
INTRODUCTION
Non-small cell lung cancer (NSCLC) accounts for the majority of diagnosed lung cancers and is dominated by the adenocarcinoma, squamous cell carcinoma (SqCC) and large cell carcinoma with or without neuroendocrine features (LCNEC and LC, respectively) histological subtypes. In NSCLC, LC and LCNEC together account for 3–9% of all cases depending on cohort demographics and classification scheme, with a generally poor prognosis compared to other NSCLC subgroups [1, 2]. In the 2004 WHO classification of lung cancer LC is defined as an undifferentiated NSCLC lacking architectural and cytologic features of small-cell carcinoma, glandular or squamous differentiation, whereas LCNEC is defined as an LC with neuroendocrine morphological features and at least one positive neuroendocrine immunohistochemical (IHC) marker [3]. LCNEC tumors share many similarities with small-cell lung cancer (SCLC) on the morphological, IHC and molecular level [4] (and references therein). Based on advances in immunomarkers for classification of adenocarcinoma and SqCC there is today significant controversy on whether LC actually represent a truly distinct biological entity, or merely a group of very poorly differentiated tumors of other NSCLC groups (adenocarcinoma and/or SqCC) [5, 6]. In fact, in the most recent 2015 WHO classification of lung cancer LCs that are mucin-positive or expresses pneumocyte markers should now be classified as adenocarcinoma, and the squamous marker-positive cases as nonkeratinizing squamous cell carcinoma [7].
In comparison to other NSCLC subgroups, LC and LCNEC tumors remain fairly uncharacterized at the molecular level by modern genomic techniques. Recent studies have investigated copy number alterations (CNAs) in LC and LCNEC [8, 9], highlighted the transcriptional similarity between LCNEC and SCLC [8], and identified a neuroendocrine DNA methylation subgroup in lung cancer [10]. In contrast, studies of the genome-wide mutational landscape in LC and LCNEC using massive parallel sequencing methods (NGS) are scarce. A recent analysis of 15 LCNEC tumors using whole-exome sequencing associated mainly mutations in TP53, RB1, and EP300 with LCNEC (and SCLC) tumor histology, with additional mutations in LCNEC also found in adenocarcinomas and SqCCs [8]. Studies of smaller gene sets have identified abnormal TP53 expression in both LC and LCNEC tumors and KRAS mutations predominantly in LCs [6, 11]. Mutations in EGFR and ALK gene fusions represent current molecular treatment predictive alterations for targeted therapy in lung cancer [12], but rarely appear in LC or LCNEC tumors with only a few reported cases in the literature [5, 6, 13–16]. Clearly, better characterization of the mutational landscape in LC and LCNEC is needed to take advantage of the growing number of targeted treatments and our emerging understanding of treatment resistance factors in lung cancer.
In this study, we aimed to investigate the mutational landscape of LC and LCNEC tumors using a panel of 26 well-established oncogenes and tumor suppressor genes in combination with ALK, RET, and ROS1 gene fusion analysis and copy number analysis of targeted genes. To this end, we analyzed 41 LC and 17 LCNEC cases by massive parallel sequencing and combined our results with 15 whole-exome sequenced LCNEC cases [8] and previously reported copy number data [8, 10].
MATERIALS AND METHODS
Patient material
DNA and total RNA were extracted from 57 early stage lung cancer patients surgically treated at the Skåne University Hospital in Lund, Sweden (Table 1 and Supplementary Table S1). Patients in this retrospective study had not received any neoadjuvant treatment before surgery. One patient harbored a mixed cancer, with one LC and one LCNEC tumor component, treated as two individual tumors in the analysis. In total, 41 LC and 17 LCNEC samples were included from this patient cohort. For all cases, relevant pathological slides were re-evaluated and clinicopathological characteristics were updated to be in line with recent international criteria and guidelines [3, 17]. Thirteen cases have been described in previous studies [10, 18]. From Seidel et al. [8], we included whole-exome sequencing and copy number data on genes investigated in the experimental Lund cohort from 15 additional LCNEC cases (Table 1).
Table 1: Patient characteristics and clinicopathological data
Lund |
CLCGP [8] |
All cases |
|
Histology |
|||
LC (basaloid) |
41 (6) *, ** |
- |
41 (6) |
LCNEC |
17* |
15 |
32 |
LC immunomarker profile |
|||
Adenocarcinoma-like |
24 (59%) |
- |
24 (59%) |
Squamous cell carcinoma-like |
5 (12%) |
- |
5 (12%) |
Marker null |
12 (29%) |
- |
12 (29%) |
Tumor stage |
|||
I |
29 |
7 |
36 |
II |
19 |
6 |
25 |
III |
8 |
2 |
10 |
IV |
2 |
0 |
2 |
Smoking history |
|||
Never-smokers |
0 |
0 |
0 |
Smokers |
34 |
11 |
45 |
Not available |
24 |
4 |
28 |
Gender |
|||
Female |
31 |
6 |
37 |
Male |
27 |
9 |
36 |
Age (median & range) |
66 (47–82) |
67 (47–80) |
66 (47–82) |
Patients evaluable for |
|||
Mutations |
58 |
15 |
73 |
ALK, RET, ROS1 fusions |
46 |
1*** |
47 |
Copy number alterations |
46 |
10 |
56 |
*One patient had a mixed tumor with both an LC and LCNEC component.
