
INTRODUCTION
It is commonly believed that major clinical phenotypes of cancer (e.g., tumorigenesis, malignancy, survival time, treatment response, metastasis) are caused by molecular aberrations - such as sequence mutations, copy number variations (CNV), epigenetic alterations - on a selected number of driver genes [1]. The list of driver genes expands substantially as new high-throughput technologies detect more rare molecular aberrations in larger patient cohorts [2]. Some scientists argue that the current list of leading driver genes is close to complete [3, 4].
Despite the rich documentation about driver genes, their specificity in terms of patient populations remains less well-known. Molecular aberrations of certain genes occur primarily on specific types of cancers in specific populations. Variations of driver molecular aberration landscapes across tumor types are widely appreciated and well-documented in cancer research [4, 5]. Characterization of drivers in race and gender specific subpopulations, in contrast, is relatively rare. Numerous studies demonstrated sex or ethnic specific prognostic outcomes in multiple cancer types. For instance, a survival advantage has been noted for younger females with cancer types such as hepatocellular carcinoma [6, 7], melanoma [8–10] and colorectal cancer [11, 12]. In contrast, a worse prognosis has been identified for female patients with bladder cancer [13, 14]. Prognostic differences between Caucasian and Asian men have also been reported in prostate cancer [15, 16]. However, the molecular aberrations underpinning these differences remain largely unclear. Characterization of drivers in race and gender specific subpopulations is a foundation of personalized medicine in oncology as it has strong implications in both diagnosis and treatments. Molecular biomarkers derived from one race/gender group may not be relevant in other populations [17, 18]. Furthermore, patients with distinct genetic backgrounds may yield drastically diverse responses to different drugs [19–21]. Identification of population-specific molecular aberration drivers bridges the gap between the demand of personalized medicine and existing knowledge and is thence highly impactful.
Lung cancer is the most commonly diagnosed and foremost cause of cancer related mortality [22]. Epidemiological studies have demonstrated significant gender and ethnic differences in trends amongst lung cancer patients. Tumors arising from the central airway compartment (predominantly small cell lung cancer and squamous cell carcinoma) are primarily associated with smoking. Tumors arising from the periphery airway compartment (predominantly adenocarcinoma) occur much more frequently in never-smokers and are attributed to poorly understood factors [23]. Lung cancers in non-smokers occur disproportionately more frequent amongst East Asian female populations [23, 24]. Moreover, lung adenocarcinoma exhibits mutually exclusive somatic mutations between patients from East Asian and European descent. East Asian patients possess predominantly EGFR mutations, whereas Western patients possess primarily KRAS mutations [25, 26].
The remarkable distinction of EGFR/KRAS mutations is already applied to screen recipients for the targeted drugs of tyrosine kinase inhibitors such as gefitinib [19, 27, 28]. Yet they are unlikely to be the only population-specific drivers of lung adenocarcinoma. A plethora of prior studies detected prognostic biomarkers of lung adenocarcinoma from high-throughput data of genome sequences [29, 30], transcriptome levels [31, 32], copy number variations [33, 34], epigenetic states [35, 36], and combinations of them [37, 38]. The majority of those studies restricted samples to certain geographic areas and/or gender groups. Without comparison and verification with other datasets, it is impossible to determine whether findings from single sources are universal, population-specific, or outliers rarely occurred elsewhere. Meta-analysis of lung adenocarcinoma has been pursued by a number of research groups [39–41]. However, those studies focused primarily on universal biomarkers common in all datasets, rather than population-specific patterns. This approach gives rise to robust findings but also fails to address the population-specific variations.
In our previous work [42], we developed an algorithm to identify association modules [43] of genes linking molecular aberrations on DNA with mRNA expressions from integrative datasets (copy number variations, DNA methylations, mRNA expressions), and employed this algorithm to the Glioblastoma multiforme data from The Cancer Genome Atlas (TCGA) [44]. In the present study, we applied the module-finding algorithm to a lung adenocarcinoma data of a cohort of female non-smoking Taiwanese patients, and examined the prognostic outcomes of the inferred modules in four additional datasets covering East Asian and Caucasian, male and female subpopulations. The novelty of this work is to investigate and demonstrate ethnic and gender specific properties of the inferred modules. Strikingly, three modules – cis-acting effects with chromosomes 7 and 18 CNVs, and methylations with UBIAD1 and VAV1 – demonstrated strong associations with prognosis amongst the East Asian or the East Asian female subpopulation. In the specified subpopulation, patients possessing high average expression levels in each association module had significantly shorter survival times than patients possessing low average expression levels. For the module with cis-acting effects on chromosome 18, the associations of module members with survival times were significant in East Asian females, but insignificant among other subpopulations (East Asian males, Western males and females). In contrast, for modules with cis-acting effects with chromosome 7 and methylations with UBIAD1 and VAV1, these demonstrated strong prognostic effects amongst the East Asian subpopulations. Extending the validation methods of our previous study, we utilized partial least squares (PLS) to analyze dependencies between the inferred association modules. Co-citation and pathway enrichment methods were also included in the analysis to identify likely putative effector genes and their possible role in related biological mechanisms. Beyond the well-known EGFR, the two cis-acting modules also harbor other likely effectors such as RAC1 (chromosome 7), CDK5 (chromosome 7), and RALBP1 (chromosome 18).
RESULTS
Association modules
Cancer cells harbor a large number of molecular aberrations such as CNV, DNA methylation and sequence mutations. Some aberrations may dysregulate gene expressions, which in turn drive the malignancy of tumors. Those mechanistic relations between molecular aberrations and gene expressions can be manifested on the statistical associations of their measured data. Previously, we encapsulated those statistical associations as modules [42, 43]. An association module consists of three components: (i) an observed effector molecular aberration on DNA; (ii) the downstream target genes with expression profiles associated with the effector molecular aberration; and (iii) regulators (transcription factors or signalling proteins) that mediate the effects between effector molecular aberrations and target gene expressions. For the data types available in this study, the association modules generated can be described by three distinct types:
- Cis-acting effects with CNVs of chromosomes The segment CNV of a chromosome is positively associated with the expressions of target genes on the same chromosomal segment location.
- Trans-acting effects with CNVs of chromosomes The segment CNV of a chromosome exhibits cis-acting effects with intermediate regulators, and both the chromosome segment CNV and regulator expressions have either positive or negative associations with the expressions of target genes on other chromosomes.
- Effects with DNA methylations The coherent DNA methylation states of a collection of genes are negatively associated with the expressions of themselves and other target genes.
A summary of the assumptions underlying these modules is illustrated in Fig. 1. We employed a layered approach to prioritize associations in terms of their mechanistic certainty, incrementally constructed the logistic regression model of each gene expression profile, and grouped the genes sharing common effectors to form association modules. The procedures of the module finding algorithm are described in Materials and Methods and S1 Text.
