
Introduction
Squamous cell carcinoma of the head and neck (SCCHN) is the sixth most common cancer globally [1]. In spite of advances in the development of therapies targeting proteins such as EGFR (epidermal growth factor receptor), other ErbB family members [2, 3] or c-MET [4], which are highly expressed in many SCCHN tumors, the mainstays of treatment for SCCHN remain surgery, cytotoxic chemotherapy, and radiation [5]. In recent decades, the emergence of SCCHN of the oropharynx related to transforming oncogenic HPV infection has been recognized. [6]. Three-year survival rates for locally advanced HPV-negative SCCHN compared to HPV-positive cases are significantly reduced (57.1% versus 82.4%; [6]). Given the poor prognosis for locally advanced HPV-negative SCCHN, the identification of prognostic markers that might inform the utilization of targeted therapies, optimize current treatment approaches, and match patients to appropriate clinical trials is necessary.
In HPV-positive disease, the virally encoded oncoproteins E6 and E7 accelerate degradation of the TP53 and RB1 tumor suppressors, respectively [7], and are essential for tumorigenesis. In HPV-negative SCCHN, the impact of RB1 levels and phosphorylation status has been less studied. RB1 is highly regulated and critical for cell cycle progression and tumor suppression [8-12]. Primary regulation of RB1 is accomplished via phosphorylation by cyclin-dependent kinases (CDKs), specifically CDK2, CDK4 and CDK6, which in the case of CDK4 and CDK6, complex with cyclin D (CDK2 complexes with cyclins E and A) [8]. Phosphorylation of RB1 is influenced by p16, encoded by CDKN2A, which disrupts CDK4 binding to cyclin D (CCND1) [13, 14]. CDKN2A is often lost in HPV-negative SCCHN, based on LOH of the locus 9p21-22, or by epigenetic silencing [15-18]. CDK4/CDK6-cyclin D dependent phosphorylation of RB1 on tyrosine residue 356 (pT356) [19], a residue located in the interdomain linker separating the RB1 N-terminal domain (RBN) region and the pocket domain, was recently defined as disrupting the interaction of RB1 with E2F1 to induce cell cycle progression, and indicates functional inactivation of the protein (CDK4/CDK6-cyclin D definitively phosphorylates RB1 at T356, while it is less clear if CDK2 also phosphorylates RB1 at T356 [19-23]). This finding suggested that measurement of phospho-T356RB1 might strongly predict RB1 activity and given the crucial role of RB1, pT356RB1 levels may be informative in terms of patient outcome for cases of HPV-negative SCCHN.
From a therapeutic standpoint, recent FDA approval of the CDK4/6 inhibitor palbociclib for the treatment of ER+/HER- breast cancer [24] emphasizes current interest in the CDK4/CDK6-cyclin D to RB1 signaling axis [25]. In addition to breast cancer, melanoma, non-small cell lung cancers, lymphomas, ovarian cancers, and liposarcomas are currently being treated with CDK4/6 inhibitors in clinical trials [25-30], with two clinical trials in progress for SCCHN [NCT00824343; NCT00899054] using the CDK inhibitor P276-00 (predominantly targets CDK4, CDK2 and CDK9; [31]). It is generally accepted that for CDK4/6 inhibitors to be active, functional RB1 is required [25, 27, 30]. In the context of CDK4/6 inhibition, RB1 remains de-phosphorylated and bound to E2F1 [22, 25, 28, 30]. p16 status has been proposed as a response predictive biomarker for CDK4/6 inhibitors, based on the suppressive role of p16 on CDK4/6 and the feedback loop that results in high levels of p16 when RB1 expression is suppressed [8, 30]. To date, in clinical or preclinical studies of melanoma [32], ovarian cancer [33], pancreatic cancer [34] and glioblastoma [35], low p16 expression has been associated with CDK4/6 inhibitor sensitivity, whereas in breast cancer, p16 levels were not predictive, leaving its overall value as a response biomarker unclear at present [24, 36]. Further, in the specific case of SCCHN, p16 may be non-optimal as a biomarker in HPV-negative disease, given that high levels of p16 often are used in screening for HPV-positive, virally dependent cancers [37-39].
In this study, we have evaluated phospho-T356RB1 as a possible prognostic indicator for HPV-negative SCCHN. We have also correlated expression of phospho-T356RB1 with activation of ERK1/2 (phospho-T202/Y204), which is typically associated with active cell cycle [40]; with the proliferative indicator Ki-67; and with other protein biomarkers. Finally, we examined the relationship between phospho-T356RB1 and known clinical outcome markers including T classification, N classification, and tumor grade. These data for the first time indicate a strong predictive function of pT356RB1 for prognosis in SCCHN, and a striking relation between pT356RB1, pT202/Y204ERK1/2, and Ki-67 expression.
Results
Patient characteristics and AQUA analysis
TMAs were constructed using tissue obtained from 94 HPV-negative patients who underwent surgery (Table 1). Sub-sites were predominantly oral cavity (44%), oral tongue (22%) and glottis (17%; Table 1). As is typical for HPV-negative SCCHN, high T-stage and high N-stage significantly predicted poor survival outcomes (P = 0.043 and P = 0.045 respectively), while tumor grade did not (P = 0.160; Supplementary Figure S1). T1 or T2 had a median survival of 124.0 months and N0 or N1 of 88.1 months. The median survivals for high T- and N-stage were 47.0 months and 24.5 months, respectively.