**Basaloid (n = 6) and lymphoepithelioma-like (n = 1) cases are included in the LC sample numbers.
***Evaluated for ALK/RET/ROS1 fusions by FISH.
Ethics statement
The study was approved by the Regional Ethical Review Board in Lund, Sweden (Registration no. 2004/762, 2008/702, 2007/445, and 2014/748).
Immunohistochemistry
Cases with neuroendocrine morphological features were evaluated for IHC staining of the neuroendocrine markers chromogranin A, synaptophysin and CD56 (Supplementary Methods). At least 10% positive tumor cells were required for positive staining for these markers. In addition, LC cases were analyzed for IHC staining of CK5/P40 (squamous cell markers) and TTF-1/Napsin A (adenocarcinoma markers). Staining intensities for these markers were categorized as 0 (<1% positive tumor cells), 1 (1–10%), 2 (11–25%), 3 (26–50%), and 4 (>50% positive tumor cells). Similar to the recent 2015 WHO update on lung cancer [7], we classified a categorized intensity of ≥1 as positive for TTF-1 or Napsin A, and ≥ 2 as positive for CK5 or P40. A LC sample was classified as adenocarcinoma-like if a positive TTF-1 and/or Napsin A staining was observed. A LC case was classified as squamous-like if a positive CK5 and/or P40 staining was observed. IHC analyses are further described in Brunnström et al. [19] and Supplementary Methods.
Mutational analysis
All Lund cases were analyzed by the NGS-based Illumina TruSight Tumor gene panel on a MiSeq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, US). Analyzed regions included a selected set of complete exons in 26 genes: AKT1 (exon 2), ALK (exon 23), APC (exon 15), BRAF (exons 11, 15), CDH1 (exons 8, 9, 12), CTNNB1 (exon 2), EGFR (exons 18, 19, 20, 21), ERBB2 (exon 20), FBXW7 (exons 7, 8, 9, 10, 11), FGFR2 (exon 6), FOXL2 (exon 1), GNAQ (exons 4, 5, 6), GNAS (exons 6, 8), KIT (exons 9, 11, 13, 17, 18), KRAS (exons 1, 2, 3, 4), MAP2K1 (exon 2), MET (exons 1, 4, 13, 15, 16, 17, 18, 20), MSH6 (exons 5), NRAS (exons 1, 2, 3, 4), PDGFRA (exons 11, 13, 17), PIK3CA (exons 1, 2, 7, 9, 20), PTEN (exons 1, 2, 3, 4, 5, 6, 7, 9), SMAD4 (exons 8, 11), SRC (exon 10), STK11 (exons 1, 4, 6, 8), and TP53 (exons 2, 3, 4, 5, 6, 7, 8, 9, 10, 11). DNA extraction was performed using the Qiagen GeneRead (Qiagen, Hilden, Germany) kit for formalin-fixed paraffin embedded tissue (FFPE), or by the Qiagen AllPrep kit for fresh frozen tissue (Supplementary Methods). Macrodissection of FFPE cases were performed when possible prior to DNA extraction. Alignment, quality filtering, variant calling, and variant annotation were performed using the standard MiSeq Reporter and VariantStudio analysis pipeline (Illumina). Only nonsynonymous variants with a quality score equal to 100 that passed the bi-directional sequencing quality filter in TruSight Tumor were considered. Read depths (X) for genes with detected variants varied between 3024–143140X (median 18624X, interquartile range 28450X).
Gene fusion analysis
Analysis of ALK, RET, and ROS1 gene fusions were performed using the RNA-based Archer FusionPlex ARR v2 kit (ArcherDX, Boulder, CO, US) and the MiSeq instrument (Illumina) (Supplementary Methods). The HCC78 (ROS1-SLC34A2), KARPAS-299 (ALK-NPM1), LC-2/ad (CCDC6-RET), and H2228 (EML4-ALK) cell lines were used as controls (Supplementary Methods). RNA from FFPE tissues (n = 11 samples) were extracted using the Qiagen Allprep FFPE extraction kit (Qiagen), while RNA from cell lines or fresh frozen tissue were extracted using the non-FFPE Qiagen Allprep extraction kit. Data analysis was performed using software tools provided with the Archer kit (ArcherDX). Confirmatory ALK immunohistochemistry was performed using the D5F3 antibody (Ventana Medical Systems), and confirmatory ALK FISH analysis using the Vysis ALK break apart FISH probe (Vysis) according to manufacturers’ instructions.
Copy number analysis
Calls of copy number gain, loss, amplification and focused copy number loss for genes included in the TruSight Tumor panel were made for 46 tumors in the Lund cohort based on data from ongoing or published studies on the same tumor cohort [10], and for ten cases from Seidel et al. [8] as described by [10, 20] and Supplementary Methods.