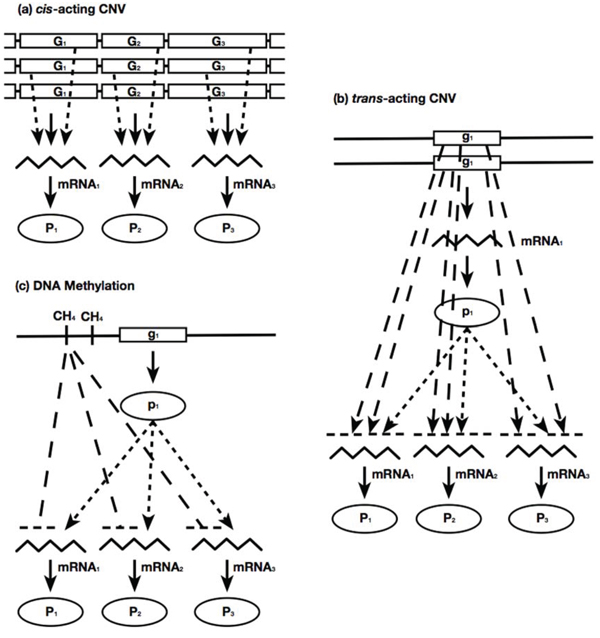
Figure 1: Three types of association module. a. Cis-acting effects with CNVs of chromosomes, b. Trans-acting effects with CNVs of chromosomes; and c. Effects with DNA methylations. Solid lines: information flows following the central dogma (DNA → mRNA → protein). Dotted lines: regulatory links from regulators to their targets on other chromosomal locations. Dashed lines: associations between observed aberrations and mRNA gene expressions. Arrowheads indicate positive associations and bar-ends indicate negative associations. The figure is adapted from [42].
Datasets
Tissue specimens were collected from non-smoking female lung cancer patients admitted to National Taiwan University (NTU) Hospital and Taichung Veterans General Hospital (details of data collection have been described previously [45]). A total of 60 tumor and adjacent normal lung tissue specimen pairs were interrogated using Affymetrix U133plus2.0 mRNA expression microarrays, Affymetrix SNP6.0 microarrays, and Illumina Infinium. A further 32 pairs of lung adenocarcinoma samples were selected with both DNA and mRNA expression data available. We pre-processed those integrative datasets and converted them into standard formats (see Materials and Methods for details), and employed the module finding algorithm to the processed data. Validation of the inferred modules was performed on the mRNA and survival data from two lung adenocarcinoma datasets of East Asian cohorts (Japanese [46] and Korean [47]) and two datasets of US Caucasian populations (US1 [48] and US2 [49]). Table 1 summarizes the information of the five datasets. In the two US datasets only lung adenocarcinoma samples from Caucasian patients were extracted. All data except the training set of the Taiwanese cohort have clinical information available to assess the prognostic value of the association modules.
Table 1: East Asian and White Caucasian datasets of lung adenocarcinoma
Key |
Ethnicity |
Subjects (Male/Female) |
Reference |
Taiwan |
East Asian |
32 (0/32) |
[45] |
Japan |
East Asian |
117 (60/57) |
[46] |
Korea |
East Asian |
63 (34/29) |
[47] |
US1 |
White Caucasian |
244 (104/140) |
[48] |
US2 |
White Caucasian |
294 (127/167) |
[49] |
A description of the data used in the study. The Taiwan mRNA, CNV and methylation integrative data were used to build the association modules. Validation of the modules was performed on mRNA and clinical data from two East Asian cohorts (Japan and Korea) and two datasets of US Caucasian populations (US1 and US2).
Discovery and validation of association modules
The module finding algorithm [42, 43] was applied to the Taiwanese dataset. A total of 44 association modules were generated: 23 modules with cis-acting effects of chromosomes’ CNVs, 15 modules with trans-acting effects of chromosomes’ CNVs, and 6 modules with DNA methylations as effectors. A complete list of association modules is provided in S1 Table. The inferred modules were passed through two validation tests on two East Asian female datasets to examine their biological and prognostic relevance. First, we checked whether the targets in each module retained coherent expression profiles in both external datasets. Expression profile coherence of an association module was gauged by statistical deviation of its pairwise correlation coefficient distribution from a background distribution derived from a random collection of genes (S2 Table (a)). Second, we checked whether the target gene expression profiles in each module possessed prognostic power. Prognostic power of an association module was assessed by two indicators. We evaluated the Cox regression coefficients of target gene expression profiles and calculated the Kolmogorov-Smirnov (KS) statistical significance of their distribution relative to a background distribution derived from a random collection of genes (S2 Table (b)). To reduce the effect of module size to the KS statistical significance, we further collapsed the expression profiles of multiple target genes into one median expression profile and evaluated the log-rank p-values of the Kaplan-Meier curves derived from the aggregate biomarker (S2 Table (c)). Validation procedures are elaborated in Materials and Methods.
Three association modules passed the validation tests: module 1 comprises cis-acting effects of chromosome 7 CNV, module 2 comprises cis-acting effects of chromosome 18 CNV, and module 3 comprises DNA methylation of UBIAD1 and VAV1 as effectors. Fig. 2 displays expression coherence and prognostic power of the three modules on two East Asian female datasets. Both expression correlation coefficients and Cox regression coefficients have significantly positive deviations from the background distributions. The false discovery rate (FDR) of randomized modules passing all the validation tests is < 0.002 (see Materials and Methods and S1 Text about FDR evaluation procedures).

Figure 2: Validation tests to examine the target gene coherence of genes and prognostic power of the modules. a. Heatmaps illustrating the distributions of correlation coefficients among target gene expressions of female East Asian datasets. b. Heatmaps illustrating the distributions of Cox coefficients of target genes for female East Asian datasets. A distribution of all genes within the external data is included to provide a background comparison to compute the Kolmogorov-Smirnov statistic.
Inferred association modules are more specific within the East Asian female population
Since the three modules were inferred from and validated on the datasets of East Asian female patients, it is unclear whether they were universal to all the patient populations or specific to the East Asian female subpopulation alone. To answer this question, we extended validation by including two additional datasets of Caucasian patients (US1 and US2) and further divided each dataset by gender. Accordingly there were four subpopulations: East Asian females, East Asian males, Caucasian females and Caucasian males. We again analyzed Kaplan-Meier curves for each inferred module to study time-to-event data. Patients were assigned to high and low expression groups if the median gene expression for the selected module was higher or lower than the average module expression respectively. All three modules demonstrated strong prognostic effects amongst female East Asian subpopulations and one module was both gender and ethnic specific.
Modules 1 and 3 show prognostic power in East Asian populations
Expression coherence of modules 1 and 3 was significant in all the four subpopulations (S1 Fig. (a) and (c)). In contrast, their prognostic outcomes were highly specific in certain subpopulations. Fig. 3 displays the Kaplan-Meier curves for modules 1 and 3, with patients assigned to low and high expression groups based on median gene expression. For module 1 we observed that the associations of module members with survival times was significant for East Asian (columns 1 and 2), but not Caucasian (columns 3 and 4) subpopulations. In both Japanese and Korean cohorts, the Kaplan-Meier curves indicated that a high expression of target genes had a negative effect on the survival prospects of the subject. The log-rank p-value of module 1 in the Japanese female data is less significant (p = 0.091), but still substantially lower than the two US counterpart datasets (p = 0.942 and 0.938 respectively). Furthermore, module 1 exhibited significant prognostic outcomes in both East Asian males (rows 1 and 3) and females (rows 2 and 4), suggesting ethnic rather than gender specificity. In contrast, module 3 demonstrated a similar significance for East Asian subpopulations, but was also significant for the female Caucasian cohorts.