For each protein of interest (pT356RB1, RB1, TP53, pT202/Y204ERK1/2, and Ki-67), antibodies for AQUA-based assays were validated as specific for the target protein (Figure 1A). In staining of primary tumor tissue, indicated by co-staining with cytokeratin to visualize epithelial tissue [41, 42], comparison of low versus high staining tissue for each marker indicated a robust dynamic range (Figure 1B). As specimens were collected over an extended period, we also performed secondary analysis to exclude the possibility that degradation of antibody epitopes over time influenced signal intensity and survival outcomes (Supplementary Figure S2 and Table S1), as has previously been reported [43, 44]. This indicated a low correlation index between age and signal intensity, indicating stability of antigens including phosphoantigens. Based on assessment by a pathologist, to ensure tissue quality and proper staining for each sample, the number of informative samples for each individual marker ranged from 55 to 94 cases (Table 1). We analyzed patients in two different categories: all patients (n = 94; includes patients who underwent surgery alone and patients who received surgery plus radiation; referred to as S/SRT), and only patients treated with surgery plus radiation (SRT; n = 69; Table 1).
Table 1: Patient characteristics for the analyzed TMA.
All (S/SRT) |
Surgery + Radiation (SRT) |
|||
Gender |
||||
Female |
35 |
37% |
24 |
37% |
Male |
59 |
63% |
45 |
65% |
Age at diagnosis |
||||
Mean |
62.8 |
61.8 |
||
Min |
25 |
25 |
||
Max |
86 |
86 |
||
SD |
12.9 |
13.3 |
||
Primary site |
||||
Glottis |
16 |
17% |
14 |
20% |
Hypopharynx |
3 |
3% |
2 |
3% |
Oral cavity |
41 |
44% |
31 |
45% |
Oral tongue |
21 |
22% |
13 |
19% |
Oropharynx |
8 |
9% |
5 |
7% |
other |
5 |
5% |
4 |
6% |
Overall stage |
||||
1 |
14 |
15% |
3 |
4% |
2 |
8 |
9% |
1 |
1% |
3 |
14 |
15% |
10 |
14% |
4 |
58 |
62% |
55 |
80% |
T stage |
||||
1 |
19 |
20% |
7 |
10% |
2 |
24 |
26% |
16 |
23% |
3 |
16 |
17% |
14 |
20% |
4 |
35 |
37% |
32 |
46% |
N stage |
||||
0 |
44 |
47% |
24 |
35% |
1 |
12 |
13% |
9 |
13% |
2 |
38 |
40% |
36 |
52% |
M stage |
||||
0 |
93 |
99% |
68 |
99% |
X |
1 |
1% |
1 |
1% |
Grade |
||||
Well diff. |
9 |
10% |
5 |
7% |
Moderately diff. |
55 |
59% |
40 |
58% |
Poor/Undiff. |
30 |
32% |
24 |
35% |
Positive staining |
||||
TB53 |
94 |
69 |
||
pT202RB1 |
79 |
61 |
||
RB1 |
55 |
38 |
||
Ki67 |
89 |
66 |
||
pT202/Y204ERK1/2 |
85 |
64 |
||
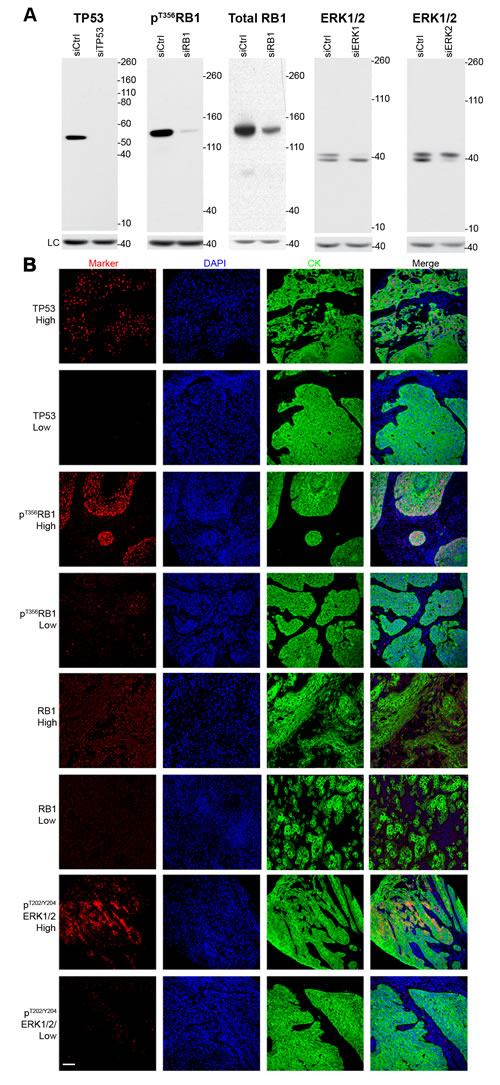
Figure 1: Validation of antibodies and immunofluorescence microscopy. A. Western blots for the relevant protein markers in the presence or absence of siRNA, B. representative high and low staining immunofluorescent microscopy images for each marker. LC = loading control (β-actin), DAPI = nuclear stain, CK = cytokeratin (epithelial tumor stain).
