
INTRODUCTION
Liver cancer is the fifth common malignancy and the second leading cause of cancer-related mortalities in the world [1]. Characterized with high rate of recurrence and metastasis, low detection rate for the curable stages and ineffective therapeutic options, liver cancer, a majority of which is hepatocellular carcinoma, is accepted as a cancer with poor prognosis. Statistics indicate that survival rate of patients after hepatectomy is 30% to 40% at 5 years [2]. Thus, there is an urgent need for research to improve our ability to diagnose, prevent, and treat this disease. Animal models are viewed as crucial tools in the study of liver cancer. Because of the physiologic and genetic similarities between rodents and humans, the short lifespan, the breeding capacity and the variety of manipulating methods, mice are often used for cancer research. While a wide range of liver cancer mouse models are currently available, the universality of each model is limited for several reasons. First, the etiology of liver cancer is rather complicated. Unlike the incidence of lung cancer, which can be reduced by limiting exposure to tobacco smoke, there are many risk factors related with liver cancer, including virus infection [3], chemical carcinogens exposure [4], alcohol abuse [5], or food contaminated with Aspergillus flavus fungus [6]. Different etiologic factors will affect different sets of target genes which result in the genetic heterogeneity of liver cancer and, therefore, make mouse models of liver cancer lack of universality. Second, mouse models can provide researchers with the opportunity to mimic the complex multistep process of liver carcinogenesis, assess tumor-host interactions, perform drug screening, and conduct various therapeutic experiments. However, no model is ideal for all purpose. Each kind of the mouse model can only recapitulate hepatogenesis in some respects. Third, other factors, such as the high requirements on instruments and lack of financial support, also limit the applications of some costly mouse models.
Owing to these facts, a knowledgeable selection should be made according to the specific situation investigators come across in their liver cancer researches. In this review, we assemble and evaluate both the currently used mouse models and the emerging ones related to liver cancer, hoping it would be helpful for researchers to make a choice.
IMPLANTATION MODELS OF HCC
Characterized by the short modeling period, the relatively lower cost and the suitability in the evaluation of various methods to treat HCC, implantation models, which can be established either by direct implant of tumor tissue fragments or by inoculation of HCC cell lines in recipient mice, have become the most widely used mouse models in current HCC researches [7–12]. According to where the grafts are implanted, implantation models can be divided into two kinds: ectopic models or orthotopic models. Besides, investigators also classify implantation models as allograft models and xenograft models based on whether the grafts and the hosts are from the same species. Since 1969, when the first evidence of human tumor growth in immunodepressed mice has been published [13], human tumor xenografts models have become the priority in preclinical studies. However, in some situations, allograft models may become a better option or even the only option. In this section, we will give out an elaborate description of xenografts models and enumerate some special cases when allograft models are needed.
Subcutaneous xenograft model
Subcutaneous xenograft model is the most commonly used implantation model in the study of HCC. In this model, human HCC cells or tumor tissue fragments are implanted subcutaneously (usually in the flanks) into immunodeficient mice including nude mice, severe combined immune deficient (SCID) mice or nonobese diabetic-severe combined immunodeficiency disease (NOD/SCID) mice. Subcutaneous xenograft model is often applied to testing the HCC inhibited factors (including new drugs or changes in gene expression). As for new drugs study, administration method could be intraperitoneal, intravenous or intratumoral injection. While intratumoral delivery is of limited value for most tumor types, transcatheter arterial chemoembolization (TACE) recommended by HCC clinical practice guidelines allows directly delivery of drugs or oligos into liver tumor cells. Therefore, intratumoral therapy with tumor suppressor may be feasible for HCC treatment.
Being more easily to access and manipulate, subcutaneous xenograft model established by HCC cell lines are preferred by researches compared with by human tumor tissue fragments. For example, to exam the therapeutic effect of cholesterol-conjugated 2′-O-methyl-modified microRNA-375 mimics (Chol-miR-375), we inoculated 4 × 106 HepG2 cells subcutaneously into the flanks of BALB/c athymic nude mice [14]. When tumor size reached approximately 100 mm3, Chol-miR-375 was injected directly into the implanted tumor. By monitored the tumor volume every four days, we demonstrated that Chol-miR-375 can significantly suppress the growth of hepatoma xenografts [14]. Besides, subcutaneous xenograft model may also be used to test the tumor suppressed effect induced by the expression alternation of certain gene. For instance, we once employed 2 × 106 SK-HEP-1 cells which were pre-infected with lentivirus-mediated-anti-miR-221 (miR-221(D)-LV) to establish subcutaneous tumors in nude mice, and found that depletion of miR-221 renders SK-HEP-1 cells less efficient in establishing tumors in vivo [15]. Furthermore, we assessed the inhibited effects of miR-221 on subcutaneous xenografts pre-established with 2 × 106 SK-HEP-1 and found that only a single intra-tumor injection with 2 × 108 TU miR-221(D)-LV reduces the growth of tumors [15]. At present, there are many human HCC cell lines which are available commercially. In general, the number of implanted cells is between 106 and 5 × 106 and the tumor formation rate is nearly 100%. As shown in Table 1, we summarized the key information (including the mouse strain, cell lines, cell number, calculation formula and the point to start the therapy) of some frequently used subcutaneous xenograft models in HCC research.