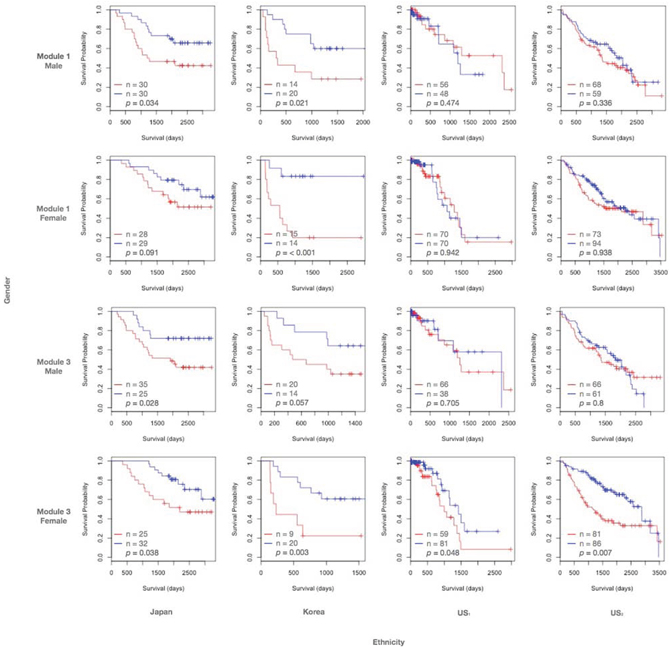
Figure 3: Kaplan-Meier curves of modules 1 and 3. Kaplan-Meier survival curves of patients divided by median gene expression amongst target genes in inferred modules. A red line indicates the survival curve of the patient group with high median expression levels in the target genes. A blue line indicates the survival curve of the patient group with low median expression levels in the target genes. Tick marks indicate censored data points; p-values are determined by log-rank tests. The size of each patient group and the log-rank p-value are reported.
Module 2 is gender and ethnic specific
Expression coherence of module 2 was also significant in all the four subpopulations (S1 Fig. (b)). Furthermore, module 2 exhibits significant prognostic outcomes in the East Asian female subpopulation alone. Fig. 4 displays the Kaplan-Meier curves for module 2, with patients assigned to low and high expression groups based on median gene expression. The log-rank p-values were significant only among the females of the Japanese and Korean datasets. Like modules 1 and 3, a high expression of target genes has a negative effect on the survival time. The Cox coefficient distributions achieved significantly positive deviation in East Asian female and Japanese male subpopulations (S2 Fig. (b)). Although the distribution was significant in Japanese males (p = 0.003), this trend was further enhanced in Japanese females (p = 0.001).
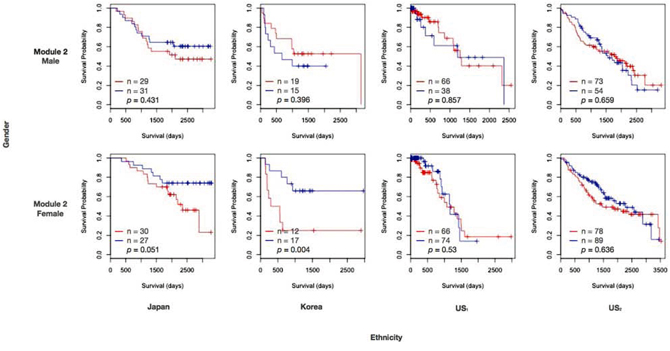
Figure 4: Kaplan-Meier curves of module 2. The legend follows Fig. 3.
Dependency between association modules
As modules 1 and 3 demonstrated a similar distribution of Cox coefficients in our tests of prognostic power (S2 Fig. (a) and (c)), we examined the possibility that chromosome 7 CNV is a candidate effector for the target gene expressions observed in module 3. A partial least squares (PLS) regression [50, 51] was employed to assess the association between target genes of modules 1 and 3.
PLS is useful for assessing the module members in this study, as we may treat the targets of two modules as X and Y blocks of genes and build a PLS model between them. This allows us to calculate an R2-type measure for the first few components in each block, that can represent a variance explained between association modules. The procedures of computing PLS are described in Materials and Methods. A correlation circle can be useful in demonstrating variable loadings on the first two PLS components. This plot demonstrates the associations between module targets, by assessing their proximity with one another. The cosine of the angles between gene locations in the plot will indicate the correlation between module members projected onto these two PLS components. In addition, the orthogonal projection of the module members onto the component axes will demonstrate the respective loading on each component.
Two sets of randomly selected genes confer no correlation on their PLS component projections (Fig. 5(a)). The correlation circles for modules 1 and 3 in Fig. 5(b) demonstrated the projection of their target genes on the first two PLS components in the Taiwanese dataset. The two clusters of genes occupied restricted regions separated by an angle less than π/2, indicating positive correlations between them. Positive correlations between target genes of modules 1 and 3 were pronounced by comparing Fig. 5(b) with the correlation circle plot between modules 2 and 3 (Fig. 5(c)). In the latter case the two clusters of genes spread over a wide range of angles and possessed a much smaller R2 for the first two components relative to the former case (0.38 versus 0.28 respectively). Similar trends were observed in Japanese and Korean datasets (S3 Fig.).
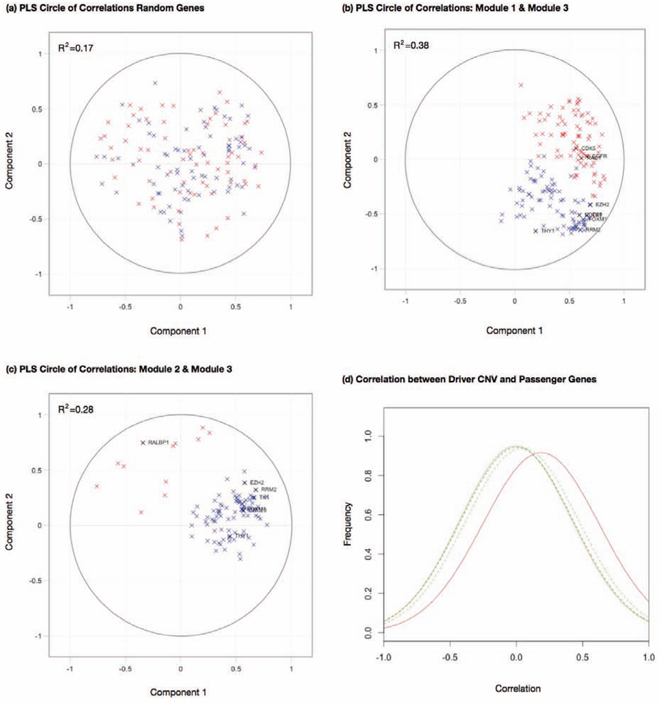
Figure 5: Partial least squares to investigate an association between modules in the Taiwan data. a. A correlation circle displaying the first two component loadings for two sets of randomly selected genes b. A correlation circle displaying the first two component loadings for target genes in modules 1 (shown in red) and 3 (shown in blue), c. A correlation circle displaying the first two component loadings for target genes in modules 2 (shown in red) and 3 (shown in blue). d. Distributions of correlation coefficients between module effectors and target genes. Solid red line: correlation coefficients between module 1 effector of chromosome 7p CNV and module 3 target gene expressions. Solid green line: correlation coefficients between module 1 effector of chromosome 7q CNV and module 3 target gene expressions. Dashed red line: correlation coefficients between module 2 effector of chromosome 18p CNV and module 3 target gene expressions. Dashed green line: correlation coefficients between module 2 effector of chromosome 18q CNV and module 3 target gene expressions.
