
Introduction
Giant cell tumor (GCT) of bone, which accounts for approximately 6% of all primary bone tumors [1], is characterized by extensive bone resorption [2], leading to regional pain and the predisposition to pathologic fractures [3]. Although considered as a non-cancerous tumor [4], GCT is known for potential recurrence following treatment with a reported incidence rate between 18% and 50% [5]. Histologically, GCT consists of three major cell types: multinucleated osteoclast-like giant cells, monocytic round-shaped macrophage-like cells, and spindle-shaped fibroblast-like stromal cells, also known as giant-cell tumor stromal cells (GCTSCs) [6]. Giant cell formation is thought to occur in a similar manner as osteoclastogenesis, where osteoblasts express the receptor activator of nuclear factor-kB ligand (RANKL), which stimulates its receptor, RANK, on osteoclast precursor cells and initiates their fusion into osteoclasts [7, 8]. Proliferating GCTSCs are believed to be the neoplastic component of GCT which is known to express RANKL [9] and thought to stimulate giant cell formation from RANK-expressing monocytic cells [10].
RANKL, a member of the tumor necrosis factor (TNF) family, triggers osteoclastogenesis by forming a complex with RANK, a member of the TNF receptor family [11]. RANKL binds to RANK expressed on the surface of osteoclast precursor cells and drives them to form active multinucleated osteoclasts for bone resorption during bone remodeling [4], while osteoprotegerin (OPG) competes with RANK, resulting in the inhibition of osteoclastic bone destruction [8]. A functional interaction between RANKL and RANK is necessary for osteoclast differentiation, survival, and activation. In addition, the up-regulation of RANKL-RANK signaling also plays an important role in other osteolysis-related diseases such as osteoporosis [12] and osteofibrousdysplasia [13], as well as bone metastasis [14]. Recent findings have revealed a number of transcription factors that regulate RANKL expression, such as parathyroid hormone (PTH) [15] and vitamin D3 [16]. However, as another vital process for the final protease activity, post-transcriptional regulation of RANKL remains poorly understood.
MicroRNAs (miRNAs) are a class of abundant, approximately 22 nucleotide non-coding RNAs that mediate post-transcriptional regulation of target mRNAs [17]. By binding to the 3’-untranslated regions (3’UTRs) of specific mRNAs, miRNAs repress translation or degrade target mRNAs [18]. Bioinformatics have predicted that miRNAs have the capacity to regulate about one-third of all mammalian genes [19], and numerous studies have linked aberrant miRNA expression to pathological conditions such as cancer [17]. Despite this, the function of miRNA-induced osteolysis in GCT has rarely been reported. Given the similar forming process of giant cells in GCT and osteoclasts, miRNAs that regulate osteoclastogenesis may play a key role in GCT. Emerging evidence has revealed a general necessity for miRNAs in osteoclastogenesis, knowing that ectopic expression of miR-148a and miR-503 inhibited the expression of V-maf musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB) [20] and RANK [21], resulting in the blockage of osteoclast differentiation. Our previous studies have confirmed that miR-126-5p played a role in osteoclastogenesis through targeting MMP13 and PTHrP in GCT [22, 23]. Although miRNAs in osteoclast differentiation are widely recognized, the function of miRNAs related to RANKL, which is pivotal for osteoclastogenesis, still remains elusive.
To explore the role of miRNAs during the process of osteolysis in GCT, miRNA expression changes between GCT and normal cancellous bone were compared, and miR-106b was found to be down-regulated in GCT. Furthermore, the cellular and molecular functions of miR-106b and one of its key targets, RANKL, were elucidated. Lastly, we validated these observations in vivo using a short short-term GCT model of chick chorio-allantoic membrane (CAM) and the OVX mice model, and provide data in support of targeting the miR-106b~RANKL axis in preventing giant cell formation and osteolysis.
Results
MiR-106b is down-regulated significantly in GCT
Human microarray assays of GCT samples (n = 17) and non-tumor infected cancellous bones (n = 4) were performed (Figure 1A, Supplemental Table 1). Bioinformatics analysis was applied to the data set of these differentially regulated miRNAs (Supplemental Figure 1). According to the results, we focused on miR-106b for its key role in tumor progression [24, 25] and its potential relevance to osteolysis. To validate the microarray data, we further detected the expression of miR-106b in 30 clinical GCT tissues and 30 normal cancellous bone tissues (Table 1, Supplemental Table 1) and the results were consistent with those of miRNA microarray (Figure 1B). The results of fluorescence in situ hybridization (FISH) further confirmed that the expression of miR-106b was down-regulated in GCT of bone (Figure 1C, 1D). Moreover, we isolated GCTSCs and bone marrow mesenchymal stem cells (BMSCs) from part of the GCT patients and detected the level of miR-106b. The result showed that the level of miR-106b in GCTSCs was significantly lower than it in BMSCs (Figure 1E, 1F).
Table 1: Characteristics of the 30 GCT patients
GCT of bone patients |
|
Age (years) |
33.2±10.9 |
Sex (male/femal) |
12/18 |
Disease history (months) |
25.4±34.9 |
Tumor size (cm) |
7.4±2.8 |
Tumor site (spine/limbs) |
29/1 |
Primary/recurrence |
17/13 |
Rseection (total/subtotal) |
24/6 |
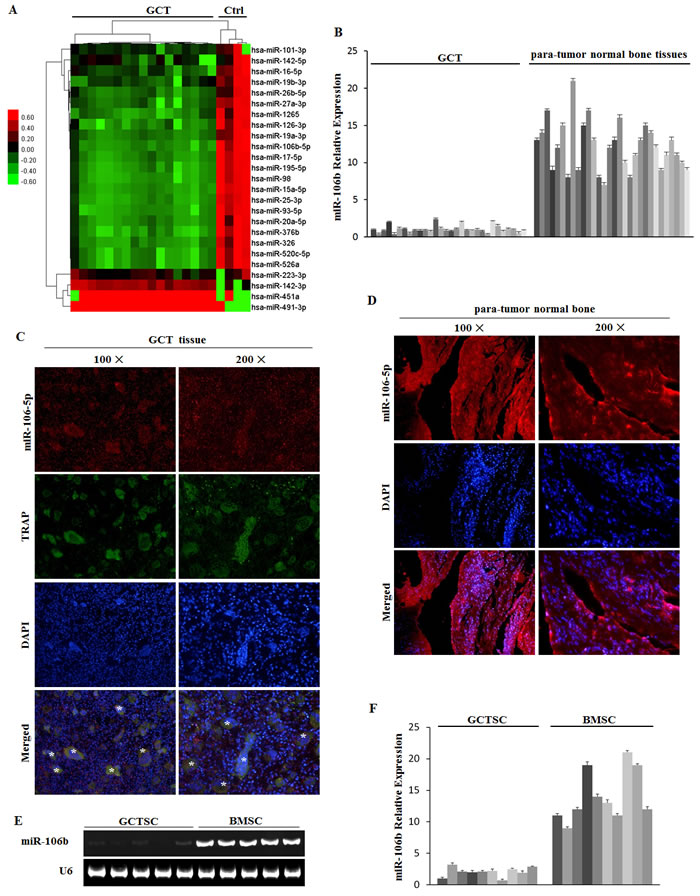
Figure 1: microRNA regulation in GCT tissue. A. Microarray assays in GCT and normal bone tissues. B. qRT-PCR measurement of miR-106b levels in tumor and normal bone tissues from the 30 GCT patients. C. MiRNA-106b (red) and TRAP (green) detected by FISH and IF in GCT specimens. D. MiRNA-106b (red) detected by FISH in para-tumor normal bone tissue specimens. E. PCR assay of miR-106b was performed in GCTSCs and BMSCs. F. The levels of miR-106b in GCTSCs and BMSCs were detected by qRT-PCR assay.