Table 1: Subcutaneous xenograft models for HCC
Mouse strain |
Cell lines |
Total volume |
Cell number |
Calculation formula |
Point to start the therapy |
References |
Female SCID mice |
PLC/PRF/5 |
200 ul |
5 × 106 |
½W2L |
tumor weight = 140 mg–160 mg |
[127] |
Balb/c nude mice(6–8 weeks) |
SK-Hep |
100 ul |
5 × 106 |
½W2L |
tumor volume = 50 mm3 |
[128] |
Balb/c nude mice |
HepG2 |
100 ul |
106 |
½W2L |
tumor volume = 200–300 mm3 |
[129] |
Female BALB/c nude mice(4–5 weeks) |
BEL7404 |
2 × 106 |
½W2L |
tumor volume = 100–150 mm3 |
[130] |
|
Balb/c nude mice |
QGY-7703 |
106 |
(π/6)W2L |
tumor volume = 100 mm3 |
[131] |
|
Male BALB/C nude mice(4–5 weeks) |
HCCLM6 |
100 ul |
3 × 106 |
[132] |
||
Male NCr athymic mice(5–7 weeks) |
Huh7 |
100 ul |
106 |
0.52 W2L |
tumor volume = 200–300 mm3 |
[133] |
Male athymic nude mice |
PLC/PRF/5 |
100 ul |
106 |
0.52 W2L |
tumor volume = 60–150 mm3 |
[20] |
Male BALB/c nude mice |
Huh7 |
100 ul |
5 × 106 |
½W2L |
tumor volume = 60–150 mm3 |
[134] |
Male BALB/c nude mice |
Hep3B |
100 ul |
5 × 106 |
½W2L |
tumor volume = 60–150 mm3 |
[134] |
Female BALB/c nude mice |
SMMC-7721 |
107 |
½W2L |
tumor volume = 100–150 mm3 |
[135] |
W, width; L, length.
On the other hand, as tumor tissue fragments retain the characters of human HCC more completely, the results tested on animal models which are established with human tumor tissues may be more reliable. Hu et al. [16] transplanted tumor tissue from each patient to 5 nude mice and assigned them to 5 experimental groups. In their experiment, each group contained 10 nude mice bearing human tumors and represented 10 patients. After the tumor xenografts had grown in size to a diameter of approximately 6–10 mm, they were used to test the antitumor effect of specific Hsp70 expression in cancer tissues combined with cytokine-induced killer (CIK) cells. The results showed that Hsp70 and CIK cells worked synergistically and had a significant inhibitory effect against the growth of HCC xenografts derived from all the 10 patients [16].
Usually, tumor volume (V) is estimated by measuring the length (L) and width (W) with calipers and calculating with the formula V= 1/2(L × W2) every a few days. Ge et al. [17], however, monitored the tumor volume by in vivo fluorescence imaging system. This method reflects tumor growth more accurately, yet would be more costly at the same time.
Being simple to establish and relatively easy to monitor the size of the tumor, subcutaneous xenograft model is widely used in most experiments aiming to discover some potential tumor suppressed factors. However, its major disadvantage is the lack of interaction between tumors and liver tissues. This is of particular concern because absence of tumor-host relationship may lead to abundance of false-positive responses with drugs. In addition, disruption of microenvironment may affect the biological behavior of malignant cells. For example, spontaneous metastasis rarely occurs when HCC cells are subcutaneously implanted, meanwhile they do metastasize when they are orthotopically implanted [18].