The effectors of modules 1 and 3 are chromosome 7 CNV and DNA methylation of UBIAD1 and VAV1 respectively. Strong correlations of their target genes suggest that module 3 targets might be affected by chromosome 7 CNV. To further verify this hypothesis, we calculated the distributions of correlation coefficients between module 3 target gene expressions and the segment CNVs of chromosome 7p and 7q in the Taiwanese data (Fig. 5(d)). The correlations between chromosome 7p CNV and module 3 target gene expressions have a significantly positive deviation (solid red line) relative to the background correlation coefficient distributions between chromosome 18 p and q arm CNVs and module 3 target gene expressions (dashed red and green lines respectively). In contrast, the correlations between chromosome 7q CNV and module 3 target gene expressions are close to the background distributions (solid green line). The results suggest that the effector genes for module 3 targets are likely located on chromosome 7p.
Identification of putative effector genes on cis-acting association modules
Prognostic effects of a cis-acting CNV module are likely caused by a few effector genes on the designated chromosome. Expression level variations of those genes may modulate activities of specific functions and eventually affect survival times. Other genes in the vicinity may possess similar CNV and expression profiles as the effectors, thus exhibit a strong correlation with survival times despite the lack of mechanistic links. To identify putative effector genes on cis-acting CNV modules, we searched co-citations of the keyword lung cancer with all the module members and examined the distributions of their chromosomal locations. Fig. 6 shows the locations of all genes on chromosomes 7 and 18 and their Cox regression coefficients. The Cox coefficients have been standardized to allow for a direct comparison across external East Asian female validation datasets. Members of association modules are flagged by red circles. Intriguingly, members of both modules 1 and 3 were clustered in a few regions of the corresponding chromosomes. Module 1 targets were located primarily on chromosome 7p and a smaller region (130–150 Mb) of chromosome 7q. Module 2 targets were located on chromosome 18p. Generally, module members possessed higher Cox regression coefficients than other genes on the same chromosomes. Yet there was no consecutive region with consistently high Cox regression coefficients among all constituent genes.
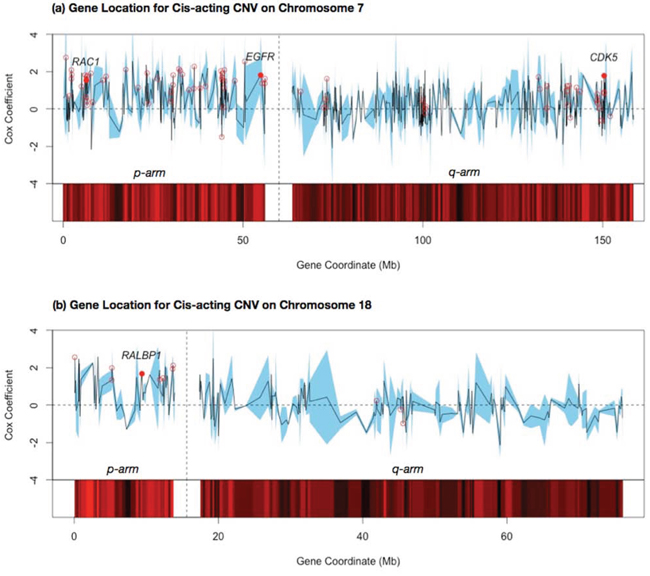
Figure 6: Cox regression coefficients of target genes within selected cis-acting CNV modules mapped by the relative locations on the chromosome. The average of the standardized Cox coefficients for the two East Asian female datasets is shown by the black line, with a confidence interval for the two datasets shown by the blue band. Each target gene in the association module is flagged by a hollow red dot. The names of genes with high-ranking PubMed citations to lung cancer key terms are highlighted (see Materials and Methods), with the mean Cox coefficient shown with a solid red dot. The p and q arms of each chromosome are separated by blank columns; a. shows the gene locations for cis-acting CNV on chromosome 7; b. shows the gene locations for cis-acting CNV on chromosome 18.
To solicit putative effector genes in the cis-acting CNV modules, we counted the number of co-citations with the keyword lung cancer and each target gene and retained the top ranking 5% of genes as described in Materials and Methods. Results of the citation count are presented in S3 Table (a). Four genes were identified in the co-citation search as putative effectors of modules 1 and 3.
- RAC1 (chromosome 7p): A signalling G protein member of the Rho family of GTPases, involved in cancer cell proliferation and invasion. The effect on cell motility may result in epithelial-mesenchymal transition, driving tumor metastasis in lung adenocarcinoma for cancer progression and drug-resistant tumor relapse [52, 53].
- EGFR (chromosome 7p): A cell surface receptor and member of the ErbB family of receptors. Overexpression of EGFR by mutation, amplifications or misregulations is considered a key factor in the uncontrolled cell division of a number of cancer types [54]. EGFR inhibitors, such as gefitinib and erlotinib, have been developed primarily to target lung adenocarcinoma [19, 27].
- CDK5 (chromosome 7q): Codes an enzyme involved in sensory signalling. CDK5 plays a role in many neurodegenerative diseases and cancers by phosphorylating the actin regulatory protein caldesmon [55]. Amplification of CDK5 has been found to play an important role in the migration and invasion of lung adenocarcinoma cancer cells.
- RALBP1 (chromosome 18p): A protein that is commonly overexpressed in malignant cells, preventing apoptosis in selected cancer cells [52]. RALBP1 is a major transporter of doxorubicin, a drug used in cancer chemotherapy, and can potentially contribute to multi-drug resistance [56].
The effector analysis was repeated on the external male East Asian validation datasets and illustrated in S4 Fig. Whilst positive Cox coefficients are again observed amongst these putative effector genes, we note that the coefficients of EGFR and RALBP1 in particular are not as strong when compared with the female subpopulations.
Whilst citation count provides a useful quantitative measure of a target genes relevance to lung adenocarcinoma, it is potentially biased against recently discovered cancer genes. To relieve this problem, we adopted an alternative approach by using the NCBI OMIM database [57] to identify cancer-related genes, and to search for their overlap with the target genes of inferred modules. Results of the OMIM analysis are presented in S3 Table (b). A total of 100 genes of the inferred modules were present in the OMIM database, including RAC1, EGFR and CDK5.
Pathway enrichment of target genes
A pathway enrichment was performed using QIAGEN’s Ingenuity pathway analysis (IPA) (http://www.qiagen.com/ingenuity) [58]. Complete results of the pathway analysis for all modules are included in S4 Table. A total of 8 pathways were found significant for the target genes in module 1 by the criteria defined in Materials and Methods. The pathways of module 1 are related to less specific processes, such as DNA transcription and translation control. In comparison, no pathways reached significance for module 2. This is likely due to the small size of the module (i.e. 12 genes) and the location restriction of target genes to chromosome 18 only.
Module 3 is the only significant association module containing target genes not restricted to a single chromosome. This reflects the nature of the majority of pathways in the IPA database. Two of the highest scoring cancer pathways in module 3 were ’uterine serous papillary cancer’ (p = 3.82 × 10−13) and ’mitosis of cervical cancer cell lines’ (p = 1.93 × 10−7). This appears relevant to the current study, as both are cancers predominantly affecting female populations. Common to both of these pathways were genes such as CDC20 and PTTG1. These genes were also significant in the PubMed search of key terms (see Materials and Methods and S3 Table), along with other members of the ’uterine serous papillary cancer’ pathway such as FOXM1, TK1, CCNA2 and UBE2C. Human Papillomavirus (HPV) has previously been linked to lung cancer in Taiwanese female non-smokers [59], and so this finding may hold some significance for module 3.