The target genes of miR-106b are overexpressed in GCT tissues
The above bioinformatics analysis also identified that some cytokines involved in osteolysis were possible target genes of miR-106b, including RANKL, IL-8, MMP2 and TWIST. Additionally, literature search convinced the regulation of IL-8, MMP2 and TWIST by miR-106b in other tumors [26-28], and their overexpression in GCT has already been confirmed [29, 30]. Western blot assay in our study confirmed that RANKL was up-regulated in GCT (Figure 2A). Using qRT-PCR assay, mRNA levels of RANKL, RANK, IL-8, MMP2 and TWIST in GCT tissues were confirmed to be evaluated in GCT compared with those in cancellous bones (Figure 2B-2F). To further investigate the distribution of these cytokines, immunohistochemistry was performed on paraffin-embedded clinical GCT samples. The results showed that RANKL, MMP2 and TWIST were predominantly localized in GCTSCs, while RANK and IL-8 were expressed both in GCTSCs and in giant cells (Figure 2G, 2H). Meanwhile, the expression of OPG, an important inhibitor of RANKL function, had no clear difference between GCT and control tissues (Supplemental Figure 2A).
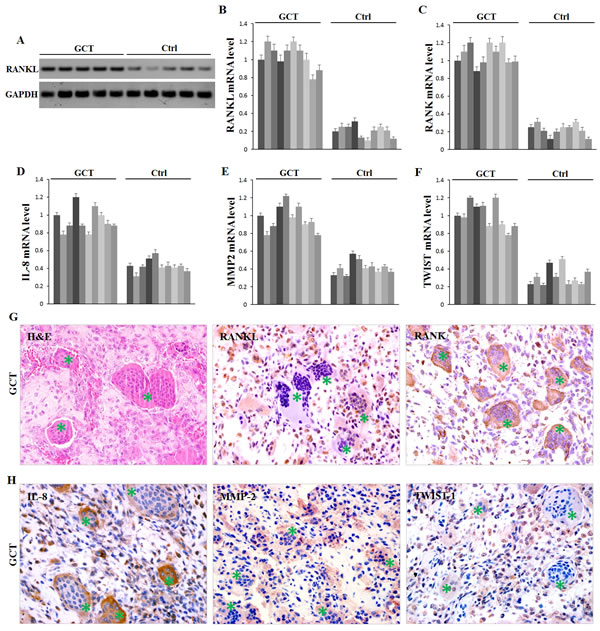
Figure 2: Expression of possible target proteins of miR-106b in GCT tissue. A. Western Blot measurement of RANKL protein. B.-F. qRT-PCR measurement of mRNA levels of RANKL, RANK, IL-8, MMP2 and TWIST1. G., H. H&E staining and IHC staining of RANKL, RANK, IL-8, MMP2 and TWIST1 in human GCT tissue specimens. Giant cells are marked with asterisks.
MiR-106b directly targets RANKL
It has been demonstrated that miRNA acts primarily through binding to the 3’untranslated regions (3’UTRs) of mRNA resulting in degradation and translation repression [17]. We explicitly predicted that the binding site of miR-106b was in 3’UTR of RANKL mRNA and examined it by luciferase assay. A luciferase reporter construct containing the 3’UTR of RANKL was generated and mutations were introduced into the predicted binding site (Figure 3A). The luciferase expression vectors containing the 3’UTR of RANKL (WT) or the mutation sequences (MUT) with agomiR-106b were introduced into HEK293 cells and the effect of miR-106b was measured on luciferase translation by the level of luciferase enzyme activity. The results showed that the luciferase activity of the RANKL 3’UTR reporter gene was significantly suppressed in WT group after being stimulated by agomiR-106b, while mutation abolished this repression, confirming that miR-106b could suppress RANKL expression by binding to this site of the 3’UTR (Figure 3B).
To identify the action of miR-106b on RANKL, IL-8, MMP2 and TWIST we transfected GCTSCs and MG63 cells, a cell line of osteosarcoma known to express these cytokines [31, 32], with agomiR-106b or antagomiR-106b and measured the mRNA and protein levels of RANKL by qRT-PCR, Western blot and ELISA. Relative to the controls, RANKL protein levels were reduced significantly after agomir transfection in both cell types, while RANKL mRNA levels showed a similar tendency (Figure 3C-3E). However, the mRNA levels exhibited less fluctuant as compared with the protein levels. By using the qRT-PCR assay, we found that the mRNA levels of IL-8, MMP2 and TWIST exhibited analogous changes after agomiR-106b and antagomiR-106b transfection (Figure 3F-3H), while the mRNA level of OPG showed no clear fluctuation after the transfection (Supplemental Figure 2B). These results suggest that miR-106b could down-regulate RANKL expression by interacting with 3’UTRs binding site of RANKL, and also inhibit MMP2, IL-8 and TWIST expression.
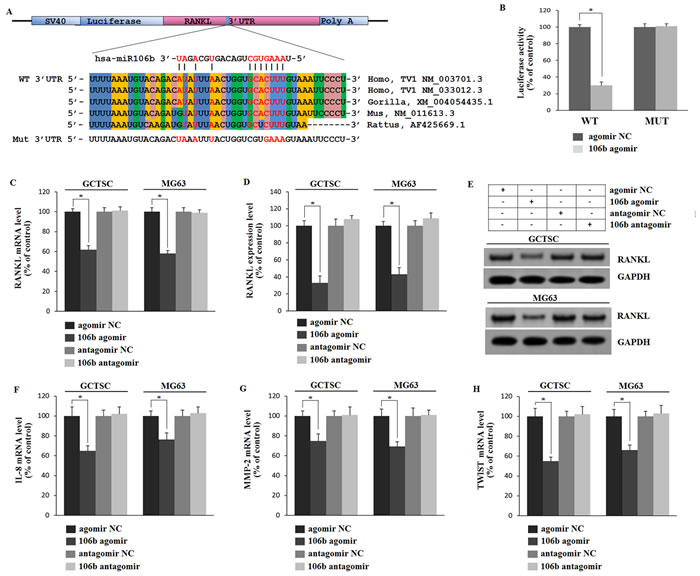
Figure 3: Mir-106b directly targets RANKL and regulates IL-8, MMP2 and TWIST. A. Prediction of the miR-106b target site in RANKL 3’UTR in humans and animals. Schematic diagram of miR-106b with WT and MUT 3’UTR regions of RANKL are shown in complementary pairing. Mutated nucleotides are labeled. B. Determination of the effect of agomir-Control, agomiR-106b on luciferase activity in HEK293 cells transfected with either WT-RANKL 3′UTR reporter or mutant RANKL 3′ UTR reporter. Firefly luciferase values are normalized for Renilla luciferase. C. qRT-PCR analysis of RANKL mRNA levels in GCTSCs and MG63 cells treated with agomiR-106b or antagomiR-106b and their corresponding negative controls. D., E. ELISA and Western blot analysis of RANKL protein level. F.-H. qRT-PCR analysis of the mRNA levels of MMP2, IL-8 and TWIST. *P < 0.05.
MiR-106b regulates osteoclastogenesis through targeting RANKL, IL-8, MMP2 and TWIST
To gain a more comprehensive understanding of the regulatory role of miR-106b in RANKL-RANK signaling and osteolysis, we established the stable cell lines (OE-miR-106b and OE-control) using TALENs targeting the PPP1R12C (the AAVS1 locus), which is considered to have no relevance to a known pathophysiology [33], and corresponding donor plasmids bearing homologous sequences. Briefly, GCTSCs were transfected with two TANEN vectors in conjunction with a targeting vector containing the EGFP gene and DNA fragments of pri-miR-106b (Figure 4A). Clones with a successful recombination in EGFP-Pri-miR-106b and EGFP-Pri-miR-106b-mutant were identified by genomic PCR and restriction digestion (Figure 4B-4D). By using qRT-PCR assay, we confirmed that OE-miR-106b cells expressed much higher level of miR-106b than OE-control cells (Figure 5A). Meanwhile, the mRNA and protein levels of RANKL were both down-regulated markedly in OE-miR-106b GCTSCs (Figure 5B-5D). Furthermore, the mRNA levels of IL-8, MMP2 and TWIST also exhibited opposite changes as compared with miR-106b expression (Figure 5E-5G), while no obvious change of OPG mRNA level was observed (Supplemental Figure 2C).