Orthotopic xenograft model
There are two approaches to establish the orthotopic xenograft model. One is called intrahepatic implantation model. Tumor fragments from patients or subcutaneous xenograft model are cut into 1 × 1 × 1 mm3 sized pieces and implanted in the liver of immunodeficient mice [19]. The other, which employed more commonly, is established by injecting tumor cells, always suspended in a volume of 10–20 ul of a serum-free medium containing 50% matrigel, directly into the left hepatic lobe of the mouse [20]. The number of cells needed in orthotopic xenograft model is also about 106 to 5 × 106. And, about one week later, liver tumors will form orthotopically (see Table 2). In some situations, liver tumor in the orthotopic model may metastasis. As the metastasis potential of each HCC cell line is different, the time needed for lung metastases to emerge is distinct. In general, MHCC97H, HCCLM3, and HCCLM6 are accepted as cell lines with high metastatic potential [21]. Yet, by alter the expression level of certain genes, the invasion and lung metastasis capacity of some low metastatic potential cell lines can be promoted. For example, Xia et al. [22] up-regulated Forkhead Box C1 (FoxC1) in SMMC7721 cells. Ten weeks after orthotopic implantation, bioluminescence molecular imaging (BLI) showed the presence of lung metastases in the mice implanted with SMMC7721-FoxC1 cells and the absence of metastasis in the control group [22].
Table 2: Orthotopic xenograft models for HCC
Mouse strain |
Cell lines |
Total volume |
Cell number |
Tumor formation time |
References |
Athymic nude mice |
PLC/PRF/5 |
20 ul |
106 |
1 wk |
[20] |
Male BALB/c nude mice |
Huh7 |
2 × 106 |
1 wk |
[134] |
|
Male BALB/c nude mice (6 weeks) |
Huh7 |
50 ul |
106 |
[136] |
|
Male BALB/c nude mice (4-5 weeks) |
Hep3B |
30–50 ul |
2 × 106 |
2 wk |
[23] |
Male nude mice |
HepG2 |
25 ul |
5 × 105 |
10 d |
[137] |
Male nude mice |
SMMC7721 |
5 × 105 |
[26] |
||
Male BALB/c nude mice |
QGY-7703 |
25 ul |
2 × 106 |
[138] |
|
Male nude mice |
HCC97L |
30 ul |
2 × 106 |
[139] |
The orthotopic xenograft model is superior to the subcutaneous xenograft model in terms of replicating the tumor microenvironment. However, its major defect is that the tumor volume cannot be measured directly unless the mice are sacrificed. To solve this problem, Yao et al. [23] employed the human HCC cell line Hep3B, which is featured by bearing the genome of hepatitis B and producing α-fetoprotein (AFP). In their experiment, they found that AFP was barely detectable in normal mice while increased dramatically and correlated directly with Hep3B tumor growth. Thus, AFP offers a minimally invasive method of monitoring Hep3B tumor progression [23]. In addition, there are also some non-invasive examinations, such as magnetic resonance imaging (MRI) [24] or even positron emission tomography (PET) [25], which can be used for the real-time and non-invasive monitoring of orthotopic HCC progression and metastasis. Optical molecular imaging, including BLI and fluorescence imaging (FLI), is another important technique developed in recent years. In general, tumors can be detected by bioluminescence molecular imaging one week after the orthotopic model has been established [20, 26]. At the same time, it is sensitive to monitor metastases in lung. Being rapid, high-throughput and straightforward as well as easily accessible with much lower instrumentation costs as compared to PET or MRI, optical molecular imaging has been valued as a favorable tool for the study of HCC once it emerged [27]. However, as liver lies in the abdomen, the poor spatial resolution caused by tissue scatter, which means that researchers cannot pinpoint the precise location of the liver tumor, could be a fatal weakness. Fortunately, information about the precise location is not so important in most experiments. These invasive or non-invasive techniques mentioned above make it possible to monitoring the growth of a tumor. Yet, they are either technically complex or instrumentally expensive. Besides, there are some other defects which would also restrict the wide application of orthotopic xenograft model. One is the possibility that inadvertent tumor cells may seed along the needle track or into the bloodstream. The other is that orthotopic xenograft model is technically more challenging compared with the ectopic model.