NGS validation analyses
Ten mutations in KRAS detected by the TruSight Tumor panel were selected for validation by the Therascreen® KRAS RGQ PCR Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. In addition, seven unrelated tumor FFPE specimens, including two melanomas, two lung adenocarcinomas, and three colon cancers were also used to validate the NGS platform. These samples had verified mutations in BRAF (the two melanomas and one colon cancer: V600E), KRAS (two colon cancers: G13D and G12S), and EGFR (the two lung adenocarcinomas: L858R and E746_A750del). Mutations in these cases were obtained from routine clinical diagnostics based on pyrosequencing or Q-PCR performed at the Skåne University Hospital in Lund, Sweden.
RESULTS
Tumor and patient characteristics
A cohort of 41 LC and 17 LCNEC cases (Lund cohort) were pooled with 15 reported LCNEC cases [8], thus rendering a total of 73 cases (Table 1). All patients with available chart data were (current or former) smokers. There were no statistical differences in the distribution of tumor stage, gender, or age of diagnosis between LC and LCNEC cases (p > 0.05, Fisher’s exact test or Wilcoxon’s test). Among the LC cases, six tumors were histopathologically subclassified as basaloid, and one as lymphoepithelioma-like. Protein expression of adenocarcinoma markers TTF-1 and Napsin A and squamous markers CK5 and P40 were investigated in the Lund cohort and the public LCNEC cohort (TTF-1 only). 77% of all analyzed LCNEC cases showed positive TTF-1 expression. 59% of LC cases in the Lund cohort were IHC positive for TTF-1, while 44% were IHC positive for Napsin A. 75% of the TTF-1 positive LC cases also showed positive Napsin A expression, while no case was Napsin A positive but TTF-1 negative. For the squamous markers CK5 and P40, 5% and 10% of LC cases showed positive staining, respectively.
The mutational spectrum of LC and LCNEC
The 73 LC and LCNEC cases were analyzed for mutations in 26 cancer-related genes through NGS-based analysis of fresh frozen or FFPE tumor tissues. In total, 117 nonsynonymous variants, with alternate variant frequencies (the fraction of all reads with the detected variant) between 3–91% (Lund cohort only), were identified in 13 genes, for which gene copy number status were also extracted (Figure 1 and Supplementary Figure S1 and Supplementary Tables S2–S3 listing explicit variant data). Median number of variants per sample was one and maximum was three. 72 out of 73 cases, including all Lund cases, showed variants in at least one gene.
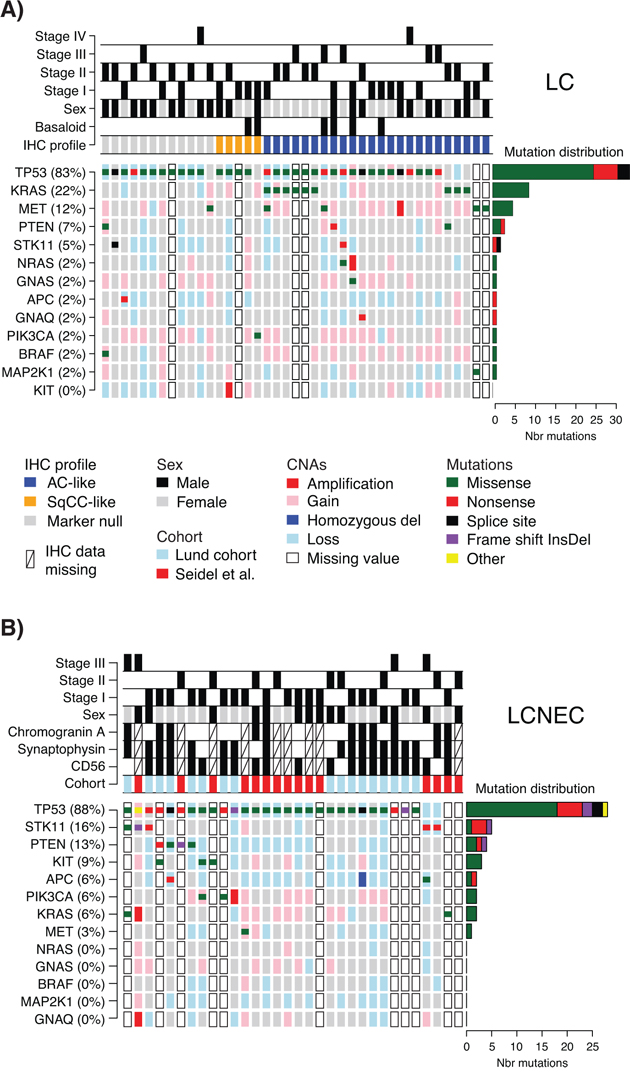
Figure 1: Detected mutations and copy number alterations in LC and LCNEC. A. Detected gene variants and copy number alterations (CNAs) (rows) in 41 LC cases (columns), ordered by immunomarker profile of adenocarcinoma-like (AC-like), squamous cell carcinoma like (SqCC-like), or marker null phenotype (TTF-1/Napsin A and CK5/P40 negative). Copy number status is shown as larger background rectangles and mutations as squares for each sample and gene. Right side bar plot summarizes the distribution of the different mutation types for each gene. B. Detected variants and copy number alterations in 32 LCNEC cases displayed as in A. Samples are ordered according to gene variant frequency.