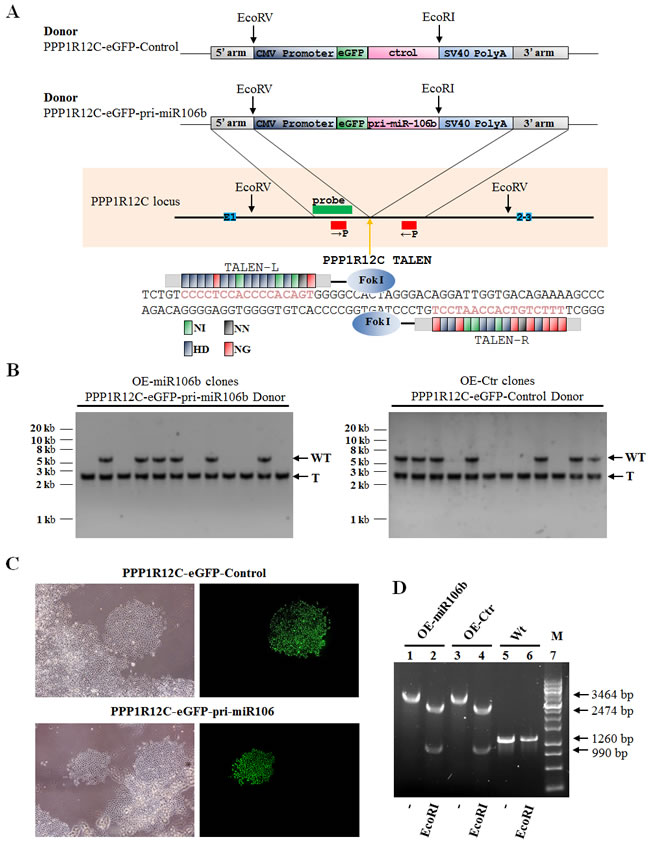
Figure 4: Genetic engineering of GCTSCs using TALENs. A. Schematic overview depicting the targeting strategy for PPP1R12C. Primers are shown as red boxes; southern blot probes as green box; exons as blue boxes. The orange arrow indicates the cut site by the TALENs. Donor plasmids: CMV Promoter,human cytomegalovirus (CMV) immediate early promoter gene; eGFP, enhanced green fluorescent protein gene; Pri-miR-106b, Pri-miR106b gene; Control,Pri-miR106 binding site mutant gene; SV40 Poly A, SV40 early mRNA polyadenylation signal gene. Below, scheme of PPP1R12C TALENs and their recognition sequence. TALE repeat domains are colored to indicate the identity of the repeat variable diresidue (RVD); each RVD is related to the cognate targeted DNA base by the following code (NI = A, HD = C, NN = G, NG = T). B. Southern blot analysis of OE-miR106b (Overexpression miR-106b) and OE-control (Overexpression miR-106b mutant) GCTSCs targeted using PPP1R12C TALENs and the PPP1R12C-eGFP-pri-miR106b or PPP1R12C-eGFP-Control donor plasmids. Genomic DNA was digested with EcoRV and hybridized with an32P-labeled probe (left). The probe detects a 5.4 kb WT and a 2.6 kb targeted fragment. WT, wild type; T, correctly targeted allele. C. Phase contrast images and eGFP fluorescence ofPPP1R12C-eGFP-pri-miR106b or PPP1R12C-eGFP-Control targeted GCTSCs clones. D. Genomic PCR and restriction digestion characterization of OE-miR106b and OE-control GCTSCs.
To investigate the role of miR-106b in osteolysis, we selected BMMs with M-CSF stimulation as in the vitro osteoclast (OC) differentiation model. OE-miR-106b and OE-control GCTSCs were used as different stimuli during bone resorption and osteoclast precursor differentiation. The two groups of BMMs were cultured with conditional medium containing M-CFS (10 ng/mL) from OE-miR-106b or OE-Control GCTSCs respectively. Seven days after, BMMs treated with the conditional medium of OE-miR-106b GCTSCs exhibited much less formation of tartrate resistant acid phosphatase (TRAP)-positive multinucleated osteoclasts as compared with the control (Figure 5H, 5I). TRAP activity was also inhibited in OE-miR-106b group (Figure 5K). Actin ring formation assays, another obvious characteristic of mature osteoclasts during osteoclastogenesis and a prerequisite for osteoclast bone resorption [34], similarly supported that the overexpression of miR-106b in GCTSCs suppressed the osteoclastogenesis (Figure 5J). In addition, the number and area of pits on the surface of the dentin slices were markedly decreased in OE-miR-106b conditional mediums group (Figure 5L, 5M). These results suggest that miR-106b played a negative role in osteoclastogenesis, and RANKL, IL-8, MMP2 and TWIST were the possible targets of miR-106b during this process.
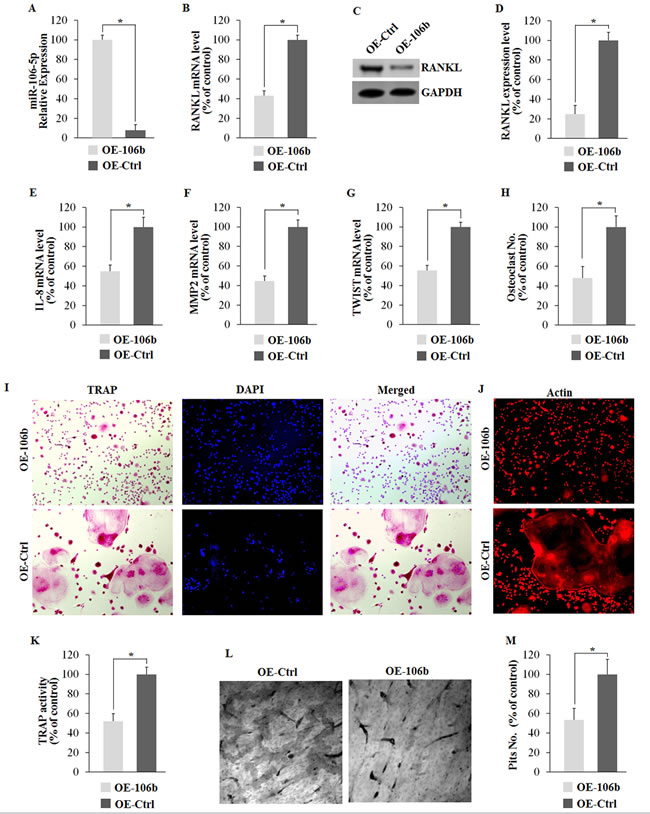
Figure 5: Overexpression of miR-106b suppresses its target genes and osteoclastogenesis in vitro. A. qRT-PCR analysis of miR-106b level 48 h after transfection. B. qRT-PCR analysis of mRNA level of RANKL. C., D. Western blot and ELISA analysis of RANKL protein level. E.-G. qRT-PCR analysis of mRNA levels of IL-8, MMP2 and TWIST. (H-M) Mouse BMMs were seeded on dentin slices or empty wells and cultured with conditional medium containing MCFS (10 ng/mL) from OE-miR106b or OE-miR106b-Control GCTSCs for 7 days. H. The number of TRAP positive osteoclasts per well. I.TRAP staining of BMM cells. J. Phalloidin-Rhodamine staining of BMM cells. K. TRAP activity assay of BMM cells. L. Dentine slices stained with Mayer’s hematoxylin after removal of cells. M. The number of pits per dentine slice. *P < 0.05.
