androgenic signalling [31, 37]. The discrepancy between our data and these findings is intriguing and may reflect key differences in experimental design and performance, as although we did detect a 3-fold and 4.5-fold enhancement of SGK1 expression in response to 10 nM DHT by quantitative PCR in the respective LNCaP-LacZ and LNCaP-wtAR cell derivatives, it was significantly less than previous reported.
Importantly, the same study provided evidence that inactivation of SGK1 with the selective small molecule inhibitor GSK650395 was able to attenuate LNCaP cell proliferation in the presence of DHT [31]. Therefore, given that LNCaP-ARW741L cells demonstrated marked up-regulation of SGK1 expression in response to bicalutamide, we speculated that they may also be sensitive to SGK1 inhibition. Consistent with the reported phenomenon in parental LNCaP cells [31], we found that ARW741L-driven SGK1 expression was down-regulated in response to GSK650395 treatment, suggesting that ectopic receptor function is dependent upon SGK1-mediated kinase activity. Furthermore, proliferation of both LNCaP and LNCaP-ARW741L cells was significantly down-regulated upon SGK1 inhibition; with similar IC50 values for both cell lines in response to GSK650395 (Supplementary Figure S16). Importantly, in the LNCaP-ARW741L cells, this anti-proliferative effect was maintained in the presence of a pro-proliferative dose of bicalutamide. We have therefore identified a strongly up-regulated ARW741L target gene that may offer an avenue for therapeutic exploitation in bicalutamide-refractory CRPC. Although these observations are based on our rescue model system, evidence from the patient-derived KUCaP xenograft and a single patient biopsy sample, that both harbour the bicalutamide-activated ARW741C mutation [30, 32], is in agreement with our findings of elevated SGK1 expression and suggests utility of our RNAi-rescue approach to define global functionality of the ARW741L mutation.
In all, we have established a novel LNCaP cell line-based strategy to model CRPC-relevant mutations to provide important information on receptor dynamics and global gene signalling in response to distinct activating ligands.
MATERIALS AND METHODS
Luciferase reporter, quantitative PCR and western analyses
Luciferase assays were performed in HEK293T cells grown in steroid-depleted media as described in [38] utilising the p(ARE)3 reporter and pFLAG-AR, -ARW741L and -ARH874Y. Quantitative PCR was used to assess expression of endogenous AR-and ARW741L-target genes (see Supplementary Table S1 for primer sequences) using cDNA generated from Trizol-mediated RNA extractions as described in [39]. Western blotting was performed as described in [40] using antibodies listed in Supplementary Table S3.
Cell proliferation assays
Sulforhodamine B (SRB) assays were performed according to [41]. Briefly, 5 × 103 LNCaP or LNCaP-ARW741L cells per well of a 96-well plate grown in steroid-depleted conditions were transfected with siScr, siART877A or siARW741L for 96 hours as described above before fixing in trichloroacetic acid for 1 hour at 4°C. Cells were washed and subsequently stained with 0.4% SRB dissolved in 1% acetic acid. Plates were air dried at room temperature, after which bound SRB was dissolved in 10 mM Tris-HCl, pH 10.8. Absorbance was measured at 570 nm using a 96-well plate reader (BioRad). For drug treatments, 10 nM bicalutamide, 1 nM DHT, or increasing doses of enzalutamide, were administered for 96 hours prior to SRB assay.
For additional Methods please refer to accompanying Supplementary Information.
ACKNOWLEDGMENTS
We would like to thank Ralf Janknecht for supplying pCMV-FLAG-AR and Osamu Ogawa for providing KUCaP cDNA.
FUNDING
The work was supported by Cancer Research UK (DO), Medical Research Council (DJ) Worldwide Cancer Research (MW) and Prostate Cancer UK (JG).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Sridhar SS, Freedland SJ, Gleave ME, Higano C, Mulders P, Parker C, Sartor O, Saad F. Castration-Resistant Prostate Cancer: From New Pathophysiology to New Treatment. Eur Urol. 2013; 65:289–299.
2. Yuan X, Cai C, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013; 33:2815–2825.
3. Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the Mechanisms of Androgen Deprivation Resistance in Prostate Cancer at the Molecular Level. Eur Urol. 2014; 67:470–479.
4. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Current opinion in pharmacology. 2008; 8:440–448.
5. Bishr M, Saad F. Overview of the latest treatments for castration-resistant prostate cancer. Nature reviews Urology. 2013; 10:522–528.
6. Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 2012; 360:38–43.
7. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009; 324:787–790.
8. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cai L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012; 72:1494–1503.
9. Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008; 26:4563–4571.
10. Nelson WG, Yegnasubramanian S. Resistance emerges to second-generation antiandrogens in prostate cancer. Cancer discovery. 2013; 3:971–974.
11. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013; 73:483–489.
12. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371:1028–1038.
13. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995; 9:401–406.
14. Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Current genomics. 2009; 10:18–25.
15. Steinkamp MP, O'Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, Hofer MD, Kuefer R, Chinnaiyan A, Rubin MA, Pienta KJ, Robins DM. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009; 69:4434–4442.
16. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011; 18:R183–196.
17. Linja MJ, Porkka KP, Kang Z, Savinainen KJ, Janne OA, Tammela TL, Vessella RL, Palvimo JJ, Visakorpi T. Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res. 2004; 10:1032–1040.
18. Chan SC, Dehm SM. Constitutive activity of the androgen receptor. Adv Pharmacol. 2014; 70:327–366.
19. Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, Fan S, Rosen E, Palvimo JJ, Janne OA, Muratoglu S, Avantaggiati ML, et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003; 23:8563–8575.
20. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W, Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer discovery. 2013; 3:1030–1043.
21. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A Clinically Relevant Androgen Receptor Mutation Confers Resistance to Second-Generation Antiandrogens Enzalutamide and ARN-509. Cancer discovery. 2013; 3:1020–1029.
22. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013; 2:e00499.
23. Duff J, McEwan IJ. Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol. 2005; 19:2943–2954.
24. Brooke GN, Parker MG, Bevan CL. Mechanisms of androgen receptor activation in advanced prostate cancer: differential co-activator recruitment and gene expression. Oncogene. 2008; 27:2941–2950.
25. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012; 367:1187–1197.
26. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013; 155:1309–1322.
27. Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002; 99:11890–11895.
28. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10:33–39.
29. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006; 10:321–330.
30. Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, Kamoto T, Ogawa O. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005; 65:9611–9616.
31. Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, Norris JD, McDonnell DP. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008; 68:7475–7483.
32. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22.
33. Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. The lancet oncology. 2013; 14:882–892.
34. Scher HI, Jia X, de Bono JS, Fleisher M, Pienta KJ, Raghavan D, Heller G. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. The lancet oncology. 2009; 10:233–239.
35. Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003; 63:149–153.
36. Gaughan L, Stockley J, Wang N, McCracken SR, Treumann A, Armstrong K, Shaheen F, Watt K, McEwan IJ, Wang C, Pestell RG, Robson CN. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011; 39:1266–1279.
37. Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ. 2007; 14:2085–2094.
38. Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005; 33:13–26.
39. Gaughan L, Stockley J, Coffey K, O'Neill D, Jones DL, Wade M, Wright J, Moore M, Tse S, Rogerson L, Robson CN. KDM4B is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013; 41:6892–6904.
40. Rajan P, Gaughan L, Dalgliesh C, El-Sherif A, Robson C, Leung H, Elliott D. The RNA-binding and adaptor protein Sam68 modulates signal-dependent splicing and transcriptional activity of the androgen receptor. J Pathol. 2008; 215:67–77.
41. Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990; 82:1107–1112.