DISCUSSION
In this study, we demonstrated that the dominant associations linking DNA molecular aberrations, mRNA expressions and prognosis in lung adenocarcinoma were population specific. Target gene expressions of chromosome 7 cis-acting CNV (module 1) and UBIAD1 and VAV1 methylations (module 3) had negative effects on survival times among East Asian patients. Furthermore, target gene expressions of chromosome 18 cis-acting CNV (module 2) had negative effects on survival times among East Asian female patients alone. Gender and ethnic specificity of biomarkers has profound implications in the diagnosis and treatment of cancer. Patients with different ethnic and gender backgrounds should be diagnosed with different molecular biomarkers and receive different treatments targeting distinct molecular aberrations. This mode of operations will constitute the foundation of personalized medicine in the post-genomic era.
Our use of the term “putative effectors” has a weaker connotation than the canonical definition of “driver genes” in cancer research. A putative effector is identified by statistical associations between DNA molecular aberrations and transcriptional profiles, and further validated by information from external datasets as well as literature co-citation and functional annotations. In contrast, the driver function of a gene cannot be affirmed without explicit intervention such as mutagenesis, RNA silencing or genome editing. Nevertheless, the list of putative effectors provides useful candidates for further experimental validation.
The salient associations of chromosome 7 cis-acting CNV (module 1) with prognosis likely reflects the effect of EGFR. EGFR mutations occur predominantly among East Asian lung cancer patients [25, 26]. Here we demonstrated that beyond mutations, copy number variations of EGFR (and chromosome 7p in general) also likely contributed to poor prognosis in East Asian cohorts.
The effectors of module 3 – DNA methylations of UBIAD1 and VAV1 – are less well-known in lung adenocarcinoma. Unlike cis-acting CNV modules, associations with DNA methylations are more difficult to verify since DNA methylations are not probed in all external datasets, and their proxies – mRNA expressions of the methylated genes – rely on a few genes. Coincidentally, we discovered that the target genes of module 3 were correlated with both effectors and targets of module 1. Such dependency suggests that module 3 may also be a trans-acting module of chromosome 7p CNV. Although no module 3 targets are located on chromosome 7p, they can be co-regulated by a common effector gene on chromosome 7p and hence exhibit strong dependency with module 1. EGFR is a likely candidate for the effector as (1) it is located on chromosome 7p, (2) it is an oncogene with a negative effect on survival duration, and (3) its molecular aberration is biased in East Asian cohorts.
Despite the strong association between chromosome 7p CNV and module 3 target gene expressions, the trans-acting module of chromosome 7 CNV inferred from the Taiwanese data (S1 Table) did not pass the validation tests nor was overlapped with module 3. As shown in Fig. 5(d), many of chromosome 7p CNV and module 3 target gene expression associations were significantly higher than the background but still weaker than the correlation coefficient threshold (0.5) in the module finding algorithm. Thus the trans-acting module of chromosome 7 CNV was not reported in our study.
Unlike chromosome 7 and EGFR, prognostic associations of chromosome 18 (module 2) and its gender and ethnic specificity are novel and striking. The negative effect on survival is manifested in East Asian female patients alone. RALBP1 is the only candidate effector with relatively extensive reports in lung cancer. The function of RALBP1 as a molecular transporter may account for its links to drug resistance and hence poor prognosis.
It is difficult to establish mechanistic links from the biomarker levels to prognostic outcomes as the latter are determined by many genetic and environmental factors. Generally, the associations with most members of cis-acting CNV modules (modules 1 and 2) are likely caused by a few effector genes in the vicinity. In contrast, the associations with members of trans-acting modules (module 3) are possibly on the genes mediating the effector molecular aberrations (chromosome 7p CNV or UBIAD1 and VAV1 methylations) and the prognostic outcomes. Indeed, the few putative effector genes of modules 1 and 2 (RAC1, EGFR, CDK5, RALBP1) and many target genes in module 3 are involved in cell division and proliferation, anti-apoptosis, and molecular transport. High activities in those processes will enhance malignancy and drug resistance and thus lead to poor prognostic outcomes. However, although the directions of prognostic associations match the functions of those genes, it remains a puzzle to explain why those apparently generic processes are gender and ethnic specific.
An equally important puzzle is the cause of gender and ethnic specificity of molecular aberrations. The majority of the molecular aberrations observed in cancer genomic data are somatic - arising after birth rather than being inherited. Why would certain types of molecular aberrations (e.g., EGFR mutations or amplifications) arise more frequently in a specific population (e.g., East Asian female patients) even though they are not inherited? Understanding the causes of population-specific bias of somatic aberrations will be a milestone to the research of tumor genome evolution.
Enrichment of pathways involved in papillary and cervical cancers in module 3 members is intriguing. Given the strong bias in East Asian female populations and correlations with other female-specific cancers, lung adenocarcinoma with EGFR aberrations has very strong links with female patients of papillary and cervical cancers.
The association modules were generated on a Taiwanese training dataset, with significance determined on two external East Asian validation datasets. The inferred modules would ideally have been verified on a third East Asian dataset independent of module discovery. However, the current study was limited by the lack of a suitable external candidate data. Nevertheless, the inclusion of East Asian and Caucasian external datasets allowed for intriguing insights to be made on the population specificity of the inferred association modules.
One counter-argument against the genetic explanation for the population-specific difference is their smoking behaviors. Male lung cancer patients have a disproportionately higher smoking rate than female counterparts [60]. Thus the male-female difference may be attributed to smoking behaviors rather than genetics and sex-specific physiology. Due the lack of smoking status reports in the external datasets, we can neither rule out nor confirm this confounding factor. A larger dataset(s) covering cases of all possible combinations of gender, ethnicity and smoking behaviors is needed in order to resolve this contention.
Likewise, the present work cannot determine whether genetic or environmental differences are the causes of ethnic specificity. An ideal study to answer this question is to collect patients of European and East Asian descent in the same locales and investigate whether their samples exhibit the same ethnic-specific trends. The datasets appeared in present work do not have sufficient ethnic diversity for the proposed study. The TCGA lung adenocarcinoma data contains only 12 Asian samples, whereas all the East Asian datasets contain samples from mono-ethnic origins.
Lung adenocarcinoma is not the only cancer exhibiting population specificity. The results in this study add to this growing body of research and further highlight gender and ethnicity as predominant risk factors in predicting the survival outcomes of patients in a range of cancer types. Identification of other cancer types demonstrating specific molecular aberrations and prognostic effects in gender, race and other attributes is an important extension of the present study.
MATERIALS AND METHODS
Data sources and processing
Table 1 summarizes the information of the five datasets analyzed in the present study. The training set of 32 Taiwanese female lung adenocarcinoma patients consists of the data of transcriptomes (Affymetrix U133plus2.0 microarrays), CNV (Affymetrix SNP6.0 microarrays), and DNA methylations (Illumina Infinium). In each patient the measurements from the tumor tissue and the adjacent normal tissue were provided. We calculated the tumor/normal ratios of each data type and used them in constructing association modules. However, survival data in the training set was not included in our analysis due to the shortage of events: 28 of the 32 subjects were censored.