Mir-106b regulates the expression of RANKL and giant cell formation in the GCT model of chick CAM
Due to the limited proliferation ability and complex composition of GCT, it is difficult to an in vivo experiment of GCT. In fact, the only animal model reported before was a short-term GCT model of chick CAM [35]. We used this animal model to detect the role of miR-106b in GCT in vivo. Tumor cells were injected into the CAM, while agomiR-106b or PBS was injected every two days. After six days, tumors were picked out for H&E staining, TRAP staining or RANKL immunohistochemical staining. The result showed that eggs with agomiR-106b had significantly fewer TRAP+ giant cells as compared with the controls (Figure 6A, 6B). Meanwhile, RANKL expression was clearly lower in tumors injected with agomiR-106b as compared with the controls (Figure 6C).
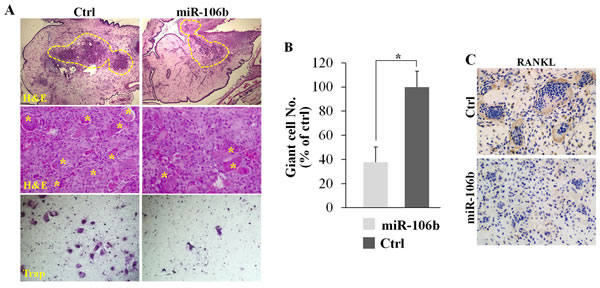
Figure 6: MiR-106b inhibits giant cell formation and the expression of RANKL in GCT in vivo. Re-suspended tumor suspensions were deposited into the CAM of eggs and the tumors were picked out for further experiments at day 16. A. H&E staining and TRAP staining of the tumors. B. The number of TRAP+ giant cells per field. C. IHC staining of RANKL of the tumors. *P < 0.05.
MiR-106b regulates bone resorption in OVX mice
To further investigate the in vivo effect of miR-106b in osteolysis, we selected ovariectomy (OVX) mice to established the animal model, due to the limited proliferation ability and complex composition led to the difficulty of establishing GCT of bone animal models, and the estrogen loss frequently promotes activation of osteoclastic bone resorption [36]. Two months after OVX operation, mice were given agomiR-106b or antagomiR-106b or their controls via a single tail vein injection (Figure 7A). Northern blotting and qRT-PCR showed that after injection, the inhibition or overexpression of miR-106b in bone lasted for at least 3 weeks in coccygeal vertebrae, while the expression of miR-106b in mice that underwent sham operation remained at a relatively high level (Figure 7B, 7C). Six weeks after the first injection, all mice were euthanized and samples were collected for further analysis.
Western Blot was used to measure the protein levels of RANKL, MMP2 and TWIST in the coccygeal vertebrae of the mice. The results showed that the protein levels of ANKL, MMP2 and TWIST were decreased significantly in the antagomiR-106b-treated mice, and increased significantly in the agomiR-106b-treated mice (Figure 7D). The mRNA levels of RANKL, TWIST and MMP2 were also detected by qRT-PCR, and the results showed that the mice treated with agomiR-106b had the minimum levels while mice treated with antagomiR-106b had the maximum levels of these mRNAs among the OVX mice (Figure 7E-7G).
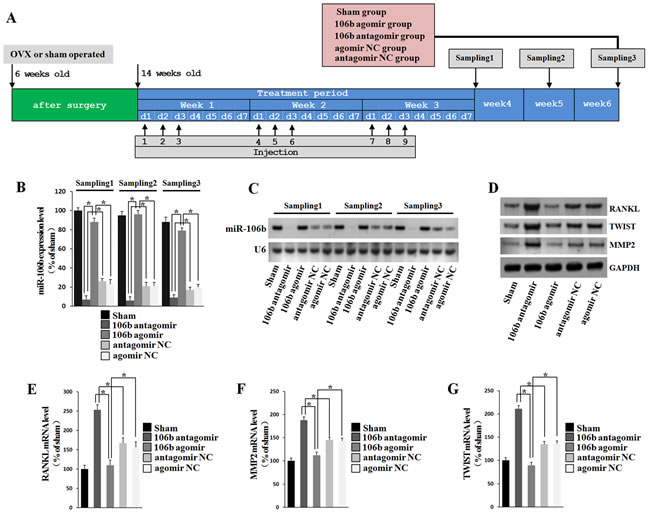
Figure 7: RANKL, MMP2 and TWIST levels are negatively correlated withmiR-106b level in mice. A. Mice were subjected to OVX operation and rest for two months, and then injected with agomiR-106b, agomirNC, antagomiR-106b or antagomir NC. MiR-106b expression was analyzed by Northern blot at 4, 5 and 6 weeks after the first injection. B., C. miR-106b level was measured using qRT-PCR and Northern blot. The result showed that the variation of miR-106b after injection could sustain for at least 6 weeks. D. Western blot determination of protein levels of RANKL, MMP2 and TWIST in bone. E.-G. qRT-PCR determination of mRNA levels of RANKL, MMP2 and TWIST in bone. *P < 0.05.
MicroCT was performed to detect the quantification of the bone volume/tissue volume ratio (BV/TV), bone mineral density (BMD), trabecular number (Tb.N), trabecular spacing (Tb.Sp) and trabecular thickness (Tb.Th). The results showed that agomiR-106b-treated mice exhibited a significant decrease in femur BMD in comparison with mice treated with con-agomiR-106b, while the BMD of the antagomiR-106b-treated mice presented an obvious increase compared with that of the mice injected mut-antagomiR-106b (Figure 8A, 8B). There was no significant difference in BMD between the two control groups (con-agomir and mut-antagomir). Similarly, overexpression of miR-106b increased BV/TV (Figure 8C) and other bone parameters, including Tb.N, Tb.Sp and Tb.Th (Supplemental Figure 3A-3C), while the absence of miR-106b decreased these parameters.
Histomorphometric analysis was performed to assess the bone resorption parameters, including the number of osteoclasts per bone perimeter (N.Oc/B.Pm) and osteoclast surface per bone surface (Oc.S/BS). The results showed that bone resorption was increased significantly in OVX mice. AgomiR-106b decreased while antagomiR-106b increased N.Oc/B.Pm and Oc.S/BS in the OVX mice (Supplemental Figure 3D, 3E).
We next measured the osteoclast activity by TRAP staining in the mouse skull. The result showed that osteoclasts were significantly activated in antagomiR-106b-injected mice (Figure 8D). By TRAP staining for osteoclasts in bone section from distal femora of mice, we foung that antagomiR-106b increased while agomir-106b decreased osteoclast formation (Figure 8E). In addition, the levels of osteoclast activity markers such as TRAP and NFATc1 mRNA in bone tissue were also elevated in antagomir group, and decreased in agomir group (Figure 8F, 8G). TRAP activity changed similarly in serum (Supplemental Figure 3F). All these results suggest that miR-106b inhibits osteolysis and osteoclastogenesis by targeting RANKL, MMP2 and TWIST in vivo.
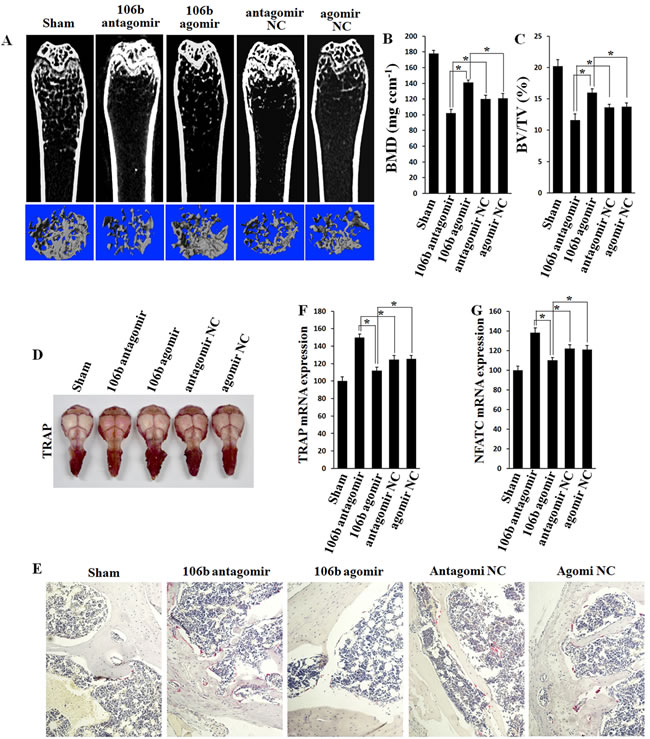
Figure 8: Bone mass is increased and bone resorption is decreased in agomiR-106b treated mice, and the effect of antagomiR-106b is opposite. A. Micro-CT section and 3D trabecular architecture of distal and middle femoral diaphysis in each group 6 weeks after the first injection. B., C. BMD and BV/TV of the femur measured on the results of micro-CT. D. TRAP staining of the mouse skull: the deeper the red color, the more the number of osteoclasts. E. Light micrographs of TRAP staining performed on trabecular bone section from tibiae. F., G. qRT-PCR analysis of TRAP and NFATc1 mRNA levels in bone. *P < 0.05.