High pT356RB1 strongly predicts reduced overall survival
We first considered individual markers as predictors of overall survival (OS; Figure 2 and Supplementary Figure S3). High pT356RB1 signal, indicating inactivated RB1 protein [22], strongly predicted reduced OS in both treatment populations, with the effect most apparent in the SRT population (SRT, 27.0 versus 198.0 months, P = 0.0078; and S/SRT, 56.1 versus 198.2 months, P = 0.0295). Total RB1 levels did not correlate with survival probability (P = 0.1110). As a benchmark, higher proliferation-associated Ki-67 staining strongly predicted extended OS in the S/SRT populations (75.1 versus 19.5 months, P = 0.0082), but was not significant in the smaller SRT population (43.0 versus 19.5 months, P = 0.07787), although the general trend was the same. Similarly, higher pT202/Y204ERK1/2 staining was near significant for improved survival in the S/SRT population (P = 0.0558) but not in the SRT population (P = 0.1477).
For contrast, we also analyzed TP53. In SCCHN, disruptive mutations in TP53 are associated with a hazard for death of 1.7 in resectable disease [45, 46]. TP53 mutations in SCCHN include frameshifts, nonsense mutations and deletions (343 out of 2022 tumors (17%); Catalogue Of Somatic Mutations In Cancer (COSMIC) [47]), and also commonly include missense mutations (870 out of 2022 tumors (43%); COSMIC [47]) that are sometimes of unknown significance, and may result in partially functional proteins [48]. Thus, retained expression of TP53 may be of functional significance, or correlate with inactivated RB1. In the TMAs examined, we observed a non-statistically significant trend towards extended OS for higher levels of TP53 (S/SRT, 130.1 versus 54.6 months, P = 0.0916, and SRT, 130.1 versus 31.5 months, P = 0.1011; Figure 2 & Supplementary Figure S3).
We additionally performed multivariate analyses using Cox proportional hazards regression to adjust for T-stage, N-stage, grade, gender, patient age, and age of specimen (Figure 2 and Supplementary Table S1). After correction, the hazard ratio (HR) for high pT356RB1 was 3.00 (adjusted P = 0.017; Figure 2). The second marker with a significant adjusted P-value was Ki67 (P = 0.030): the corresponding HR was 0.52. RB1, TP53 and pT202/Y204ERK1 did not have significant adjusted P-values (P = 0.384; P = 0.051 and P = 0.052, respectively).
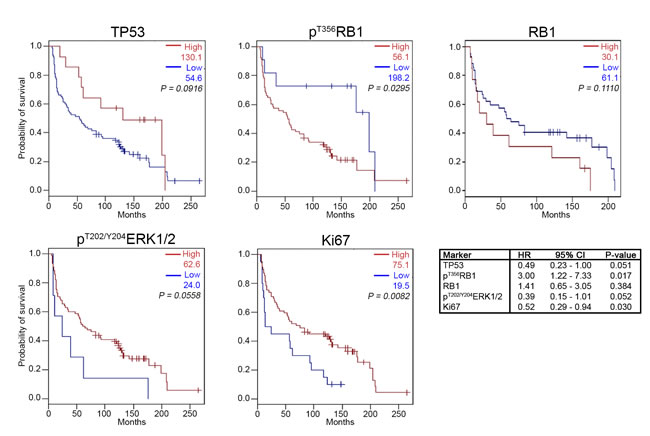
Figure 2: Kaplan-Meier survival analysis for high and low expression levels of TP53, RB1, pT356RB1, pT202/Y204ERK1/2, and Ki67. Patients treated with surgery only and patients treated with surgery and radiation therapy were included. OS = overall survival; HR = hazard ratio; CI = confidence interval. See Supplementary Table S1 for additional details regarding the HR.
High pT356RB1 correlates with elevated pT202/Y204ERK1/2 and Ki67
We next considered the possibility that significant correlations might be observed among the expression of the five assessed biomarkers, reflecting involvement in common cellular processes. In order to specifically analyze the relationship between pT356RB1 and total RB1, only samples with available RB1 staining (n = 55) were considered. The most striking correlations seen were noted between high expression of pT356RB1 and pT202/Y204ERK1/2 (ρ = 0.64; P < 0.0001) and between pT356RB1 and Ki67 (ρ = 0.42; P = 0.002). Additionally, high Ki67 expression correlated with high pT202/Y204ERK1/2, although to a lesser degree (ρ = 0.29; P = 0.039). No significant correlation between total RB1 and pT356RB1 was detected, and TP53 expression did not correlate with any of the other biomarkers examined (Figure 3A). In correlating markers to pathological properties of tumors (Figures 3B and 3C), high expression of pT202/Y204ERK1/2 (P = 0.047) and pT356RB1 (P = 0.010) in each case correlated with low T-stage (T1/2; Figure 3B). High pT356RB1 levels also strongly correlated with poorly differentiated or undifferentiated disease (P = 0.005; Figure 3B and 3C).