TP53 mutations were the most dominant alteration in both LC and LCNEC tumors (83% and 88% of cases, respectively). TP53 mutations typically manifested as missense mutations in active protein domains and nonsense mutations in between active domains (Figure 1 and Supplementary Figure S1). Remaining alterations were found in considerably lower numbers in both subgroups. In LC, KRAS and MET were the second and third most frequently mutated genes (22% and 12%, respectively), while corresponding genes in LCNEC were STK11 and PTEN (16% and 13%, respectively) (Figure 1, Supplementary Table S3). These alterations highlight a more general difference between the two subgroups, regarding alterations in oncogenes versus tumor suppressors. Here, LC cases typically showed more alterations in oncogenes compared to LCNECs. Specifically, 20 oncogene alterations in BRAF, GNAQ, GNAS, KRAS, KIT, MET, NRAS, MAP2K1/MEK1, and PIK3CA were found in 44% of the LC cases as compared to eight alterations affecting 22% of LCNEC cases (Supplementary Table S3). Notably, for the two KRAS alterations observed in LCNEC cases, one was not in the active RAS protein domain (a KRAS M1I mutation). The second, a G12C mutation, was found in both the LCNEC and the LC component of the included multicomponent tumor, with different alternate allele frequencies (40.3% in the LCNEC and 7.9% and in the LC component of the tumor). Together, this suggests that activating KRAS mutations are in fact rare in LCNEC. Consistent with a general idea of a limited number of oncogene hits required to activate a tumorigenic pathway, we observed only one case in each histological subgroup with >1 mutation in any of the eight oncogenes.
Two additional differences regarding oncogene mutations may be noted. Firstly, KIT mutations were exclusively found in LCNEC cases (n = 3, 9%). Secondly, all TP53 wild type LC cases (17% of all LC cases) harbored oncogene mutations in KRAS, MET or PIK3CA (Figure 1A). This observation suggests that these TP53 wild type tumors may be more dependent on oncogene activation alone for sustained tumor development. However, in this relatively small retrospective cohort we did not find support for differences related to tumor stage or gender (p = 0.69 and p = 0.42, respectively, Fisher’s exact test), or patient outcome (overall survival, log-rank p > 0.05) between these tumors and TP53-mutated LCs.
Finally, high-level copy number gain (amplifications), or low copy deletions (putative homozygous deletions) were very scarce in analyzed cases (only single cases with NRAS, KRAS, GNAQ, MET, KIT, or PIK3CA amplifications, Figure 1), and no distinct cases of monoallelic amplification of mutated oncogene alleles were observed. For several tumor suppressors (TP53, STK11, PTEN, and APC), in especially LCNEC cases, we observed apparent support of Knudson’s multiple-hit hypothesis [21], with DNA mutation and associated copy number loss (Figure 1).
DNA mutations in histopathological and immunohistological subgroups of LC and LCNEC
Basaloid tumors represent a rare histopathological subgroup of LC characterized by specific cytological and tissue architectural characteristics [3]. In our LC cohort, six cases were subclassified as basaloid cancer. When viewed as two subgroups, i.e., basaloid versus non-basaloid LC, there were no differences in mutation frequencies between the groups for two of the most commonly mutated genes, TP53 and MET. In contrast, no KRAS mutations were observed in basaloid cases, consistent with Rossi et al. [6].
Recently, patient outcome and specific oncogene mutations in LC tumors have been associated with tumor subgroups defined by positive expression of adenocarcinoma (TTF-1/Napsin A) or squamous cell carcinoma (CK5/P40) immunohistochemistry markers [5]. In our LC cohort, 24 tumors (59%) were positive by immunohistochemistry for TTF-1/Napsin A (referred to as adenocarcinoma-like), 5 tumors (12%) were CK5/P40 positive (squamous-like), whereas 12 tumors (29%) did not express any of these IHC markers (marker null cases). Stratification of identified mutations by IHC subgroup revealed a striking enrichment of oncogene mutations in adenocarcinoma-like LC tumors (85% of all oncogene mutations, affecting 63% of these cases), including all nine KRAS mutations and the single NRAS (G12D) and MAP2K1/MEK1 (K57N) mutations (Figure 1A, Supplementary Tables S2 and S3). These KRAS mutations were all typical driver mutations located in codon 12 (one G12S, two G12C, three G12V mutations), 13 (one G13C and one G13D mutation), and 61 (one Q61K mutation), suggesting that these represent likely driver events in the affected tumors. In contrast, 92% of marker null cases carried a TP53 mutation, but only 17% of cases had an oncogene mutation (one BRAF Q456K and one MET T1010I mutation). Moreover, marker null cases showed a poorer overall survival compared to adenocarcinoma-like cases (p = 0.007, log-rank test, Figure 2). The poorer outcome of marker null LC cases compared to adenocarcinoma-like cases was significant also in multivariate analysis including immunomarker stratification, tumor stage, and gender as covariates and overall survival as clinical endpoint (Hazard ratio = 4.4, 95% Confidence interval = 1.5–12.5, p = 0.006 for marker null stratification).