Discussion
Giant cell tumor of bone presents with numerous osteoclast-like multinucleated giant cells that are principally responsible for the extensive bone resorption by the tumor [2]. However, GCTSCs were considered as the neoplastic components expressing RANKL and other cytokines that induce giant cell differentiation [9]. Here, we demonstrated that miR-106b was down-regulated in GCT and directly targeted RANKL and suppressed the expression of IL-8, MMP2 and TWIST. By overexpressing miR-106b using TALENs, osteoclastogenesis and osteolysis induced by GCTSCs were significantly suppressed in vitro. In addition, by using the GCT model of chick CAM, we found that miR-106b could suppress the expression of RANKL and giant cell formation in vivo. We further showed that a single miRNA could negatively dictate osteoclast differentiation and bone resorption by targeting RANKL, MMP2 and TWIST in the OVX mice model. Therefore, we conformed that miR-106b is a suppressor of osteolysis both in vitro and in vivo.
MiR-106b, located at Chr7, is a member of miR-106b-25 cluster [37]. It is known to regulate stem cell proliferation and neuronal differentiation, causing tumor formation or Alzheimer’s disease [37, 38]. Other studies demonstrated that ectopic expression of miR-106b inhibited the expression of P21, RB and Smad3 participated in the regulation of G1-to-S cell cycle transition, thereby leading to excessive proliferation and tumor development [37, 39, 40]. Additionally, miR-106b was reported to induce epithelial-mesenchymal transition (EMT), which is essential for tumor metastasis, by suppressing RUNX3 [41]. Consistent with these mechanisms, miR-106b overexpression has been detected in breast, prostate, gastric, lung and renal cancers [25, 37, 40-42]. However, we found that miR-106b was significantly decreased in GCT. Similarly, Slaby et al [42] demonstrated that miR-106b level was decreased in metastatic renal cell carcinoma and considered a potential predictive marker of early metastasis after nephrectomy. Ni et al [26] also reported a negative correlation between breast cancer bone metastasis and miR-106b levels in orthotopic tumor tissue. Nonetheless, the function of miR-106b decrease in these tumors has not been well defined. Our data indicate that declined miR-106b levels could enhance osteoclast differentiation and bone resorption by targeting RANKL, TWIST and MMP2, which might partly explain the role of miR-106b down-regulation in GCT and bone metastasis.
Previous studies have reported the evidence of direct regulation of IL-8 [27], MMP2 [26] and TWIST [28] by miR-106b in other tumors. In addition, some other studies showed that IL-8, MMP2 and TWIST could promote RANKL production, leading to osteoclast differentiation [26, 43-45]. However, there is still no study reporting the role of regulation of miR-106b in osteoclastogenesis or osteolysis. In the present study, we for the first time demonstrated that miR-106b could suppress IL-8, MMP2 and TWIST expression in GCTSCs and further inhibit bone resorption.
RANKL-induced activation of the NF-kB pathways is essential for osteoclast differentiation [46]. RANKL produced by tumor cells or the bone marrow microenvironment in response to tumor cells played important roles in activation of osteoclastic bone resorption in bone tumors and bone metastases [48]. In bone marrow microenvironment, RANKL was mainly secreted by osteoblast, a cell differentiated from mesenchymal stem cells (MSC), while in GCT, it was mainly produced by GCTSC which also derived from MSC. A few miRNAs has been confirmed to play key roles in osteoblasts [49, 50]. However, the role of miRNAs in GCTSCs has rarely been reported. Our study exhibited the decrease of miR-106b in GCTSCs and indentified RANKL as a new target of miR-106b. In addition, we revealed the binding site of miR-106b in the 3’UTR of RANKL mRNA, and further demonstrated the decrease of RANKL expression induced by over-expression miR-106b in GCTSCs, leading to the inhibition of osteoclastogenesis or osteolysis.
Inhibition of bone resorption has been confirmed to be an effective therapeutic strategy to reduce recurrence of GCT [51]. Bisphosphonate has been widely used in the clinical treatment of GCT for its protective effect against osteolysis [51, 52]. Recently, denosumab, a monoclonal antibody for RANKL, was considered an effective therapeutic option for GCT patients in clinical study [53]. Our results showed an inhibiting effect of miR-106b to osteoclastogenesis or osteolysis in vitro. By using the GCT model of chick CAM, we found that miR-106b could suppress RANKL expression and giant cell formation in vivo. Due to the limitation of no GCT of bone animal model, we chose OVX model to detect the role of miR-106b during the process of osteolysis. It was found that agomiR-106b could partly recover the osteoporosis induced by OVX via suppressing the expression of RANKL, while antagomiR-106b aggravated osteoporosis. Thus, our study revealed that miR-106b might be a new potential therapeutic target of GCT for its inhibiting effect to osteolysis via targeting RANKL.
In the OVX mice experiments, we also found that miR-106b was significantly down-regulated in this postmenopausal osteoporosis mice model compared with it in mice underwent sham operation, which has never been reported before. RANKL/RANK axis plays a crucial role in osteoporosis and offers important therapeutic targets [54], such as denosumab in the clinical treatment of osteoporosis via targeting RANKL [55]. Our data showed that miR-106b could negatively regulate RANKL in OVX mice and partly inhibit osteoporosis. These results indicate that miR-106b may also play a key role in postmenopausal osteoporosis and be a new possible diagnostic and therapeutic target in this disease, though further investigation is necessary.
In conclusion, our results showed that miR-106b was down-regulated in GCT of bone and negatively regulated the expression of RANKL, IL-8, MMP2 and TWIST, resulting in the inhibition of osteoclastogenesis and osteolysis both in vitro and in vivo. These results suggest that miR-106b may be a driver of bone resorption via targeting multiple genes including RANKL, and may provide a new possible approach to the diagnosis and treatment of GCT and other diseases associated with bone destruction, such as postmenopausal osteoporosis and bone metastasis.
Materials and Methods
Clinical samples
The study population comprised 30 Chinese GCT patients aged 12-56 years who underwent resection for primary GCT in our hospital between July 2011 and July 2012. Patients with metastases and those with hepatopathy, nephropathy, diabetes mellitus, hematologic diseases, rheumatoid arthritis, and diseases of the thyroid or parathyroid were excluded from the analysis, knowing that these conditions may affect bone metabolism. Patients who received radiotherapy/chemotherapy, or used bone-modifyingagents such as zoledronic acidbefore surgery were also excluded. Primary GCT tissues were isolated from tumor samples derived from tumor resections. The non-tumor infected cancellous bones from the same GCT patients were used as normal controls. The tissues were snap-frozen and stored in liquid nitrogen within two hours after surgical excision. The clinical study was approved by the Ethnic Committee of Changzheng Hospital of the Second Military Medical University (Shanghai, China), and written informed consent was obtained from all participants.
miRNA microarray assay
Total RNA was extracted from the GCT tissue (n = 17) and cancellous bone (n = 4) using Trizol (Invitrogen, Carlsbad, CA, USA). Microarrays were performed by utilizing the miRCURY LNA™ microRNA Array (Version 14.0 Exiqon, Vedbaek,Denmark). After RNA measurement on the NanoDrop2000 (Thermo Scientific, Wilmington, USA) instrument, the samples were labeled using the miRCURY™ Array Power Labeling kit (Cat #208032-A, Exiqon) and hybridized on the miRCURY LNA™ Array (v.14.0). After the labeling procedure, the RNA sample was mixed pair wise and hybridized to the mercury LNA™ Array version 14.0 (Exiqon). The hybridization and subsequent wash steps were performed according to the miRCURY LNA™ array manual. The microarray slides were scanned using the Axon Gene Pix 4000B scanner (Axon Instruments, USA) and image analysis was carried out using the Gene Pix pro V6.0 software (Axon Instruments, USA). Statistical comparisons were performed by analysis of variance (ANOVA).