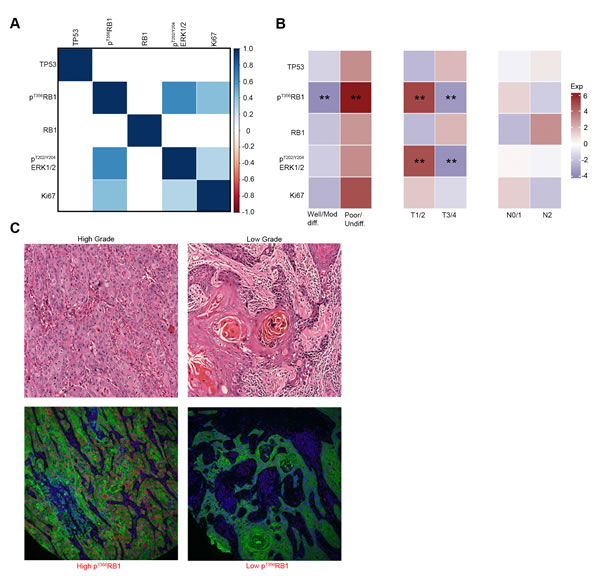
Figure 3: Expression correlation between markers and between markers and tumor stage and grade. A. Statistically significant correlations between marker expression levels, increasing saturation of blue indicates higher correlation, correlations with P > 0.05 are suppressed, B. correlations between marker expression levels and tumor grade, T-stage and N-stage, blue squares indicate low marker expression levels and red squares indicate high maker expression levels, C. hematoxylin and eosin (H&E) stained samples for high and low grade tumors with the corresponding pT356RB1 staining. (**) P < 0.05.
Genomic and transcriptomic analysis supports relevance of high pT356RB1 as an indicator of poor outcome
If high pT356RB1 predicts poor outcome, then increased activity of CDK4/6 should also predict poor survival, and may be detectable by genomic biomarkers. Increased CDK4/6 activity can be caused by loss of expression or function of the kinase inhibitor CDKN2A (p16) or elevated mRNA expression and/or activating mutation of CDK4, CDK6, and CCND1 (cyclin D1). Inversely, reduced activity of CDK4/6 might be linked to overexpression of CDKN2A. To extend our analysis, we examined the expression and mutational status of RB1 and functionally interacting proteins, including CDKN2A (p16), CDK4, CDK6 and CCND1 in a TCGA dataset of 243 cases of HPV-negative SCCHN. The dominant detected genetic alteration for CCND1 and CDK6 was gene amplification, with most amplifications affecting CCND1 (76/243); while most alterations affecting CDKN2A resulted in shallow deletions reflecting loss of heterozygosity (LOH), deep deletions associated with homozygous deletions, and truncating mutations (192/243; Figure 4A). Protein-damaging RB1 mutations or deletions were detected in only 7 out of 243 specimens, without any discernible effect on survival (Supplementary Figure S4A). A high number of shallow deletions were also detected for RB1, again without indication of any impact on survival (Supplementary Figure S4A). We next investigated whether loss of CDKN2A or RB1 is associated with significant survival implications and found that patients with mutations or LOH of the CDKN2A locus (78/243 tumors) had a strong tendency towards a reduction in survival (P = 0.072; Figure 4B). Amplification of CCND1 was independently not predictive of response (P = 0.75; Supplementary Figure S4B); however, further analysis indicated significant co-occurrence between CCND1 amplification and homozygous deletion of CDKN2A (co-occurrence ratio: 0.817; P = 0.041), with significant impact on survival (Figure 4C).
We also analyzed changes in mRNA expression. The most commonly overexpressed genes in the group were CDKN2A (44/243 with a 2-fold and 28/243 cases with a >3-fold expression increase) and CCND1 (29/243 with a 2-fold expression increase), with lower levels of overexpression seen for RB1, CDK4 and CDK6 (Supplementary Figure S4C and S4D). Overexpression (z > 3-fold) of CDKN2A was robustly associated with increased survival (Figure 4D). In contrast, overexpression of CCND1 (z > 2-fold) correlated with reduced survival (P = 0.013; Figure 4E). Finally, CDK6 overexpression did not significantly change survival (Supplementary Figure S4E) and significant overexpression of CDK4 and RB1 was not present in sufficient cases for meaningful analysis (Supplementary Figure S4C and S4D).