Validation data were identified and downloaded from open-source locations. These included the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/) and caArray (https://array.nci.nih.gov/caarray/home.action). The four validation sets consist of transcriptomic and survival data. The Japanese dataset contains the transcriptomic data of Agilent G4112F microarrays on 117 patients. The Korean dataset contains the transcriptomic data of Affymetrix U133plus2.0 microarrays on 63 patients. The US1 source is the TCGA lung adenocarcinoma (LUAD) dataset. We selected the transcriptomic data of Illumina RNAseq on 244 Caucasian patients. The US2 data also includes patients from diverse ethnic backgrounds. We selected the transcriptomic data of Affymetrix U133A on 294 Caucasian patients.
Probe information for the specific microarray platform was used to assign gene information (i.e. gene name, chromosome number, genomic location) to each probe level data. For CNV and mRNA expression data, we normalized the measurements into a compatible scale. For each probe in a microarray or each gene in an RNAseq assay, we converted the vector of observed data over a patient cohort into the vector of cumulative distribution function (CDF) values. The normalized values ranged between 0 and 1. DNA methylation data ranged in [0, 1] thus did not require normalization. The normalized value of a gene in a subject was the median over its probe values. Furthermore, the CDF value was converted into a probability vector of trinary states (up, down regulation and no change):
where y denotes the CDF value and x the hidden discrete variable of the expression (CNV) state. Derivation of the formulation is described in [42]. Following the CDF transformation, we split the patients of each transcriptome data by gender to generate separate datasets for the Japan, Korea and US studies.
The elementary subunits of CNV data are segments bounded by amplification and deletion events. To identify segmentation regions, the raw CNV data were visualized using heatmaps. We observed that CNV data were mostly coherent within each chromosomal arm. To simplify computation, a decision was therefore made to use chromosomal arms as natural segmentation boundaries. We aggregated the measurements of probes on each chromosomal arm and used the median over the corresponding probe values as the proxy CNV value of a chromosome segment.
Purity and ploidy of the tumors
We employed the ABSOLUTE algorithm [61] to estimate both ploidy and purity of samples in the training set. The inferred results are reported in S5 Table. The inferred purity indicates most samples retain a significant fraction of cancer DNA. Therefore, the association modules reflect information in cancer genomes. The inferred ploidy indicates many cancer samples undergo copy number amplifications in most spots in the genomes. The fluctuations of CNVs provide a necessary condition to build associations between CNV and mRNA data.
Module finding algorithm
We established a logistic regression model to fit the mRNA expression profile of each gene in terms of CNV and DNA methylation data. Denote y the expression of a gene and x the molecular aberrations that explain y. The conditional probability is
where fi(x)’s are scalar feature functions specifying the relations of x and y. λi’s are non-negative parameters, and Z(x) is the partition function that normalizes the conditional probabilities.
Candidate covariates (molecular aberrations) included the CNV of each chromosome and DNA methylations of selected genes with sufficient variation across the subjects. For each gene expression profile, we pre-selected a list of candidate covariates with sufficient pairwise association scores. Threshold values for incorporating associations into the model are reported in S6 Table.
We incrementally added the covariates to fit the expression data and employed a model selection procedure to balance goodness of fit and model complexity. Inclusion of covariates was prioritized with the following order: cis-acting CNV, trans-acting CNV, and DNA methylation.
After the logistic regression model of each gene expression profile was established, we grouped genes in terms of their covariates in the models. These groups of genes were the association modules. The dependent variables of gene expressions constituted the targets, while the independent variables of molecular aberrations constituted the effectors. Details about the module finding algorithm are described in our previous work [42, 43].
Validation tests
Expression coherence of target genes
The expression coherence of target genes was evaluated using the correlations amongst target gene expressions within each association module. The distribution of correlation coefficients was compared to a background of correlations of 1000 randomly selected genes from the chosen dataset. A Kolmogorov-Smirnov (KS) test was applied to examine the difference in correlations between the module and background distributions. We applied both two-sided and one-sided KS tests to identify if a deviation exists, and if so, whether it is skewed in a common direction amongst East Asian female validation datasets. To determine the significance of a module for this test, an adjusted p-value was developed to account for differences in module size. Smaller modules would tend to have larger p-values, whereas larger modules are more likely to be identified as significant. To mitigate this bias, an adjusted p-value was calculated by selecting a random group of genes of the same dimension as the test set and comparing each to the background distribution. This process was repeated 1000 times for each module, with the original p-value ranked within this set. The final position of the p-value relative to the 1000 random samples would determine the significance (with threshold p < 0.05) of the module.
Prognostic power of association modules
We validated prognostic power of association modules with both Cox regression coefficients [62] and log-rank p-values of the Kaplan-Meier curves [63]. Cox regression coefficients gauged the dependency of a patients hazard function on observed variables. Negative associations with survival times exhibit positive Cox regression coefficients. For each module, we evaluated the distribution of Cox regression coefficients of its target gene expression profiles. The prognostic power of an association module was quantified by the KS statistic of its Cox regression coefficient distribution against a background distribution of coefficients for all genes in the selected dataset. Modules with an adjusted p-value < 0.05 were selected as significant.
In addition to the distribution of Cox regression coefficients, we also intended to demonstrate that an aggregate index derived from the data of an association module could quantify its prognostic power. To fulfill this goal we used the median over the expression profiles of target genes as the aggregate biomarker. In each dataset, the patients were divided into two groups in terms of whether their aggregate biomarker values exceeded the average expression values over all genes in the data. The log-rank p-value of the Kaplan-Meier curves of the two groups was used to measure prognostic power (p < 0.1).
Validation of segment boundaries
Chromosomal arms were used as natural segmentation boundaries for the CNV data. To verify this decision, we applied the Circular Binary Segmentation algorithm [64] to identify segments of consistent CNV on the individual samples of the training data. We visually inspected the segment boundaries on individual samples to identify candidate boundary partitions across all samples. The results of the Circular Binary Segmentation are illustrated using heatmaps of the CNV data in S5 Fig.
For the majority of chromosomes we found that either no clear segment boundaries were identified, or the chromosomal arm partitions were sufficient. However, chromosomes 4, 7, 8 and 12 demonstrated evidence of additional boundaries. We therefore ran the module finding algorithm again on the new segment CNV data to identify whether using these additional boundaries would affect our final results in the study. None of the cis-acting CNV or trans-acting CNV modules for chromosomes 4, 8 and 12 passed all of the validation tests. In contrast, the chromosome 7 cis-acting CNV module was still found to be significant. The Kaplan-Meier curves for this module are presented in S6 Fig.
False-discovery rates of pairwise associations and module discovery
A large number of pairwise models were generated between effector aberrations (cis-acting CNV, trans-acting CNV and methylations), and target mRNA gene expressions. It is likely that some of these significant associations have arisen by chance in the data. To assess the proportion of such discoveries, we calculated a false-discovery rate (FDR) for each effector aberration type [65, 66]. The data was permuted for each sample, and pairwise calculations were assessed. Significant associations were identified using the thresholds previously employed. The process was repeated 100 times for each molecular aberration type, with the number of significant associations recorded for each repetition. The expected number of false discoveries was calculated over the repetitions and the FDR evaluated as follows:
[66]. We also assessed the significance of a large number of association modules using validation tests of passenger coherence and prognostic value. To assess the FDR of the validation tests, we first randomly assigned genes to modules of equal sizes as the original 44 reported in the study. The validation tests of passenger coherence and prognostic value were then applied to the randomly generated modules. The number of significant modules was recorded for each individual test, and also for how many pass all validation tests. This process was repeated over 200 runs and the expected value of false negatives calculated by averaging the significant modules over all repetitions.