Cell lines and culture
For primary cell culture, bone marrow-derived macrophages (BMM) isolated from C57/BL6 mice and GCTSCs isolated from GCT samples were cultured as described previously [4]. BMSCs were isolated from bone marrow of the GCT patients as described previously [56]. This had been approved by the Ethnic Committee of Changzheng Hospital of the Second Military Medical University and specimens were taken with patients’ written consent. HEK293 (ATCC, Manassas VA) and MG63 (ATCC, Manassas VA) cells were maintained in DMEM (GIBCO) supplemented with 10% fetal bovine serum (HyClone) in the cell incubator (37 °C, 5% CO2). BMM and GCTS cells were maintained in α-MEM (GIBCO) supplemented with 10% fetal bovine serum.
qRT-PCR for mRNA and miRNA analysis
qRT-PCR for mRNA was performed by iTaq™ Universal SYBRGreen (Bio-Rad Laboratories, CA, USA) on 7900HT Fast Real-Time PCR System (Life Technologies Corporation, USA). All PCR primers are listed in Supplemental Table 2. The expression of miRNA-106b and U6 was examined by TaqManmiRNA Assay system (Life Technologies Corporation, USA).
In situ hybridization (ISH) and immunofluorescence (IF)
ISH was performed using the miR-106b locked nucleic acid probe (5′-digoxigenin-ATCTGCACTGTCAGCACTTTA-3′-digoxigenin) and the microRNA ISH Optimi-zation Kit (Exiqon, Vedbaek, Denmark) according to the manufacturer’s instructions, as described previously [23]. The signals of IF were examined with a BX51 fluorescence microscope (Olympus).
Western blot and ELISA
For western blot, the antibodies against RANKL (ab124797) were purchased from Abcam. The antibodies against IL-8 (sc-73321), MMP2 (sc-53630) and TWIST (sc-15393) were purchased from Santa (Santa Cruz, USA). ELISA assay was performed with Uscn (Uscn Life Science Inc., Wuhan, China) ELISA kit for human RANKL (SCA855Hu) following the manufacturer’s instructions.
Histology and immunohistochemistry
The tissue sections were stained with hematoxylin-eosin and by immunohistochemistry using an indirect immunoperoxidase technique, with the antibodies against RANKL, IL-8, MMP2, TWIST, RANK (sc-34249, Santa Cruz, USA) and OPG (sc-8468, Santa Cruz, USA). Results were analyzed by standard light microscopy.
Plasmids
RANKL 3’-UTR constructs were PCR amplified using cDNA encoding RANKL (NM_033012.3) as templates, and subcloned into pGL3-Basic vector using In-Fusion™ advantage PCR cloning Kit (Clontech, USA) for Luciferase reporter gene assay. The construction of the RANKL 3’-UTR mutant was done by PCR using the Quickchange™ site directed mutagenesis kit from Qiagen (Qiagen, USA).
Transfection of agomiR-106b and antagomiR-106b
The agomiR-106b, antagomiR-106b and negative controls of miR-106b were purchased from RiboBio (RiboBio, Guangzhou, China). Cells were transfected with agomiR-106b, antagomiR-106b and negative controls at a final concentration of 50 nM. The FuGENE HD transfection agent (Promega, USA) was used according to the manufacturer’s instructions.
Luciferase reporter assay
Dual luciferase assays were conducted in a 24 well plate format. pGL-RANKL 3’ UTR report / pGL-RANKL 3’ UTR Mutant report + TK100 Renilla report were transfected into 70% confluent HEK293 cells, along with agomiR-106b, antagomiR-106b (antago-miR-106b) or each control. After 48-h transfection, firefly and renilla luciferase were quantified sequentially using the Dual Luciferase Assay kit (Promega, USA) following the manufacturer’s recommendations.
Construction of stable cell lines using TALENs
The plasmid of TALENs targeting the PPP1R12C (the AAVS1 locus) was constructed using Fast TALETM TALEN Assembly kit from SiDanSai biotechnology (SiDanSai, China). The PPP1R12C locus homologous sequences were PCR amplified using genomic DNA extracted from HEK293 cells as template, and cloned in pEASY-T1 vector(TransGen, China) as Donor-1. The following primer sets were used: forward, 5’-ATGCCGTCTTCACTCGCTGGGTT-3’; reverse,5’-CTCCTGGGCTTGCCAAGGACTCAA-3’. Then, Donor-HindIII mutant vector was constructed using Donor-1 vector as template by PCR, and using Quickchangesite directed mutagenesis kit using following primer sets: forward, 5’-GGCCACTAGGGACAAGCTTGGTGACAGAA-3’; reverse, 5’-GCTTGTCCCTAGTGGCCCCACTGTGG-3’. After that, human cytomegalovirus (CMV) immediate early promoter gene, enhanced green fluorescent protein (EGFP) gene, pri-mir106 gene, SV40 early mRNA polyadenylation signal gene were inserted into Hind III site from Donor-Hind III vector in turn. All clones were constructed using In-Fusion TM advantage PCR cloning Kit (Clontech, USA).
The two TALENs and corresponding Donor plasmids were transfected into GCTSCs. After selection with puromycin, resistant colonies with green fluorescence were picked up, and examined by genomic PCR (the following primers were used: forward, 5’-ATGCCGTCTTCACTCGCTGGGTT-3’; reverse, 5’-CTCCTGGGCTTGCCAAGGACTCAA-3’.) and digestion with EcoRI restriction enzyme.
Sourthern blot
Each sample of total DNA was extracted from OE-106b and OE-Ctrl cells using the Puregene kit (Qiagen, Valencia, CA). Approximately 10 μg of total DNA were digested with restriction endonucleases EcoRV and EcoRI for 8 h at 37 °C. DNAs were denatured and separated by electrophoresis on a 0.8% agarose gel and transferred onto BioBond Plus nylon membrane (Sigma, USA). Blots were hybridized with a α32P dCTP-(3000 Ci/mmol) (PerkinElmer) labeled probe corresponding to the 411 bp left arm of PPP1R12C locus DNA generated with the Random Primed DNA Labeling Kit (Roche, Mannheim, Germany). Hybridizations were performed overnight at 50 °C.
TRAP staining assays, actin ring-formation and osteoclastogenesis assays
For TRAP staining, cells were fixed and stained using the TRAP activity kit (Sigma, USA). TRAP-positive multinucleated cells containing three or more nuclei were counted as mature osteoclasts. For actin ring formation assay, cultured BMM cells were first fixed with 4% PFA in PBS for 10 min, permeabilized with 0.1% Triton-X 100 in PBS for 5 min, and then incubated with rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR, USA). The ring structure of F-actin dots that indicates osteoclastogenesis was observed under a fluorescent microscope [34]. For osteoclastogenesis assay, BMMs were seeded on a dentin slice and cultured with the conditional mediumcontaining MCFS (10 ng/mL) for 7 days, with the medium changed every 2 days.