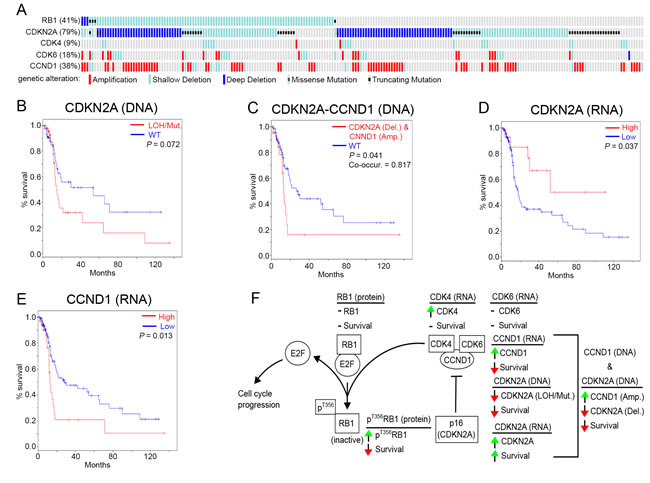
Figure 4: Genomic and transcriptomic analysis of RB1, CDK4, CDK6, CDKN2A (p16) and CCND1. A. Genomic alterations of RB1, CDKN2A (p16), CDK4, CDK6 in 243 HPV negative SSCHN TCGA specimens (only tumors with changes affecting at least one of these genes are shown), B. Kaplan-Meier survival analysis of CDKN2A with deletions (Del.) compared to WT, C. Kaplan-Meier survival analysis for cases of simultaneous CCND1 amplification (Amp.) and CDKN2A homozygous deletion (Del.), D. Kaplan-Meier survival analysis of high versus low RNA expression for CDKN2A (p16) and E. CCND1. F. Summary of RB1-pathway alterations and the survival impact thereof. See Supplementary Figure S4 for additional data. Shallow Deletion = LOH = loss of heterozygosity; Deep Deletion = homozygous deletion; Mut. = mutation; WT = wild type; green arrow = increase; red arrow = decrease; horizontal line = no change; co-occur. = trend towards co-occurrence.
Discussion
While genomic profiling of tumors yields insight into tumor subclasses with distinct prognoses [49], identification of protein expression patterns is useful in cases where significant biological effects are associated with post-translational modification of functionally important proteins [41, 50]. RB1 activity has been much investigated in HPV-positive SCCHN and cervical cancer, but less so in HPV-negative disease. Our research for the first time highlights the potential clinical value specifically of pT356RB1 as a biomarker in HPV-negative SCCHN. Patients harboring tumors with low levels of pT356RB1 had a median survival nearly four times the median survival for patients with high pT356RB1 expression (198.2 versus 56.1 months; P = 0.0295) when treated with surgery or surgery plus radiation (Figure 2). The difference was even more pronounced when only patients who received surgery plus radiation, a population with more adverse clinical risk factors, were considered (198.0 months versus 27.0 months; P = 0.0078; Supplementary Figure S2). The observed differences were mediated at the post-translational level, as expression levels of total RB1 did not correlate with survival (Figure 2 and 3). These findings can potentially be explained by the observation that alterations in the RB1-pathway are associated with increased sensitivity to ionizing radiation [51] and furthermore suggest that phosphorylation status of RB1 is critical. We further found that low levels of pT356RB1 correlated with low-grade disease (Figure 3B and 3C).
As RB1 is infrequently mutated in HPV-negative SCCHN (Figure 4A; [49]), these data suggest that phosphorylation of T356 may serve an important role in functionally inactivating tumor suppression in aggressive disease. The discrepancy between total RB1 and pT356RB1 in terms of survival probability highlights the importance of interrogating post-translational events that control protein activity. TCGA databases currently do not include information on T356RB1 and, although proteomics-based analyses are in progress, no systematically validated data is currently available, underscoring the value of complementary analysis of protein expression on TMAs. It is possible that on a biological level, a low level of pT356RB1 is very different from a complete loss of RB1, based on interactions of RB1 with partners other than E2F1. While multiple potential factors can result in low levels of T356 phosphorylation, all, as far as is known, are linked to CDK4/6-CCND1, which are necessary to phosphorylate this residue of RB1 and are negatively regulated by CDKN2A [19, 22, 23, 51-53]. In this study, TCGA data analysis of RB1, CCND1, CDK4, CDK6, and CDKN2A corroborated the potential value of considering inhibition of T356RB1 phosphorylation status. Most significantly, overexpression of CCND1 (cyclin D) mRNA, or loss of CDKN2A expression or function, particularly in conjunction with CCND1 overexpression, significantly correlated with worse outcome (Figure 4B-4E). Of note, the CCND1 and CDKN2A loci show some of the highest frequencies of amplification and deletion rates seen in SCCHN and several other cancer types [30]; this further supports the idea that it may be important to restrain RB1 function post-translationally. Larger cohort studies are required to investigate and confirm these ideas.
As summarized in Figure 4F, our data are compatible with a model in which control of RB1 activity is regulated by inhibitory CDK4/6 phosphorylation that varies in tumors based on regulation at the level of CCND1, CDKN2A, or both. Our findings also align with the recognition that oncogenic signaling changes can target multiple points in tumor pathways rather than single critical genes, such as activation of the RAS pathway by mutation or overexpression of upstream receptor tyrosine kinases (RTKs), RAS, RAF, or PI3K or loss of PTEN [54-57]. Similarly, acquisition of resistance to DNA damaging cytotoxic drugs can occur by mutation or expression changes of any one of a group of DNA damage response pathway genes [58, 59]. Consideration of these factors in sum may have considerable value in improving prognostic and diagnostic accuracy. While RNA and DNA were not available for the specimens analyzed by TMA in this study, our results suggest the value of a prospective study integrating measurement of pT356RB1, CDKN2A loss, and CCND1 amplification or overexpression
Our study has several limitations: particularly the retrospective nature, lack of cause-specific survival data, and the small sample size available for this analysis call for larger prospective studies in the future to validate our findings. It is also important to consider the inherent limitations of TCGA data. TCGA data is based on samples collected and processed at different institutions; molecular characterization is performed at different centers; and varying degrees of clinical annotation for samples has been noted [60]. Furthermore, differences in acquisition platforms and data processing pipelines may be confounding factors. In spite of these limitations, our work presents substantial evidence that pT356RB1 could be exploited to evaluated activity of the CDK4/6-cyclin D axis and thus may serve as a valuable predictive biomarker.