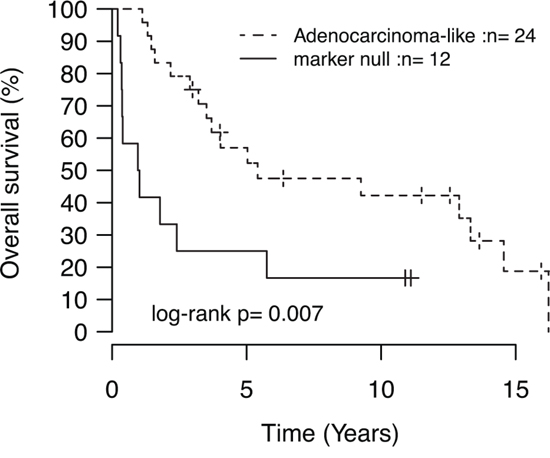
Figure 2: Kaplan-Meier analysis of the association with overall survival for immunomarker-defined subgroups of LC. P-value calculated using the log-rank test.
For LCNEC cases, we observed no association between DNA mutations and IHC expression of the chromogranin A, synaptophysin or CD56 neuroendocrine markers.
Validation analyses of NGS DNA mutation results
To validate the NGS platform we first analyzed seven independent tumor FFPE samples from lung, colon and melanoma with known mutations in BRAF (V600E), KRAS (G12S, G13D), and EGFR (L858R and E746_A750del). The variant allele frequency for these alterations by pyrosequencing ranged between 27.7–41.8%. All mutations could successfully be identified by the NGS platform.
Secondly, we selected the ten LC and LCNEC cases from the Lund cohort harboring KRAS mutations (variant allele frequencies between 3.1–41%), and validated them using quantitative PCR (Qiagen Therascreen). Eight KRAS mutations were correctly identified, while the remaining two, Q61K and G13C, were not covered by the Therascreen assay.
ALK, RET, and ROS1 gene fusion analysis in LC and LCNEC
The ability of the Archer FusionPlex assay to identify gene fusions in ALK, RET and ROS1 was successfully validated in four cell lines with known fusion gene rearrangements, HCC78 (ROS1-SLC34A2), KARPAS-299 (ALK-NPM1), LC-2/ad (CCDC6-RET), and H2228 (EML4-ALK).
Fusion gene analysis was performed on all 58 Lund cases using RNA from fresh frozen (n = 47) or FFPE (n = 11) tumor tissues. However, only 46 fresh frozen tumors passed the initial Archer data quality analysis steps after sequencing (35 LC, 11 LCNEC). The failure of the FFPE cases is likely due to extensive RNA degradation in the tissue blocks caused by the fixation process and subsequent storage. In the 46 analyzable cases, we identified no RET or ROS1 fusions. Only one analyzed tumor, a LCNEC case, showed a candidate ALK gene fusion event (DNBL-ALK) based on NGS data, however just with the minimum number of reads required for reporting (Supplementary Methods). However, confirmatory ALK IHC and FISH analysis could not confirm protein overexpression or an actual gene fusion event in this case.
DISCUSSION
In the current study, we have explored the mutational spectrum of 26 well-established cancer-related genes and ALK, RET, and ROS1 gene fusions by massive parallel sequencing in a large panel of thoroughly histopathologically classified primary LC and LCNEC lung cancers. In comparison to existing methods, NGS-based methods for DNA variant detection generally offer higher sensitivity in detecting low-frequency variants. Together with the specific feature of bi-directional sequencing in the Illumina TruSight Tumor assay this allows for sensitive variant detection also in FFPE samples. Besides the presented molecular characterization of LC and LCNEC tumors, the current study also supports the feasibility of using NGS-based methods for analysis of treatment predictive DNA alterations in routine clinical lung FFPE tumor tissues.
In LC as a whole, our findings of frequent KRAS mutations and less frequent alterations in BRAF, MAP2K1, and PIK3CA are in agreement with previous studies [5, 6]. Similarly, in LCNEC the high mutation rate of TP53 and the scarcity of KRAS mutations have also been reported before (see, e.g., Rossi et al. [4]). The similar frequency of TP53 mutations between the LC and LCNEC group mimics findings of similar p53 protein expression by Iyoda et al. [11]. Due to a paucity of studies, the roles of PTEN, STK11 and MET mutations in LC and LCNEC are largely unknown. In our study, alterations in the tumor suppressors PTEN and STK11 were mainly observed together with TP53 mutations in both LC and LCNEC, while MET mutations were more often found in TP53 wild type LC cases. Although LCNEC tumors have been shown to strongly express receptor tyrosine kinases such as KIT, PDGFRA, PDGFRB and MET, compared to other NSCLC groups, there is less support of mutations being the underlying cause for the elevated expression [4, 18]. Supporting these results, we identified no PDGFRA mutations in any of the tumors, only one MET mutation in the LCNEC group, whereas three KIT mutations were found exclusively in LCNEC cases. However, the impact of some of these mutations is difficult to assess without functional characterization, as all do not occur in active protein domains (see Supplementary Figure S1).