Allograft model
In recent decades, accumulating studies reveal that immunotherapy may act as a potentially beneficial option for HCC patients, especially for those who suffer from tumor recurrence [28]. In these cases, immunodeficient mice cannot be used and allograft models which are established by implanting murine HCC cell lines or murine tumor fragments in immunocompetent mice (not necessarily syngeneic) become crucial. As the most appropriate subjects for immunotherapy are patients with relapsed HCC, investigators usually inject tumor cells at a low dose from portal vein [29, 30] or splenic vein [7]. To mimic tumor metastasis, Avella et al. [7] developed an allograft model through seeding of tumorigenic hepatocytes (5 × 105 cells) from Simian Virus 40 T-antigen (SV40 T-Ag) transgenic MTD2 mice into the livers of C57BL/6 mice by intrasplenic injection. Then, they tested the efficacy of sunitinib combined with adoptive transfer of tumor antigen-specific CD8+T cells using this model. They found that sunitinib provide immune-enhancing effects by interrupting signal transducer and activator of transcription 3 (STAT3) signaling and regulating the function of distinct immune cells. Sunitinib together with adoptive transfer of tumor antigen-specific CD8+ T cells led to elimination of established tumors without recurrence [7]. Hepatic stellate cells (HSCs) have long been considered to contribute to the occurrence and development of HCC. In order to study its immunosuppressive properties, Zhao et al. [31] established a model by injecting a mixture of 1 × 106 mouse hepatoma cells (H22) and 2 × 105 HSCs directly in the liver of BALB/c mice. They found that the activated HSCs promoted HCC growth not only by inducing tumor angiogenesis and lymphangiogenesis, but also by significantly increasing the suppressive immune cell population of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in the spleen, bone marrow, and tumor tissues of the tumor-bearing mice. This finding provides a new concept in adjuvant immunotherapy. In order to modify the existing orthotopic allograft model to a more reliable one which is accordant to HCC patients with liver fibrosis, Kornek et al. generated liver fibrotic mice by intraperitoneal injection of thioacetamide and oral intake of alcohol. Then, orthotopic allograft model was established in these mice [32, 33]. Subsequently, they found that tumors in fibrotic livers grew significantly larger and more rapidly than those in normal liver. In addition, they even had the capacity to metastasize and form satellite nodules [34].
Allograft model provide us with another choice in some specific research fields. However, as murine liver tumors may differ from human liver tumors, it is doubtful whether the real situation in clinic will agree with those experiment results.
Xenograft model for liver cancer stem cells (CSCs)-mediated drug resistance
HCC patients even with resection often have a high frequency of recurrence. Unfortunately, systemic chemotherapy is often offered with limited success for those inoperable patients. The reason is that these anticancer therapies mainly kill rapidly growing differentiated tumor cells, thus reducing tumor mass. However, CSCs are resistant to current anti-cancer therapies. They will be left behind and result in relapse of therapy-resistant and more aggressive tumors. For this reason, a mouse model for screening selective agents which can target and eradicate CSCs is needed. To generate such a model, liver CSCs isolated from human HCC fragments or human HCC cell lines [35] will be implanted ectopically or orthotopically in immunodeficient mice. Tumors generated in this way show no major histological differences from the original patient’s tumors and, most importantly, drug responsiveness of these tumors better correlates with clinical outcome [36, 37]. This characteristic facilitates the applying of CSCs xenograft models in the liver cancer research. On the other hand, subcutaneous implantation models established by HCC cell lines can also be used in the study of CSCs-mediated drug resistance. Lee et al. [38] establish a HCC nude mouse model with highly chemoresistant MHCCLM3 cells, and treated with either lupeol, large dose of cisplatin and doxorubicin or lupeol plus small dose of cisplatin and doxorubicin. The result showed that lupeol exerted a synergistic effect with low-dose chemotherapeutic drugs. In the following research, they found that lupeol can down-regulate CD133 expression and sensitize HCC cells to chemotherapeutic agents through the phosphatase and tensin homolog (PTEN)-Akt-ABCG2 pathway [38]. The former is a significant marker for hepatic CSCs [39], the latter plays a crucial role in the self-renewal and chemoresistance of tumor-initiating cells [40, 41]. Haraguchi et al. [42] established subcutaneous xenograft models by injecting Huh7 or PLC/PRF/5 into NOD/SCID mice. Then, they demonstrated that combination of a CD13 inhibitor (ubenimex) and the genotoxic chemotherapeutic fluorouracil (5-FU) drastically reduced tumor volume compared with either agent alone. 5-FU inhibited CD90+ proliferating CSCs, some of which produce CD13+ semiquiescent CSCs, while CD13 inhibition suppressed the self-renewing and tumor-initiating ability of dormant CSCs. Therefore, combining a CD13 inhibitor with a reactive oxygen species (ROS)-inducing chemo/radiation therapy may improve the treatment of liver cancer [42]. The discovery of CSCs-mediated drug resistance brings new vision to the treatment of HCC. Yet, identification and functional characterization of CSCs have been mainly performed in cultured cell lines rather than in vivo [43]. Additional validation studies are needed by using animal models, primary tumor specimens and circulating blood cells, which will provide further clinical relevance to support the exploration of CSC knowledge in HCC clinical diagnosis and prognosis.