As noted in the introduction, recent data also link functional RB1 to responsiveness to pharmacological CDK4/6 inhibition, adding interest to our findings and raising the possibility that pT356RB1 may have utility in stratifying SCCHN patients for clinical trials of CDK4/6 inhibition. This is of particular interest as high levels of CDKN2A (p16) protein, as seen in HPV-positive SCCHN, are associated with limited response to CDK4/6 inhibition in several tumor types [25, 30, 32, 34, 35, 61]. Maximal possible benefit of these inhibitors to patients will greatly depend on reliable biomarkers to guide patient selection. In sum, our findings suggest that pT356RB1 status can function as an important survival predictor in HPV-negative SCCHN and should be tested as a potential marker for selection of patients for clinical trials.
Patients and Methods
Patients
Tumor samples were obtained from archival formalin-fixed paraffin-embedded FCCC pathology specimens, collected at the time of initial surgery between 1990 and 2007. Institutional Review Board-approved consent forms were signed prior to sample collection. Five TMAs were constructed with tumor cores represented in duplicate and a selection of normal controls. Clinical data were extracted from FCCC clinical databases in an anonymized fashion. A total of 94 HPV-negative (all oropharyngeal primary tumors with either positive or unknown status of p16 were excluded) surgical SCCHN specimens were analyzed (Table 1).
Fluorescence immunohistochemistry, image acquisition and AQUA analysis
Immunohistochemistry was performed as previously described [41]. Tissue sections were blocked with Background Sniper (BS966, Biocare Medical). Antigen Retrieval was performed in Tris/EDTA pH 9 Buffer for 20 minutes (S2367, Dako). The sections were incubated overnight with the appropriate primary antibody: pT356RB1 (1:200, 2223-1, Epitomics), RB1 (1:200, #9309, Cell Signaling Technology), pT202/Y204ERK1/2 (1:100, #9101S, Cell Signaling Technology), N-terminal TP53 (1:200, M7001, DAKO), or Ki-67 (1:800, AC-0009, Epitomics), and pan-cytokeratin (Rabbit 1:400, Z0622, Dako or Mouse, 1:100, M3515, Dako; tumor mask) in Da Vinci Green antibody diluent (PD900, Biocare Medical) at 4°C overnight. Signals were intensified with Envision reagents (DAKO). Pan-cytokeratin primary antibody was probed with an Alexa Fluor 555 dye-labeled secondary antibody (Invitrogen). Primary antibody visualization was accomplished using a Cy-5-tyramide signal amplification system (TSA; AT705A, PerkinElmer). Tissue nuclei were stained using Prolong Gold mounting medium (P36931; Molecular Probes) containing 4,6-diamidino-2-phenylindole (DAPI). HistoRx PM-2000 (HistoRx) with AQUAsition software was used for automated image capture as previously described [41].
Statistical analysis
Patients eligible for analysis had a valid read (defined as detectable staining intensity within the dynamic range of the AQUA acquisition software, in the absence of staining artifacts) for at least one of the assessed proteins (Table 1). Associations between each marker’s expression level, and grade, stage and survival, were assessed after choosing the optimal cutpoint using Classification and Regression Trees (CART; Supplementary Table S2; [62]), fit was determined using the rpart procedure in R software (version 3.0.2). Survival curves were generated using the methods of Kaplan and Meier [63], and tested for significance using Log-Rank tests. We further assessed the relationship between overall survival and marker expression levels by performing multivariate analysis using Cox proportional hazards regression [64], adjusting for T-stage, N-stage, grade, gender, the patient’s age, and the specimen’s age. The relationships between markers and stage/grade were analyzed using Spearman’s correlation [65]. Correlations were presented graphically using the corrplot procedure in R.
Cell culture, siRNA and western blot
FaDu and SCC61 cells from the ATCC were cultured as recommended by the suppliers. Transfection of cells with siRNA was accomplished using DharmaFECT1 (GE Healthcare) at a dilution ratio of 1:100 with serum free media. Depletion of proteins was accomplished using siRNA SMARTpools (four combined siRNAs per target) from GE Healthcare/Dharmacon: RB1 (NM_000321; cat.# M-003296-03), TP53 (NM_000546; cat.# M-003329-03), ERK2 (NM_138957; cat.# M-003555-04), and ERK1 (NM_001109891; cat.# M-003592-03). Scramble siRNA control was purchased from GE Healthcare/Dharmacon. Cells were plated in six well plates with the siRNA transfection mixture. After 48 hours, cells were lysed using M-PER Mammalian Protein Extraction Reagent (Thermo Scientific; #78501) supplemented with protease/phosphatase inhibitor cocktail (Thermo Scientific; #1861282). Western blotting was performed using standard procedures and was developed using SuperSignal West Pico Stable Peroxidase and Luminol/Enhancer solutions (Thermo Scientific; #1856135 & #1856136). Primary antibodies used were the same as described above, plus anti-β-actin conjugated to horseradish peroxidase (HRP; ab49900) from Abcam. All primary antibodies were used at a dilution of 1:1000; except anti-β-actin, which was used at 1:50,000. Secondary anti-rabbit and anti-mouse HRP-conjugated antibodies from GE Healthcare were used at dilutions of 1:10,000.