In this study, alterations in oncogenes, with exception of KIT alterations, are generally more frequent in LCs when considered a single entity compared to LCNEC. However, it is becoming apparent that the mutational spectrums in LC and LCNEC are different based on recent whole-exome sequencing studies. Specifically, LCNEC has been suggested to be more similar to SCLC [8], in line with the similarity of LCNEC and SCLC on the morphological, immunohistochemical, transcriptional, copy number, and epigenetic levels [4, 8–10]. Consistently, SCLC have recently been reported to harbor high frequencies of TP53 mutations and similar frequencies of KIT, PIK3CA and KRAS alterations as for the LCNEC cases in the current study [22]. Together, our observations further support that LC and LCNEC follow different evolutionary paths.
Stratification of LC cases based on immunomarkers for adenocarcinoma (TTF-1/Napsin A) and squamous cell carcinoma (CK5/P40) revealed that 71% of the cases could be classified as variants of adenocarcinoma or SqCC. This observation is in line with previous reports (59–90%) [5, 23–27], although the observation of 29% of LC as marker null is on the higher end compared to the literature. One reason for this could be that we in this retrospective cohort used the TTF-1 8G7G3/1 clone that compared to the SPT24 clone is slightly less sensitive, whereas the SPT24 clone yields more cases positive for both CK5/P40 and TTF-1. Likewise, we used CK5 and P40 as markers of squamous cell carcinoma, while P63 may be more sensitive (but also less specific). Irrespectively, our data demonstrates the value of using multiple immunomarkers for undifferentiated lung cancers. While no apparent differences in oncogene amplification frequency could be observed between immunomarker-defined subgroups, the subgroups showed a distinctively different spectrum of especially oncogene mutations. Adenocarcinoma-like LCs (59% of all analyzed LC cases) harbored the overwhelming majority of detected oncogene mutations (85%), affecting 63% of these tumors. By comparison, only 17% of marker null LC cases showed oncogene mutations. Thus, adenocarcinoma-like LC appears to represent a more oncogene driven subgroup compared to CK5/P40 positive tumors and marker null LCs. These findings are in excellent agreement with Rekhtman et al. [5], including the observation of the single PIK3CA mutation in a CK5/P40 positive case, and a poorer overall survival for marker null patients compared to TTF-1/Napsin A positive LC patients. Despite the retrospective nature of the patient material, our results in combination with other recent molecular studies clearly challenge LC as an independent tumor entity on the molecular level, supporting that the current LC definition rather includes a heterogeneous collection of poorly differentiated tumors from other NSCLC subgroups [5, 6, 8]. In fact, supported by both clinicopathological and molecular studies the recent 2015 WHO classification of lung cancer now stress that the term LC should now only be used for undifferentiated tumors not expressing pneumocyte or squamous markers [7]. Importantly, in the 2004 WHO classification the LC definition provides little molecular information for a predictive molecular testing strategy to guide individualized treatment for this patient cohort [5]. A refined stratification of LC based on molecular characteristics may therefore have considerable impact on diagnosis, predictive molecular testing and in the end, therapy selection [5, 6, 8].
In contrast to the immunomarker-defined LC subgroups, less is known whether LCNEC tumors may be divided into similar subgroups. In the current study we found no associations of the neuroendocrine markers used to identify LCNEC tumors with specific mutations. This lack of association may be because these markers do not represent putative subgroups at all, and/or, as indicated by our analyses, that the mutational landscape in LCNEC is different in respect to, especially, oncogene drivers compared to non-neuroendocrine NSCLC. Clearly, further genomic studies of both marker null LC (undifferentiated LC) and LCNEC tumors including large scale sequencing approaches, gene expression profiling, and DNA methylation profiling are needed to further characterize these tumor groups.
EGFR mutations and ALK gene fusions are the key molecular treatment predictive alterations for targeted therapy in lung cancer today [12]. However, both alterations are scarce in LC and LCNEC tumors [5, 6, 14–16], consistent with our findings of no EGFR mutations or validated ALK gene fusions in either LC or LCNEC tumors. Specifically, the absence of ALK rearrangements in our LC cohort compared to the few ALK rearranged cases reported by Rekhtman et al. [5] is consistent with that our cohort comprises only of known smokers, while ALK rearrangements in the former study were found in never or light smokers. In recent studies, lung cancer patients with tumors harboring ROS1 or RET gene fusions have shown notable responses to ALK or other multi-target kinase inhibitors [28, 29]. Similar to ALK fusions, the frequency of these alterations in LC and LCNEC is largely unknown, but may be expected to be very low. Consistently, we found no RET or ROS1 gene fusions in LC or LCNEC tumors based on targeted RNA sequencing.