GENETICALLY ENGINEERED MOUSE MODELS (GEM) OF LIVER CANCER
Development of transgenic and gene targeting technologies facilitated the generation of GEM to study tumor biology. The most common ways to generate mouse models of cancers are activating oncogenes or inactivating tumor-suppressor genes (or both) in vivo through the use of transgenic and gene-targeting approaches. There are numerous works describe the related methods and conclusions in details [44–46]. For this reason, we will explain most of the models briefly and focus on GEM associated with microRNA studies.
Constitutive gene expression system
Constitutive transgenic mouse is created by transmitting a foreign DNA fragment into a single-cell embryo of the mouse. Thus, when the embryo develops into a mouse, it can express the target gene in all its cells and even transmit the target gene to its offsprings.
Chronic HBV infection is one of the major causes of HCC. The most studied proteins among the compositions of HBV are hepatitis B virus X (HBx) protein and hepatitis B virus surface antigen (HBsAg). Transgenic mice habouring the entire HBx gene succumbed to progressive histopathological changes in the liver, beginning with multifocal areas of altered hepatocytes at the age of about 4 months and followed by the appearance of benign adenomas at the age of about 8–10 months. Over 80% males died with hepatocellular carcinoma at between 11 and 15 months while over 60% females at between 17 and 21 months. In this model, AFP can be detected once adenomas formed [47]. HBx protein has been suspected to be a transcriptional transactivator that can stimulate expression of a broad range of proto-oncogenes including c-fos, c-myc and c-jun [48–50], and thus, involved in hepatocarcinogenesis. In addition, HBx protein is found to bind to and inactivates p53 [51], stimulate the expression of insulin-like growth factor II [52] and the insulin-like growth factor I receptor [53], and compromise DNA repair [54].
On the other hand, transgenic mice with a high expression of HBsAg do develop distinctive inflammation and HCC, especially male mice [55, 56]. When HBsAg is knocked into the p21 locus, 53.3% male p21HBsAg/+ heterozygotes and 72.7% p21HBsAg/HBsAg homozygotes developed liver tumors between the ages of 15 and 24 months, yet, no female mice developed tumors at the same ages [55]. In contrast to this model, transgenic mice that overproduce the hepatitis B virus large envelope polypeptide and HBsAg within the hepatocyte developed severe, prolonged hepatocellular injury which processed to neoplasia within 18 months. Consistent with the situation in patients infected with chronic hepatitis B viral, males in the experiment had more tumors than females and at an earlier age [56, 57]. It indicated that HBV acts as a complete carcinogen that causes HCC by initiating a complex series of events in response to chronic hepatocyte injury. Further studies showed that pre-S-1/S-2 mutant HBsAg can induce oxidative DNA damage and mutations in hepatocytes in the late stages of HBV infection and cause hepatocarcinogenesis [58].
Hepatitis C virus (HCV) is also the main cause of HCC worldwide. To demonstrate the chief role of the HCV core protein, Koike et al. [59, 60] generated transgenic mice containing the complete core gene of HCV. These transgenic mice developed hepatic steatosis without inflammation as early as 3 months of age. Then, at 16 months, gross hepatic nodules with the characteristics of hepatocellular adenoma formed. These nodules developed into hepatocellular carcinoma later. Approximately one-fourth of the male transgenic mice had hepatic nodules, while none of the female did (see Table 3). These results were consistent with the epidemiological data that men chronically infected with HCV are more likely to develop to HCC than women [61]. In their further study [62], they found that in core gene transgenic mice over 16 months old the levels of hydroperoxides of phosphatidylcholine was increased by 180%. Interaction of core protein with mitochondria and subsequent oxidation of the glutathione pool and complex I inhibition is an important cause of the oxidative stress seen in chronic hepatitis C [63]. Oxidative Injury would in turn acts as a direct effect of core protein on mitochondria [64] which may contribute to the development of HCC in the absence of inflammation.