FDR’s of 20% and 46% were calculated for intra-CNV and inter-CNV pairwise associations respectively. A relatively high FDR of 75% reported for the methylation pairwise associations is likely attributed to the small sample size of the data and large number of features. The validation procedures at module level were designed to introduce larger external datasets into the analysis to verify the inferred modules of the Taiwan data. An overall FDR < 0.002 indicates that we can reduce the impact of false positives from pairwise models at the module level of the analysis. The complete results of the validation FDR rates for pairwise associations and validation tests are reported in S7 Table.
Partial least square analysis of dependency between two groups of variables
Partial least squares (PLS) is a dimension reduction methodology, with certain similarities to principal component analysis (PCA). Like PCA, PLS seeks to obtain linear combinations of manifest covariates that are orthogonal to one another (i.e. components). A bilinear decomposition of the predictors X can be formed such that,
where TM = t1, t2, …, tM are the m latent variables (or scores), PM = p1, p2, …, pM are the weights (or loadings) on the components and E is a residual error term. In contrast with the global variance maximizing goal that defines PCA components (XTX), PLS attempts to maximize X and response Y covariance (i.e. XTY). For PLS, a linear combination has to be obtained for both the predictors and the response [67]. This is achieved by an iterative process to compute the optimal weight vectors to maximize covariance between X and Y. The components are ordered by their maximal covariance with the response. As such, removing weaker components from the model will often maintain explanatory power in a PLS regression.
We used PLS to analyze the dependency between the target genes of our inferred association modules. PLS allows us to gain an insight into how the modules behave as a complete set of genes, rather than limit the study to pairwise correlations only. With PLS we can summarize modules in a few latent components that maximize the covariance between two sets of target genes (considered as X and Y blocks). If we retain only the first two PLS components, we are able to plot the results on a two-dimensional ’correlation circle’ (e.g. S3 Fig). Each point on the circle demonstrates the correlation between the gene expression and the PLS component axes. The cosine of the angle between the gene locations in the plot will indicate the correlation between module members projected onto these two component axes. In addition, the orthogonal projection of the module members onto the component axes will demonstrate the respective loading on each component. We can also indicate the variance explained by one block on the other by calculating a cumulative R2 for each component retained in the model. This measure gives some indication of the dependency between target genes in the comparative modules.
Pubmed co-citation and OMIM analysis of selected genes
To find candidate effector genes on inferred association modules, we examined whether some genes were more frequently co-cited with variations of the keyword lung cancer from prior studies. We wrote a Pearl script to query the NCBI PubMed database and counted the number of publications where the keyword and the name of a gene were co-present in the text. We then sorted all the genes according to their co-citation numbers and identified the intersection of the top-ranking (5%) genes and members in each module.
Whilst providing a useful indication of more established oncogenes in the literature, the co-citation approach is susceptible to false negatives that have been more recently discovered. To relieve this problem, we used the NCBI OMIM database of cancer-related genes to search for intersection with the target genes of the three significant modules.
QIAGEN’s Ingenuity pathway analysis
A pathway enrichment was performed on the target genes of all significant modules using QIAGEN’s Ingenuity pathway analysis (IPA) (http://www.qiagen.com/ingenuity) [58]. The IPA software provides matches of functions or pathways with a significant overlap with the target genes. For each pathway, a p-value was calculated using a right tailed Fisher’s exact test to assess the likelihood that any identified overlap of genes was statistically significant (i.e. not due to random chance). As the software performs tests on a large number of pathways, it is appropriate to adjust for multiple testing and provides an adjusted p-value using the Benjamini-Hochberg method [65]. Significance was determined by p < 0.01. The results were further filtered to report only pathways with an intersection > 4 genes.
ACKNOWLEDGMENTS
We thank the advice and technical support of Ker-Chau Li, Shin-Sheng Yuan, Chen-Hsin Chen and Yuh-Chyuan Tsay from the Institute of Statistical Science, Academia Sinica.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
2. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature Reviews Genetics. 2010; 11:685–696.
3. Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative Haploinsufficiency and Triplosensitivity Drive Aneuploidy Patterns and Shape the Cancer Genome. Cell. 2013; 155:948–962.
4. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer Genome Landscapes. Science. 2013; 339:1546–1558.
5. Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318:1108–1113.
6. Kaseb AO, Shama M, Sahin IH, Nooka A, Hassabo HM, Vauthey JN, Aloia T, Abbruzzese JL, Subbiah IM, Janku F, Curley S, Hassan MM. Prognostic indicators and treatment outcome in 94 cases of fibrolamellar hepatocellular carcinoma. Oncology. 2013; 85:197–203.
7. Ng IO, Ng MM, Lai EC, Fan ST. Better survival in female patients with hepatocellular carcinoma. Possible causes from a pathologic approach. Cancer. 1995; 75:18–22.
8. Scoggins CR, Ross MI, Reintgen DS, Noyes RD, Goydos JS, Beitsch PD, Urist MM, Ariyan S, Sussman JJ, Edwards MJ, Chagpar AB, Martin RC, Stromberg AJ, et al. Gender-related differences in outcome for melanoma patients. Annals of Surgery. 2006; 243:693–698.
9. Joosse A, de Vries E, Eckel R, Nijsten T, Eggermont AM, Hölzel D, Coebergh JW, Engel J. Gender differences in melanoma survival: female patients have a decreased risk of metastasis. Journal of Investigative Dermatology. 2011; 131:719–726.
10. Gamba CS, Clarke CA, Keegan TH, Tao L, Swetter SM. Melanoma survival disadvantage in young, non-Hispanic white males compared with females. JAMA Dermatology. 2013; 149:912–920.
11. Hendifar A, Yang D, Lenz F, Lurje G, Pohl A, Lenz C, Ning Y, Zhang W, Lenz HJ. Gender disparities in metastatic colorectal cancer survival. Clinical Cancer Research. 2009; 15:6391–6397.
12. Majek O, Gondos A, Jansen L, Emrich K, Holleczek B, Katalinic A, Nennecke A, Eberle A, Brenner H. Sex differences in colorectal cancer survival: population-based analysis of 164, 996 colorectal cancer patients in Germany. PLoS ONE. 2013; 8:e68077.
13. Mungan NA, Aben KK, Schoenberg MP, Visser O, Coebergh JW, Witjes JA, Kiemeney LA. Gender differences in stage-adjusted bladder cancer survival. Urology. 2000; 55:876–880.
14. Fajkovic H, Halpern JA, Cha EK, Bahadori A, Chromecki TF, Karakiewicz PI, Breinl E, Merseburger AS, Shariat SF. Impact of gender on bladder cancer incidence, staging, and prognosis. World Journal of Urology. 2011; 29:457–463.
15. Robbins AS, Yin D, Parikh-Patel A. Differences in prognostic factors and survival among White men and Black men with prostate cancer, California, 1995–2004. American Journal of Epidemiology. 2007; 166:71–78.
16. Kimura T. East meets West: ethnic differences in prostate cancer epidemiology between East Asians and Caucasians. Chinese Journal of Cancer. 2012; 31:421–429.
17. Zahm SH, Fraumeni JF Jr. Racial, ethnic, and gender variations in cancer risk: considerations for future epidemiologic research. Environmental Health Perspectives. 1995; 103:283–286.
18. Rozek LS, Herron CM, Greenson JK, Moreno V, Capella G, Rennert G, Gruber SB. Smoking, gender, and ethnicity predict somatic BRAF mutations in colorectal cancer. Cancer Epidemiology Biomarkers & Prevention. 2010; 19:838–843.