In summary, the current study adds further insights into the mutational landscape of LC and LCNEC, supporting that these tumor subgroups follow different tumorigenic paths. Moreover, our study supports that LC may be refined by molecular and immunomarkers into clinically relevant subgroups that may have implications for diagnosis, and therapy decisions. Despite the identification of adenocarcinoma-like LC as a subset of tumors with a potentially high frequency of forthcoming therapeutically relevant driver mutations, a continued search for additional molecular targets for therapeutic inhibition in non-adenocarcinoma NSCLC is warranted.
Authors’ contributions
JS, AK and MP conceived of the study. AK, JS, FR, and CR performed the mutational analyzes and gene fusion experiments. HC performed the cell culturing. KJ and KEL provided validation samples and data. MJ and KEL assisted in validation experiments. AK and JS performed statistical analyses with support from GJ. HB performed pathological evaluations and IHC analyses. PJ and MP included patients. JS drafted and wrote the manuscript with support from AK and GJ. The study was methodologically and scientifically supported by ÅB. All authors approved the final manuscript.
FUNDING
Financial support for this study was provided by the Swedish Cancer Society, the Knut & Alice Wallenberg Foundation, the Foundation for Strategic Research through the Lund Centre for Translational Cancer Research (CREATE Health), the Mrs Berta Kamprad Foundation, the Gunnar Nilsson Cancer Foundation, the Swedish Research Council, the Lund University Hospital Research Funds, the Gustav V:s Jubilee Foundation, and governmental funding (ALF).
CONFLICTS OF INTEREST
Authors declare that they have no competing interests.
REFERENCES
1. Asamura H, Kameya T, Matsuno Y, Noguchi M, Tada H, Ishikawa Y, Yokose T, Jiang SX, Inoue T, Nakagawa K, Tajima K, Nagai K. Neuroendocrine neoplasms of the lung: a prognostic spectrum. J Clin Oncol. 2006; 24:70–76.
2. Battafarano RJ, Fernandez FG, Ritter J, Meyers BF, Guthrie TJ, Cooper JD, Patterson GA. Large cell neuroendocrine carcinoma: an aggressive form of non-small cell lung cancer. J Thorac Cardiovasc Surg. 2005; 130:166–172.
3. Travis W.D. BE, Muller-Hermelink H.K., Harris C.C. (Eds.). (2004). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. (Lyon: IARC Press).
4. Rossi G, Cavazza A, Marchioni A, Longo L, Migaldi M, Sartori G, Bigiani N, Schirosi L, Casali C, Morandi U, Facciolongo N, Maiorana A, Bavieri M, et al. Role of chemotherapy and the receptor tyrosine kinases KIT, PDGFRalpha, PDGFRbeta, and Met in large-cell neuroendocrine carcinoma of the lung. J Clin Oncol. 2005; 23:8774–8785.
5. Rekhtman N, Tafe LJ, Chaft JE, Wang L, Arcila ME, Colanta A, Moreira AL, Zakowski MF, Travis WD, Sima CS, Kris MG, Ladanyi M. Distinct profile of driver mutations and clinical features in immunomarker-defined subsets of pulmonary large-cell carcinoma. Mod Pathol. 2014; 26:511–522.
6. Rossi G, Mengoli MC, Cavazza A, Nicoli D, Barbareschi M, Cantaloni C, Papotti M, Tironi A, Graziano P, Paci M, Stefani A, Migaldi M, Sartori G, et al. Large cell carcinoma of the lung: clinically oriented classification integrating immunohistochemistry and molecular biology. Virchows Arch. 2014; 464:61–68.
7. Travis W.D. BE, Burke A.P., Marx A, Nicholson A.G. (Eds.). (2015). WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. (Lyon: IARC Press).
8. CLCGP. A genomics-based classification of human lung tumors. Sci Transl Med. 2013; 5: 209ra13.
9. Staaf J, Isaksson S, Karlsson A, Jonsson M, Johansson L, Jonsson P, Botling J, Micke P, Baldetorp B, Planck M. Landscape of somatic allelic imbalances and copy number alterations in human lung carcinoma. International journal of cancer. 2012; 1:2020–2031.
10. Karlsson A, Jonsson M, Lauss M, Brunnstrom H, Jonsson P, Borg A, Jonsson G, Ringner M, Planck M, Staaf J. Genome-wide DNA Methylation Analysis of Lung Carcinoma Reveals One Neuroendocrine and Four Adenocarcinoma Epitypes Associated with Patient Outcome. Clin Cancer Res. 2014; 20:6127–6140.
11. Iyoda A, Hiroshima K, Moriya Y, Mizobuchi T, Otsuji M, Sekine Y, Shibuya K, Iizasa T, Saitoh Y, Fujisawa T. Pulmonary large cell neuroendocrine carcinoma demonstrates high proliferative activity. Ann Thorac Surg. 2004; 77:1891–1895; discussion 1895.