Table 3: GEM models of liver cancer
System |
Transgene |
Promotor |
Strain |
Percentage HCCs |
References |
Constitutive expression system |
HBx |
HBV |
CD1 |
> 80% in males |
[47] |
HBx + pre C-C sequence |
HBV |
C57BL/6xDBA |
75% in TG mice at > 15 months |
[140] |
|
p21 + HBsAg |
HBV |
C57BL/6 |
53.3% in p21HBsAg/+males, 72.7% in p21HBsAg/HBsAg males at > 15 months |
[55] |
|
p21 + HBx |
HBV |
C57BL/6 |
60% in p21HBx/+males, 45.4% in p21HBx/+females, 63.6% in p21HBx/HBx males and 42.9% in p21HBx/HBxfemales at > 15 months |
[55] |
|
HCV core, E1, E2 |
HBV |
C57BL/6 |
>25.9% in males at >16 months |
[59] |
|
Conditional expression system |
HBV |
albumin |
C57BL/6 × SJL |
100% at 20 months |
[56] |
HCV core, E1, E2 |
albumin |
C57BL/6 × FVB |
100% at 32 weeks (treated with DEN) |
[69] |
|
c-myc |
albumin |
C57BL/6J × CBA/J |
65% in males at 20 months |
[141] |
|
c-myc + E2F1 |
albumin |
C57BL/6J × CBA/J |
100% at 9 months |
[142] |
|
TGF-α |
metallothionein |
CD1 |
50% in males > 12 months |
||
TGF-α + c-myc |
albumin |
C57BL/6J × CBA/J |
100% in males at 8 months |
[141] |
|
SV40 T-antigen |
antithrombin III |
C57BL/6 × DBA2 |
100% at 8 months |
[144] |
|
Inducible expression system |
myc |
liver activator protein (LAP) |
FVB/N × NMRI |
Tumors regress within 3 days after myc inactivation and completely regressed within 30 days |
[72] |
Constitutive gene expression is technically straightforward and appeared early. However, it has several disadvantages. The major one is the embryonic death phenomenon. Constitutive, especially bilateral expression of some genes may lead to death during embryonic period, which makes it impossible to study the effects of mutations on tumor development in adult mice. Besides, it may induce various types of injury outside of tissues of interest or compensate from related gene products or those in the same pathway [65–67]. Another caveat to this approach is the inability to control the level and pattern of transgene expression on account of the randomness of the transgenic copy number and the integration sites. Random integration of the transgene is of particular concern because it can result in a lack of transgene expression due to positional effects or an unexpected phenotype resulting from secondary effects of transgene integration into sensitive genomic sites. Constitutive transgenic mouse models fail to mimic sporadic multistep tumorigenesis because the initiating mutation is present throughout the body and germ line from the beginning of development. With the advent of conditional and inducible system, investigators have obtained smarter approaches allowing for the induction of somatic mutations in a tissue-specific and time-controlled manner.
Conditional and inducible gene expression systems
Conditional gene expression system
In conditional systems, some liver-specific promoter elements, such as those for albumin [68, 69], metallothionein [70], transthyretin [71], and liver-activating protein (LAP) [72] are utilized. Thus, certain genes can be expressed individually or in combinations only in liver. Sandgren et al. generated a mouse model by directing the expression of c-myc to the liver of transgenic mice using the albumin enhancer/promoter [73]. In their experiment, c-myc expression can cause mild to severe hepatic dysplasia in young mice, and focal hepatic adenomas in mice over 15 months of age. Further study showed that c-myc can activate somatic mutations within the β-catenin gene. These alterations lead to a disregulation of the signaling function of β-catenin and thus to hepatocarcinogenesis [74]. In addition, to study the interaction between c-myc and transforming growth factor α (TGF-α) in hepatic oncogenesis, double transgenic mice bearing fusion genes consisting of mouse albumin (Alb) enhancer/promoter-mouse c-myc complementary DNA and mouse metallothionein 1 promoter-human TGF-α complementary DNA was generated [70, 75–78]. Compared with Alb/c-myc transgenic mice and TGF-α transgenic mice, time needed to induce dysplastic lesions were reduced significantly. Co-expression of TGF-α and c-myc began to induce persistent proliferation of the hepatocytes as early as the first weeks of life. This continuous replication lead to neoplastic lesions by the second month of age [75]. At 10 weeks of age, the production of ROS was significantly elevated [79] and a high rate of genomic instability and loss of heterozygosity was observed [80] in c-myc/TGF-α mice when compared with c-myc lesions. Factor et al. [77] even showed that nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-induced survival signaling is activated in preneoplastic and neoplastic lesions of c-myc/TGF-α double transgenic mice. However, activation of β-catenin, which was most frequent in liver tumors from c-myc transgenic mice, was very rare in hepatocellular carcinomas developed in c-myc/TGF-α mice [81]. It is a shift of profound importance for it provides a general paradigm for characterizing the interaction of nuclear oncogenes/transcription factors and dissecting the genetic and molecular pathways leading to human HCC [75]. Afterwards, c-myc/E2 promoter-binding factor 1 (E2F1) conditionally transgenic mice [78, 82] and c-myc/epidermal growth factor (EGF) conditionally transgenic mice [83] were established successively (see Table 3). The conditional expression system can circumvents embryonic death effect to a great extent. Yet, it is irreversible strategy and cannot mimic the roles of oncogenes in different stages of tumor progression accurately.