TCGA data analysis
243 HPV-negative SCCHN specimens from the TCGA set [Head and Neck Squamous Cell Carcinoma (TCGA, in revision); [66]] were analyzed using cBioPortal (http://www.cbioportal.org; [67, 68]). Datasets reporting mRNA expression (RNA Seq V2 RSEM) and mRNA expression z-scores (RNA Seq V2 RSEM), mutations, putative copy-number alterations from GISTIC, as well as protein/phosphoprotein levels (RPPA) were retrieved. For mRNA, fold expression over the average was calculated and the corresponding z-scores were used as input for cBioPortal analysis. Kaplan-Meier survival curves and maps indicating DNA/RNA status were generated using cBioPortal.
Acknowledgments
The authors would like to thank all the patients who participated in this study. We would also like to thank the facilities and clinical staff at Fox Chase Cancer Center.
conflicts of interest
The authors have declared no conflict of interest.
Funding
The authors were supported by U54 CA149147 and R21CA181287 from the NIH (to EAG); F30 CA180607 from the NIH (to TNB); NIH core grant CA06927 (to Fox Chase Cancer Center); subsidy from the Russian Government to support the program of competitive growth of Kazan Federal University (to IGS); and support from the Fox Chase Cancer Center Head and Neck Keystone.
References
1. Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest 2012; 122: 1951-1957.
2. Burtness B, Bauman JE, Galloway T. Novel targets in HPV-negative head and neck cancer: overcoming resistance to EGFR inhibition. Lancet Oncol 2013; 14: e302-309.
3. Liu H, Cracchiolo JR, Beck TN et al. EGFR Inhibitors as Therapeutic Agents in Head and Neck Cancer. In Burtness B, Golemis EA (eds): Molecular Determinants of Head and Neck Cancer. New York: Springer 2014.
4. Seiwert TY, Beck TN, Salgia R. The Role of HGF/c-MET in Head and Neck Squamous Cell Carcinoma. In Burtness B, Golemis EA (eds): Molecular Determinants of Head and Neck Cancer. New York: Springer 2014.
5. Marur S, Burtness B. Oropharyngeal squamous cell carcinoma treatment: current standards and future directions. Curr Opin Oncol 2014; 26: 252-258.
6. Ang KK, Harris J, Wheeler R et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 2010; 363: 24-35.
7. Munger K, Baldwin A, Edwards KM et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol 2004; 78: 11451-11460.
8. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 2013; 14: 297-306.
9. Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 1989; 58: 1097-1105.
10. Chen PL, Scully P, Shew JY et al. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 1989; 58: 1193-1198.
11. DeCaprio JA, Ludlow JW, Lynch D et al. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 1989; 58: 1085-1095.
12. Mihara K, Cao XR, Yen A et al. Cell cycle-dependent regulation of phosphorylation of the human retinoblastoma gene product. Science 1989; 246: 1300-1303.
13. Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8: 714-724.
14. Lukas J, Parry D, Aagaard L et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 1995; 375: 503-506.
15. Ai L, Stephenson KK, Ling W et al. The p16 (CDKN2a/INK4a) tumor-suppressor gene in head and neck squamous cell carcinoma: a promoter methylation and protein expression study in 100 cases. Mod Pathol 2003; 16: 944-950.
16. Gonzalez MV, Pello MF, Lopez-Larrea C et al. Loss of heterozygosity and mutation analysis of the p16 (9p21) and p53 (17p13) genes in squamous cell carcinoma of the head and neck. Clin Cancer Res 1995; 1: 1043-1049.
17. O’Regan EM, Toner ME, Finn SP et al. p16(INK4A) genetic and epigenetic profiles differ in relation to age and site in head and neck squamous cell carcinomas. Human Pathology 2008; 39: 452-458.
18. Mountzios G, Psyrri A. Molecular Features and Treatment Modalities of “Classic” Versus Human Papilloma Virus-Associated Head and Neck Cancer. In Burtness B, Golemis EA (eds): Molecular Determinants of Head and Neck Cancer. New York: Springer New York 2014; 7-21.
19. Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem 1997; 272: 12738-12746.
20. Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell 1997; 8: 287-301.
21. Adams PD. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochimica Et Biophysica Acta-Reviews on Cancer 2001; 1471: M123-M133.
22. Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev 2012; 26: 1156-1166.
23. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323-330.
24. Finn RS, Crown JP, Lang I et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015; 16: 25-35.
25. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14: 130-146.