12. Leighl NB, Rekhtman N, Biermann WA, Huang J, Mino-Kenudson M, Ramalingam SS, West H, Whitlock S, Somerfield MR. Molecular Testing for Selection of Patients With Lung Cancer for Epidermal Growth Factor Receptor and Anaplastic Lymphoma Kinase Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Guideline. J Clin Oncol. 2014.
13. Nakamura H, Tsuta K, Yoshida A, Shibata T, Wakai S, Asamura H, Furuta K, Tsuda H. Aberrant anaplastic lymphoma kinase expression in high-grade pulmonary neuroendocrine carcinoma. J Clin Pathol. 2013; 66:705–707.
14. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, Iafrate AJ, Arcila ME, Ladanyi M, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1, 683 patients with non-small cell lung cancer. Clin Cancer Res. 2013; 19:4273–4281.
15. De Pas TM, Giovannini M, Manzotti M, Trifiro G, Toffalorio F, Catania C, Spaggiari L, Labianca R, Barberis M. Large-cell neuroendocrine carcinoma of the lung harboring EGFR mutation and responding to gefitinib. J Clin Oncol. 2011; 29:e819–822.
16. Aroldi F, Bertocchi P, Meriggi F, Abeni C, Ogliosi C, Rota L, Zambelli C, Bna C, Zaniboni A. Tyrosine Kinase Inhibitors in EGFR-Mutated Large-Cell Neuroendocrine Carcinoma of the Lung? A Case Report. Case Rep Oncol. 2014; 7:478–483.
17. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, Powell CA, Beer D, Riely G, Garg K, Austin JH, Rusch VW, Hirsch FR, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: international multidisciplinary classification of lung adenocarcinoma: executive summary. Proc Am Thorac Soc. 2011; 8:381–385.
18. Salomonsson A, Jonsson M, Isaksson S, Karlsson A, Jonsson P, Gaber A, Bendahl PO, Johansson L, Brunnstrom H, Jirstrom K, Borg A, Staaf J, Planck M. Histological specificity of alterations and expression of KIT and KITLG in non-small cell lung carcinoma. Genes Chromosomes Cancer. 2013; 52:1088–1096.
19. Brunnstrom H, Johansson L, Jirstrom K, Jonsson M, Jonsson P, Planck M. Immunohistochemistry in the differential diagnostics of primary lung cancer: an investigation within the Southern Swedish Lung Cancer Study. Am J Clin Pathol. 2013; 140:37–46.
20. Karlsson A, Ringner M, Lauss M, Botling J, Micke P, Planck M, Staaf J. Genomic and transcriptional alterations in lung adenocarcinoma in relation to smoking history. Clin Cancer Res. 2014; 20:4912–4924.
21. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971; 68:820–823.
22. Ross JS, Wang K, Elkadi OR, Tarasen A, Foulke L, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Chmielecki J, Ali SM, Elvin J, et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol. 2014; 67:772–776.
23. Au NH, Cheang M, Huntsman DG, Yorida E, Coldman A, Elliott WM, Bebb G, Flint J, English J, Gilks CB, Grimes HL. Evaluation of immunohistochemical markers in non-small cell lung cancer by unsupervised hierarchical clustering analysis: a tissue microarray study of 284 cases and 18 markers. The Journal of pathology. 2004; 204:101–109.
24. Barbareschi M, Cantaloni C, Del Vescovo V, Cavazza A, Monica V, Carella R, Rossi G, Morelli L, Cucino A, Silvestri M, Tirone G, Pelosi G, Graziano P, et al. Heterogeneity of large cell carcinoma of the lung: an immunophenotypic and miRNA-based analysis. Am J Clin Pathol. 2011; 136:773–782.
25. Monica V, Ceppi P, Righi L, Tavaglione V, Volante M, Pelosi G, Scagliotti GV, Papotti M. Desmocollin-3: a new marker of squamous differentiation in undifferentiated large-cell carcinoma of the lung. Mod Pathol. 2009; 22:709–717.
26. Monica V, Scagliotti GV, Ceppi P, Righi L, Cambieri A, Lo Iacono M, Saviozzi S, Volante M, Novello S, Papotti M. Differential Thymidylate Synthase Expression in Different Variants of Large-Cell Carcinoma of the Lung. Clin Cancer Res. 2009; 15:7547–7552.
27. Pardo J, Martinez-Penuela AM, Sola JJ, Panizo A, Gurpide A, Martinez-Penuela JM, Lozano MD. Large cell carcinoma of the lung: an endangered species? Applied immunohistochemistry & molecular morphology: AIMM/official publication of the Society for Applied Immunohistochemistry. 2009; 17:383–392.
28. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014; 371:1963–1971.
29. Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, Ross J, Miller V, Ginsberg M, Zakowski MF, Kris MG, Ladanyi M, Rizvi N. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013; 3:630–635.