Inducible gene expression systems
Inducible system allows for temporal control over genetic changes. Currently, there are three widely used types of inducible systems: (a) tetracycline (Tet)-controlled system (b) tamoxifen-controlled system and (c) virus-mediated Cre delivery system.
Tetracycline-controlled system consists of two complementary components. One is Tet-Off system, conditionally expressing tetracycline controlled transactivator (tTA); the other is Tet-On system, conditionally expressing reverse tetracycline controlled transactivator (rtTA) (see Figure 1A and 1B). In the Tet-Off system, doxycycline (Dox) prevents binding of tTA to the promoter of Tet (Ptet), and thus abolishes transcription. By contrast, in the Tet-On system, Dox is needed for rtTA to bind to and activate Ptet. This inducible system has been used to study liver tumors induced by c-myc. Catherine et al. [72] crossed mice with rTA driven by the LAP promoter and myc under the control of the tetracycline-responsive minimal promoter (tet-o-Myc). Thus, progeny possessing both transgenes expressed MYC, whereas mice with either transgene alone or mice with both transgenes and treated with Dox did not express myc. Subsequently, all transgenic mice that overexpressed myc succumbed to liver tumors with a mean latency of tumor onset of 12 weeks. Then, HCC-harboring mice were treated with Dox. Within 4 days, the liver tumors differentiated into normal liver cells accompanied by apoptosis. Within 2 weeks, most of the tumors had grossly regressed. This same system of inducible myc expression was used to study the role of microRNA-26a (miRNA-26a) [84] and miRNA-122 [85] in HCC in subsequent studies. MiRNA-26a and miRNA-122 are found to be suppressed in c-myc induced liver tumors. Tet regulatory systems have some favorable properties: (a) it can be set up in a way that allows extremely tight control and, at the same time, regulation over a wide range, (b) Dox concentrations required to quantitatively control expression are far below the toxicity threshold, and (c) Due to the excellent cell and tissue penetration properties of Dox, these concentrations can be readily achieved, even in different compartments of the mouse including the placenta and the milk of lactating mothers. Thus, Tet regulation can be imposed on the developing embryo as well as on the offspring before and during weaning [86].
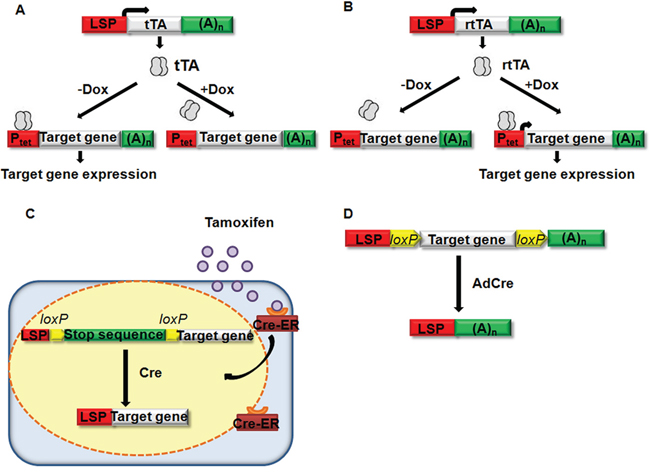
Figure 1: Inducible gene expression system. A. Tet-Off system, the tet-transactivating protein (rTA), which is driven by LSP, will bind to tet-O promoter sequenes in Ptet to activate the expression of a target gene (myc, for example) when Dox is not existed. Doxycycline can prevent tTA from binding and abolish the transcription of the target gene [72]. B. Tet-On system, the reverse tet-transactivating protein rtTA, which is driven by a LPS, will bind to tet-O promoter sequences and activate transcription of the target gene if doxycycline is existed [86]. C. Tamoxifen-controled system, Cre will translocate into nuclear and mediate the recombination of flanked target DNA sequence (stop sequence of HCV, for example) if tamoxifen is present and bind to Cre-ER [88]. D. Virus-mediated Cre delivery system, Cre recombinase encoding by adenovirus will inactivate the target gene (Acp, for example) which is flanked by loxP sequences [95].