19. Wang L, McLeod HL, Weinshilboum RM. Genomics and Drug Response. The New England Journal of Medicine. 2011; 364:1144–1153.
20. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, et al. Erlotinib in previously treated non—small-cell lung cancer. The New England Journal of Medicine. 2005; 353:123–132.
21. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500.
22. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA: A Cancer Journal for Clinicians. 2014; 64:9–29.
23. Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers - a different disease. Nature Reviews Cancer. 2007; 7:778–790.
24. Lam WK. Lung cancer in Asian women - the environment and genes. Respirology. 2005; 10:408–417.
25. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G, Yang PC. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). Journal of Thoracic Oncology. 2014; 9:154–162.
26. Zhou W, Christiani DC. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chinese Journal of Cancer. 2011; 30:287–292.
27. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Medicine. 2005; 2:e17.
28. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England Journal of Medicine. 2010; 362:2380–2388.
29. Murakami Y, Nobukuni T, Tamura K, Maruyama T, Sekiya T, Arai Y, Gomyou H, Tanigami A, Ohki M, Cabin D, Frischmeyer P, Hunt P, Reeves RH. Localization of tumor suppressor activity important in nonsmall cell lung carcinoma on chromosome 11q. Proceedings of the National Academy of Sciences. 1998; 95:8153–8158.
30. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150:1107–1120.
31. Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Research. 2012; 22:2109–2119.
32. Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, Chan WK, Li HN, Liu CC, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. The New England Journal of Medicine. 2007; 356:11–20.
33. Massion PP, Kuo WL, Stokoe D, Olshen AB, Treseler PA, Chin K, Chen C, Polikoff D, Jain AN, Pinkel D, Albertson DG, Jablons DM, Gray JW. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization implications of the phosphatidylinositol 3-kinase pathway. Cancer Research. 2002; 62:3636–3640.
34. Lu TP, Lai LC, Tsai MH, Chen PC, Hsu CP, Lee JM, Hsiao CK, Chuang EY. Integrated analyses of copy number variations and gene expression in lung adenocarcinoma. PLoS ONE. 2011; 6:e24829.
35. Tsou JA, Galler JS, Siegmund KD, Laird PW, Turla S, Cozen W, Hagen JA, Koss MN, Laird-Offringa IA. Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Molecular Cancer. 2007; 6:70.
36. Zhang Y, Wang R, Song H, Huang G, Yi J, Zheng Y, Wang J, Chen L. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Letters. 2011; 303:21–28.
37. Son JW, Jeong KJ, Jean WS, Park SY, Jheon S, Cho HM, Park CG, Lee HY, Kang J. Genome-wide combination profiling of DNA copy number and methylation for deciphering biomarkers in non-small cell lung cancer patients. Cancer Letters. 2011; 311:29–37.
38. Kim SC, Jung Y, Park J, Cho S, Seo C, Kim J, Kim P, Park J, Seo J, Kim J, Park S, Jang I, Kim N, et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS ONE. 2013; 8:e55596.
39. Chen R, Khatri P, Mazur PK, Polin M, Zheng Y, Vaka D, Hoang CD, Shrager J, Xu Y, Vicent S, Butte AJ, Sweet-Cordero EA. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Research. 2014; 74:2892–2902.
40. Botling J, Edlund K, Lohr M, Hellwig B, Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Pontén F, König A, Fernandes O, Karlsson M, et al. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clinical Cancer Research. 2013; 19:194–204.
41. Kim B, Lee HJ, Choi HY, Shin Y, Nam S, Seo G, Son DS, Jo J, Kim J, Lee J, Kim J, Kim K, Lee S. Clinical validity of the lung cancer biomarkers identified by bioinformatics analysis of public expression data. Cancer Research. 2007; 67:7431–7438.
42. Sintupisut N, Liu PL, Yeang CH. An integrative characterization of recurrent molecular aberrations in glioblastoma genomes. Nucleic Acids Research. 2013; 41:8803–8821.
43. Li SD, Tagami T, Ho YF, Yeang CH. Deciphering causal and statistical relations of molecular aberrations and gene expressions in NCI-60 cell lines. BMC Systems Biology. 2011; 5:186.
44. Cancer Genome Atlas Research Network: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068.
45. Lu TP, Tsai MH, Lee JM, Hsu CP, Chen PC, Lin CW, Shih JY, Yang PC, Hsiao CK, Lai LC, Chuang EY. Identification of a Novel Biomarker, SEMA5A, for Non—Small Cell Lung Carcinoma in Nonsmoking Women. Cancer Epidemiology Biomarkers & Prevention. 2010; 19:2590–2597.
46. Takeuchi T, Tomida S, Yatabe Y, Kosaka T, Osada H, Yanagisawa K, Mitsudomi T, Takahashi T. Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. Journal of Clinical Oncology. 2006; 24:1679–1688.
47. Lee ES, Son DS, Kim SH, Lee J, Jo J, Han J, Kim H, Lee HJ, Choi HY, Jung Y, Park M, Lim YS, Kim K, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clinical Cancer Research. 2008; 14:7397–7404.
48. Cancer Genome Atlas Research Network: Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550.
49. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nature Medicine. 2008; 14:822–827.
50. Wold S, Ruhe A, Wold H, Dunn WJ III. The collinearity problem in linear regression. The partial least squares (PLS) approach to generalized inverses. SIAM Journal on Scientific and Statistical Computing. 1984; 5:735–743.
51. Tenenhaus M, Pagès J, Ambroisine L, Guinot C. PLS methodology to study relationships between hedonic judgements and product characteristics. Food Quality and Preference. 2005; 16:315–325.
52. Stallings-Mann ML, Waldmann J, Zhang Y, Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields AP, Radisky DC. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Science Translational Medicine. 2012; 4:142ra95.
53. Akunuru S, Palumbo J, Zhai QJ, Zheng Y. Rac1 targeting suppresses human non-small cell lung adenocarcinoma cancer stem cell activity. PLoS ONE. 2011; 6:e16951.
54. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. The Lancet Oncology. 2005; 6:279–286.
55. Quintavalle M, Elia L, Price JH, Heynen-Genel S, Courtneidge SA. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Science Signaling. 2011; 4: ra9.
56. Awasthi YC, Sharma R, Yadav S, Dwivedi S, Sharma A, Awasthi S. The non-ABC drug transporter RLIP76 (RALBP-1) plays a major role in the mechanisms of drug resistance. Current Drug Metabolism. 2007; 8:315–323.
57. Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Research. 2005; 33:D514–D517.
58. IPA®, QIAGEN Redwood City.
59. Cheng YW, Chiou HL, Sheu GT, Hsieh LL, Chen JT, Chen CY, Su JM, Lee H. The association of human papillomavirus 16/18 infection with lung cancer among nonsmoking Taiwanese women. Cancer Research. 2001; 61:2799–2803.
60. Zang EA, Wynder EL. Differences in lung cancer risk between men and women: examination of the evidence. Journal of the National Cancer Institute. 1996; 88:183–192.
61. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnology. 2012; 30:413–421.
62. Cox DR, Oakes D. Analysis of Survival Data. 1984; 21.
63. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958; 53:457–481.
64. Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004; 5:557–572.
65. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological). 1995; 57:289–300.
66. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences. 2003; 100:9440–9445.
67. Phatak A, De Jong S. The geometry of partial least squares. Journal of Chemometrics. 1997; 11:311–338.