The chick chorio-allantoic membrane (CAM) assay
A short-term model of GCT in vivo was set up in the chick CAM as described previously [35]. Fertilized white leghorn chicken eggs (Valo-SPF eggs, LohmannTierzucht GmbH, Cuxhaven, Germany) were incubated at a humidity of 70% and 37°C. At embryonic day 3, 2-3 ml albumen was removed with a syringe. After 10-day incubation, small plastic rings made of Thermanox™ cover discs were placed on the CAM. After gentle laceration of the CAM surface, 20 μl re-suspended tumor suspension with 2nmol agomiR-106b or PBS was deposited into the rings (8 eggs per group). After 24h suspension grafting, a solid tumor became apparent. Then 2nmol agomiR-106b or PBS were injected into the tumor every two days. Until day 16, CAMs were measured and collected for further analysis. All embryos that died before day 16 were excluded from further analysis.
Mice
Thirty-six female WT C57BL/6 mice aged 6 weeks were equally randomized to 6 groups, of which 5 groups underwent OVX operation, and the remaining group underwent sham operation. Two months after operation, the 5 OVX groups of mice respectively received 10nmol/per mouse of agomiR-106b, con-agomiR-106b, mut-antagomiR-106b, antagomiR-106b or 0.2ml PBS through the tail vein on day 1-3 for 3 consecutive weeks. A section of coccygeal vertebrae were get out from all the mice before injection and at 3, 4.5 and 6 weeks after the first injection (Figure 7A). Six weeks after the first injection, mice were euthanized. Bone and serum samples were collected for further experiments. The agomir, con-agomir, mut-antagomir and antagomir were all purchased from RiboBio (RiboBio, Guangzhou, China). All procedures involving the mice were approved by the Animal Management Committeeof the Second Military Medical University.
Northern blot
20μg RNA was loaded onto 15% ureapolyacrylamide gel with 0.5×Tris borate-EDTA for electrophoresis. Then separated RNA was transferred to a Hybond-N+ nylon membrane (Amersham Biosciences) using a semidry transfer cell (Bio-Rad). Hybridization was performed according to a standard protocol. P-Labeled oligonucleotide probes complementary to the mature miR-106b and U6 were used in hybridization. The blots were processed using Typhoon™ FLA 7000 biomolecular imager.
Micro-CT analysis
Bone samples were removed and the distal left femur of each mouse was scanned by microCT (SCANCO viva 4.0). The Image-Pro+ 7.0 and SCANCO microCT software packages were used to analyze BMD and several structural parameters of the proximal femur, including BV/TV, Tb.Th, Tb.N, and Tb.Sp.
Statistics
SPSS 19.0 statistical software (SPSS Inc., Chicago, IL) was used for statistical analysis. All data are presented as mean ± standard error of the mean (SEM). Statistics were assessed using both Student t test and ANOVA, assuming double-sided independent variance. All experiments were repeated at least three times, and representative experiments are shown. P values of < 0.05 were considered statistically significant.
Acknowledgments
This work was jointly supported by grants from the National Natural Science Foundation of China (81201556, 811330059, 81372874, 81402222, 81401355), Shanghai R&D public service platform Foundation (12DZ2295103) and China Postdoctoral Science Foundation (43302).
Conflicts of interest
The authors indicate no potential conflict of interest.
References
1. Hoch B, Inwards C, Sundaram M, Rosenberg AE. Multicentric giant cell tumor of bone: clinicopathologic analysis of thirty cases. J Bone Joint Surg Am. 2006; 88: 1998–2008.
2. Cowan R W, Singh G. Giant cell tumour of bone: A basic science perspective. Bone. 2013; 52: 238–246.
3. Wu Z, Yang X, Xiao J, Feng D, Huang Q, Zheng W, Huang W, Zhou Z. Aneurysmal bone cyst secondary to giant cell tumor of the mobile spine: a report of 11 cases. Spine (Phila Pa 1976). 2011; 36: E1385–1390.
4. Ng PK, Tsui SK, Lau CP, Wong CH, Wong WH, Huang L, Kumta SM. CCAAT/enhancer binding protein beta is up-regulated in giant cell tumor of bone and regulates RANKL expression. J Cell Biochem. 2010; 110: 438–446.
5. ArbeitsgemeinschaftKnochentumoren, Becker WT, Dohle J, Bernd L, Braun A, Cserhati M, Enderle A, Hovy L, Matejovsky Z, Szendroi M, Trieb K, Tunn PU. Local recurrence of giant cell tumor of bone after intralesional treatment with and without adjuvant therapy. J Bone Joint Surg Am. 2008; 90: 1060–1067.
6. Branstetter DG, Nelson SD, Manivel JC, Blay JY, Chawla S, Thomas DM, Jun S, Jacobs I. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. 2012; 18: 4415–4424.
7. AsagiriM, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007; 40: 251–264.
8. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch BiochemBiophys. 2008; 473: 139–146.
9. Atkins GJ, Haynes DR, Graves SE, Evdokiou A, Hay S, Bouralexis S, Findlay DM. Expression of osteoclast differentiation signals by stromal elements of giant cell tumors. J Bone Miner Res. 2000; 15: 640–649.
10. Atkins GJ, Kostakis P, Vincent C, Farrugia AN, Houchins JP, Findlay DM, Evdokiou A, Zannettino AC. RANK expression as a cell surface marker of human osteoclast precursors in peripheral blood, bone marrow, and giant celltumors of bone. J Bone Miner Res. 2006; 21: 1339–1349.
11. William C, Dougall. Molecular Pathways: Osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer Res. 2012; 18: 326–335.
12. To TT, Witten PE, Renn J, Bhattacharya D, Huysseune A, Winkler C. Rankl-induced osteoclastogenesis leads to loss of mineralization in a medaka osteoporosis model. Development. 2012; 139: 141–150.
13. Taylor RM, Kashima TG, Ferguson DJ, Szuhai K, Hogendoorn PC, Athanasou NA. Analysis of stromal cells in osteofibrous dysplasia and adamantinoma of long bones. Modern Pathol. 2012; 25: 56–64.
14. Pardeep Bhatia, M. Melinda Sanders, Marc F. Hansen. Expression of receptor activator of nuclear factor-KB ligand is inversely correlated with metastatic phenotype in breast carcinoma. Clin Cancer Res. 2005; 11: 162–165.
15. Fu Q, Jilka RL, Manolagas SC, O’Brien CA. Parathyroid hormone stimulates receptor activator of NF kappa B ligand and inhibits osteoprotegerin expression viaprotein kinase A activation of cAMP response element-binding protein. J Biol Chem. 2002; 277: 48868–48875.
16. Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone-related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res. 2002; 17: 1667–1679.
17. Croce C M. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009; 10: 704–714.
18. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011; 12: 99–110.
19. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010; 11: 252–263.
20. Cheng P, Chen C, He HB, Hu R, Zhou HD, Xie H, Zhu W, Dai RC, Wu XP, Liao EY, Luo XH. MiR-148a regulates osteoclastogenesis via targeting V-maf musculoaponeurotic fibrosarcoma oncogene homolog B. J Bone Miner Res. 2013, 28:1180-1190.
21. Chen C, Cheng P, Xie H, Zhou HD, Wu XP, Liao EY, Luo XH. MiR-503 regulates osteoclastogenesis via targeting RANK. J Bone Miner Res. 2014; 29: 338–347.
22. Wu Z, Yin H, Liu T, Yan W, Li Z, Chen J, Chen H, Wang T, Jiang Z, Zhou W, Xiao J. MiR-126-5p regulates osteoclast differentiation and bone resorption in giant cell tumor through inhibition of MMP-13. Biochem Biophys Res Commun. 2013; 443: 944–949.
23. Zhou W, Yin H, Wang T, Liu T, Li Z, Yan W, Song D, Chen H, Chen J, Xu W, Yang X, Wu Z, Xiao J. MiR-126-5p regulates osteolysis formation and stromal cell proliferation in giant cell tumor through inhibition of PTHrP. Bone. 2014; 66: 267–276.
24. Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, Ford HL. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene. 2012; 31: 5162–5171.