26. Dickson MA, Tap WD, Keohan ML et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol 2013; 31: 2024-2028.
27. Flaherty KT, Lorusso PM, Demichele A et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res 2012; 18: 568-576.
28. Leonard JP, LaCasce AS, Smith MR et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012; 119: 4597-4607.
29. Schwartz GK, LoRusso PM, Dickson MA et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer 2011; 104: 1862-1868.
30. Choi YJ, Anders L. Signaling through cyclin D-dependent kinases. Oncogene 2014; 33: 1890-1903.
31. Mishra PB, Lobo AS, Joshi KS et al. Molecular mechanisms of anti-tumor properties of P276-00 in head and neck squamous cell carcinoma. J Transl Med 2013; 11: 42.
32. Young RJ, Waldeck K, Martin C et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014; 27:590-600.
33. Konecny GE, Winterhoff B, Kolarova T et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 2011; 17: 1591-1602.
34. Heilmann AM, Perera RM, Ecker V et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res 2014; 74: 3947-3958.
35. Cen L, Carlson BL, Schroeder MA et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol 2012; 14: 870-881.
36. DeMichele A, Clark AS, Tan KS et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res 2015; 21: 995-1001.
37. Keller LM, Galloway TJ, Holdbrook T et al. p16 status, pathologic and clinical characteristics, biomolecular signature, and long-term outcomes in head and neck squamous cell carcinomas of unknown primary. Head Neck 2014; 36: 1677-1684.
38. Pfister DG, Spencer S, Brizel DM et al. Head and neck cancers, Version 2.2014. Clinical practice guidelines in oncology. J Natl Compr Canc Netw 2014; 12: 1454-1487.
39. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 2010; 10: 550-560.
40. Roskoski R, Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 2012; 66: 105-143.
41. Mehra R, Zhu F, Yang DH et al. Quantification of excision repair cross-complementing group 1 and survival in p16-negative squamous cell head and neck cancers. Clin Cancer Res 2013; 19: 6633-6643.
42. Hass R, Bertram C. Characterization of human breast cancer epithelial cells (HBCEC) derived from long term cultured biopsies. J Exp Clin Cancer Res 2009; 28: 127.
43. Vassilakopoulou M, Parisi F, Siddiqui S et al. Preanalytical variables and phosphoepitope expression in FFPE tissue: quantitative epitope assessment after variable cold ischemic time. Lab Invest 2015; 95:334-41.
44. Siddiqui S, Rimm DL. Pre-analytic variables and phospho-specific antibodies: the Achilles heel of immunohistochemistry. Breast Cancer Res 2010; 12: 113.
45. Poeta ML, Manola J, Goldwasser MA et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007; 357: 2552-2561.
46. Masica DL, Li S, Douville C et al. Predicting survival in head and neck squamous cell carcinoma from TP53 mutation. Hum Genet 2015; 134:497-507.
47. Forbes SA, Beare D, Gunasekaran P et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43(Database issue):D805-11.
48. Peltonen JK, Helppi HM, Paakko P et al. p53 in head and neck cancer: functional consequences and environmental implications of TP53 mutations. Head Neck Oncol 2010; 2: 36.
49. Stransky N, Egloff AM, Tward AD et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011; 333: 1157-1160.
50. Psyrri A, Lee JW, Pectasides E et al. Prognostic biomarkers in phase II trial of cetuximab-containing induction and chemoradiation in resectable HNSCC: Eastern cooperative oncology group E2303. Clin Cancer Res 2014; 20: 3023-3032.
51. Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res 2010; 16: 1094-1099.
52. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. Journal of Clinical Oncology 2006; 24: 1770-1783.
53. Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol 1994; 14: 2077-2086.
54. Johnson GL, Stuhlmiller TJ, Angus SP et al. Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res 2014; 20: 2516-2522.
55. Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev 2012; 26: 641-650.
56. Vogelstein B, Papadopoulos N, Velculescu VE et al. Cancer genome landscapes. Science 2013; 339: 1546-1558.
57. Beck TN, Gabitova L, Serebriiskii IG. Targeted Therapy: Genomic Approaches. In Meyers RA (ed) Cancer Translational Medicine (Current Topics from the Encyclopedia of Molecular Cell Biology and Molecular Medicine), 1 Edition. Wiley-Blackwell 2015; 1100.
58. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012; 12: 104-120.
59. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13: 714-726.
60. Yuan Y, Van Allen EM, Omberg L et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nature Biotechnology 2014; 32: 644-+.
61. Dean JL, Thangavel C, McClendon AK et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010; 29: 4018-4032.
62. Breiman L. Classification and regression trees. Belmont, Calif.: Wadsworth International Group 1984.
63. Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association 1958; 53: 457-481.
64. Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society Series B-Statistical Methodology 1972; 34: 187-+.
65. Best DJ, Roberts DE. Algorithm AS 89: The Upper Tail Probabilities of Spearman’s Rho. Journal of the Royal Statistical Society 1975; 24: 377-379.
66. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015; 517: 576-582.
67. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1.
68. Cerami E, Gao J, Dogrusoz U et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data (vol 2, pg 401, 2012). Cancer Discovery 2012; 2: 960-960.