25. Hudson RS, Yi M, Esposito D, Glynn SA, Starks AM, Yang Y, Schetter AJ, Watkins SK, Hurwitz AA, Dorsey TH, Stephens RM, Croce CM, Ambs S. MicroRNA-106b-25 cluster expression is associated with early disease recurrence and targets caspase-7 and focal adhesionin human prostate cancer. Oncogene. 2013; 32: 4139–4147.
26. Ni X, Xia T, Zhao Y, Zhou W, Wu N, Liu X, Ding Q, Zha X, Sha J, Wang S. Downregulation of miR-106b induced breast cancer cell invasion and motility in association with overexpression of matrix metalloproteinase 2. Cancer Sci. 2013; 105: 18–25.
27. Chuang TD, Luo X, Panda H, Chegini N. MiR-93/106b and their host gene, MCM7, are differentially expressed in leiomyomas and functionally target F3 and IL-8. Mol Endocrinol. 2012; 26: 1028–1042.
28. Dong P, Kaneuchi M, Watari H, Sudo S, Sakuragi N. MicroRNA-106b modulates epithelial–mesenchymal transition by targeting TWIST in invasive endometrial cancer cell lines. Mol Carcinog. 2014; 53: 349–359.
29. Singh S, Mak IW, Cowan RW, Turcotte R, Singh G, Ghert M. The role of TWIST as a regulator in giant cell tumor of bone. J Cell Biochem. 2011; 112: 2287–2295.
30. Nishimura M, Yuasa K, Mori K, Miyamoto N, Ito M, Tsurudome M, Nishio M, Kawano M, Komada H, Uchida A, Ito Y. Cytological properties of stromal cells derived from giant cell tumor of bone (GCTSC) which can induce osteoclast formation of human blood monocytes without cell to cell contact. J Orthop Res. 2005; 23: 979–987.
31. Wu J, Liao Q, He H, Zhong D, Yin K. TWIST interacts with β-catenin signaling on osteosarcoma cell survival against cisplatin. Mol Carcinog. 2012; 53: 440–446.
32. Ho YC, Yang SF, Huang FM, Chang YC. Up-regulation of osteolyticmediators inhuman osteosarcoma cells stimulated with nicotine. J Periodontal Res. 2009; 44: 760–766.
33. Smith JR, Maguire S, Davis LA, Alexander M, Yang F, Chandran S, ffrench-Constant C, Pedersen RA. Robust, Persistent Transgene Expression in Human Embryonic Stem Cells Is Achieved with AAVS1-Targeted Integration. Stem Cells. 2008; 26: 496–504
34. Koide M, Kinugawa S, Ninomiya T, Mizoguchi T, Yamashita T, Maeda K, Yasuda H, Kobayashi Y, Nakamura H, Takahashi N, Udagawa N. Diphenylhydantoin inhibits osteoclast differentiation and function through suppression of NFATc1 signaling. J Bone Miner Res. 2009; 24: 1469–1480.
35. Maurice Balke, Anna Neumann, KárolySzuhai, Konstantin Agelopoulos, Christian August, Georg Gosheger, Pancras CW Hogendoorn, Nick Athanasou, Horst Buerger, Martin Hagedorn. A short-term in vivo model for giant cell tumor of bone. BMC cancer. 2011; 11: 241.
36. Ominsky MS, Li X, Asuncion FJ, Barrero M, Warmington KS, Dwyer D, Stolina M, Geng Z, Grisanti M, Tan HL, Corbin T, McCabe J, Simonet WS, Ke HZ, Kostenuik PJ. RANKL inhibition with osteoprotegerin increases bone strength by improving cortical and trabecular bone architecture in ovariectomized rats. J Bone Miner Res. 2008; 23: 672–682.
37. KeminCai, Yu Wang, XueliBao. MiR-106b promotes cell proliferation via targeting RB in laryngeal carcinoma. J Exp Clin Canc Res. 2011; 30: 73–78.
38. Wang H, Liu J, Zong Y, Xu Y, Deng W, Zhu H, Liu Y, Ma C, Huang L, Zhang L, Qin C. MiR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer’s disease targets TGF-beta type II receptor. Brain Res. 2010; 1357: 166–174.
39. Zhang W, Edwards A, Fan W, Flemington EK, Zhang K. miRNA-mRNA correlation-network modules in human prostate cancer and the differences between primary and metastatic tumor subtypes. PLoS One. 2012; 7: e40130.
40. Kan T, Sato F, Ito T, Matsumura N, David S, Cheng Y, Agarwal R, Paun BC, Jin Z, Olaru AV, Selaru FM, Hamilton JP, Yang J, Abraham JM, Mori Y, Meltzer SJ. The miR-106b-25 polycistron, activated by genomic amplification, functions as an oncogene by suppressing p21 and Bim. Gastroenterology. 2009; 136: 1689–1700.
41. Xu Y, Wang K, Gao W, Zhang C, Huang F, Wen S, Wang B. MicroRNA-106b regulates the tumor suppressor RUNX3 in laryngeal carcinoma cells. FEBS Lett. 2013; 587: 3166–3174.
42. Slaby O, Jancovicova J, Lakomy R, Svoboda M, Poprach A, Fabian P, Kren L, Michalek J, Vyzula R. Expression of miRNA-106b in conventional renal cell carcinoma is a potential marker for prediction of early metastasis after nephrectomy. J Exp Clin Canc Res. 2010; 29: 90–96.
43. Hwang YS, Lee SK, Park KK, Chung WY. Secretion of IL-6 and IL-8 from lysophosphatidic acid-stimulated oral squamous cell carcinoma promotes osteoclastogenesis and bone resorption. Oral Oncol. 2012; 48: 40–48.
44. Yuen HF, Kwok WK, Chan KK, Chua CW, Chan YP, Chu YY, Wong YC, Wang X, Chan KW. TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis. 2008; 29: 1509–1518.
45. Kitazawa R, Mori K, Yamaguchi A, Kondo T, Kitazawa S. Modulation of mouse RANKL gene expression by Runx2 and vitamin D3. J Cell Biochem. 2008; 105: 1289–1297.
46. Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006; 12: 17-25.
47. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003; 3: 537–549.
48. Roodman G D. Genes associate with abnormal bone cell activity in bone metastasis. Cancer Metastasis Rev. 2012; 31: 569–578.
49. Li H, Xie H, Liu W, Hu R, Huang B, Tan YF, Xu K, Sheng ZF, Zhou HD, Wu XP, Luo XH. A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J Clin Invest. 2009, 119: 3666-3677.
50. Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY, Xia ZY, Zhou HD, Cao X, Xie H, Liao EY, Luo XH. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest. 2015; 125:1509-22.
51. Xu W, Li X, Huang W, Wang Y, Han S, Chen S, Xu L, Yang X, Liu T, Xiao J. Factors affecting prognosis of patients with giant cell tumors of the mobile spine: retrospective analysis of 102 patients in a single center. Ann Surg Oncol. 2013; 20: 804–810.
52. Yin H, Yang X, Xu W, Li B, Li B, Wang T, Meng T, Wang P, Liu T, Zhou W, Xiao J. Treatment and outcome of primary aggressive giant cell tumor in the spine. Eur Spine J. 2015; DOI 10.1007/s00586-015-3777-5.
53. Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, Roudier M, Smith J, Ye Z, Sohn W, Dansey R, Jun S. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010; 11: 275–280.
54. Jules J, Ashley JW, Feng X. Selective targeting of RANK signaling pathways as new therapeutic strategies for osteoporosis. Expert Opin Ther Targets. 2010; 14: 923–934.
55. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011; 377: 1276–1287.
56. Leyh M, Seitz A, Dürselen L, Schaumburger J, Ignatius A, Grifka J, Grässel S. Subchondral bone influences chondrogenic differentiation and collagen production of human bone marrow-derived mesenchymal stem cells and articular chondrocytes. Arthritis Res Ther. 2014; 16: 453.