In the tamoxifen-controlled system, transgenic (tg) mice were produced that express conditionally HCV structural proteins (core, E1, E2 and p7) in the liver following Cre-mediated DNA recombination. Then, Cre-estrogen receptor fusion protein (Cre-ER) induction strategy was used. As Cre-ER can only response to tamoxifen instead of other endogenous estrogen, Cre nuclear translocation, which led to the expression of HCV protein in the liver, can be induced by intraperitoneal injection of tamoxifen (see Figure 1C) [87–89]. The major disadvantage of this system is the toxicity of tamoxifen and the fusion protein. Excess amounts of tamoxifen can be lethal or can cause damage to certain tissues, such as the uterus [90]. In addition, the Cre-ER fusion protein is toxic in the hematopoietic system [91].
Virus-mediated Cre delivery system is another genetic manipulation. In this system, replication-deficient recombinant adenovirus is commonly used to deliver liver-specific Cre gene to mouse carrying loxP sequence (see Figure 1D) [92, 93]. Colnot et al. constructed a mutant mouse strain in which exon 14 of the tumor-suppressor gene adenomatous polyposis coli (Apc) is flanked by loxP sequences (Apclox/lox). By intravenous injection of adenovirus encoding Cre recombinase (AdCre), the Apc gene was inactivated in the liver. 8 months after administration of 0.5 × 109 pfu of AdCre, 67% of Apclox/lox mice developed HCC. β-catenin signaling was found to be strongly activated in these Apc-inactivated HCCs [94–96]. Compared with other inducible system, adenovirus-mediated expression system is easier to carry out with a relatively low technical difficulty. Moreover, investigators can achieve not only precise temporal control but also tightly regulation of the expression level of certain gene. However, the adenovirus does not integrate into the host chromosome and thus is unsuitable for stable gene expression.
GEM involved in HCC-related microRNA (miRNA)
MiRNAs are small, noncoding RNAs, with lengths of 19–25 nucleotides. Recently, numerous researches confirm that liver cancer had an abnormal expression pattern of miRNAs [97]. Modulation of miRNA was considered as a promising therapeutic strategy due to the ability of these small RNAs to potently influence cellular behavior [84]. For these reasons, genetic manipulation animal models for the study of HCC-related miRNA are eagerly awaited. In this section, we will outline several emerging models associated with miRNA.
Constitutive and conditional miR-122 knockout mice
MiR-122 is the predominant liver miRNA, making up 70% of the total miRNA population [98]. To elucidate the relevance of miR-122 depletion and HCC development, mutant mice with germ line knockout (KO) or liver-specific knockout (LKO) of the miR-122 locus were generated based on the Cre/loxP recombinase system in two studies [85, 99, 100]. Both mir122-KO (5-week-old) and -LKO (8- to 10-week-old) mice developed microsteatosis and liver inflammation due to triglyceride (TG) accumulation. The microsteatosis progressed to steatohepatitis, fibrosis, and spontaneous HCC. 89% male KO mice and 23% female KO mice developed HCC at the age of 10 months [99], while 50% male LKO mice and 10% female LKO mice did at the age of 12 months. In addition, both KO and LKO mice developed moderately to poorly differentiated AFP-positive HCCs with age. Among the tumor-bearing mice, 3 were also found to have lung metastases. In addition, miR-122 played a protective role against DEN by down-regulating genes involved in proliferation, growth factor signaling, neovascularization, and metastasis [101]. At 35 weeks following DEN exposure, LKO mice exhibited a higher incidence of macroscopic liver tumors (71%) and cysts (86%) compared to a 21.4% and 0% incidence of tumors and cysts, respectively, in control mice. The tumors in LKO mice were bigger and predominantly hepatocellular carcinoma, whereas control mice mostly developed hepatocellular adenoma. DEN treatment also reduced survival of LKO mice compared to control mice. Moreover, miR-122 exhibited tumor suppressor activity when delivered to livers of a non-inflammatory myc-driven HCC mouse model [100]. In molecular level, it was confirmed that miR-122 negatively regulates Ccl2 expression by binding to the 3′UTR of Ccl2 mRNA. Without the inhibition of miR-122, Ccl2 expression increased [102]. As a result, the hepatic population of CD11bhiGr1+ cells was found to be higher in 10-month-old KO mice than in controls. They produced a high level of IL-6 and TNF-α, which have been shown to promote HCC development by activating the oncogenic transcription factor STAT3 [103]. Besides, several pathways involved in the immune response, fibrogenesis, EMT, signal transduction, cell survival, cell death, and cancer phenotypes, including MAPK, KEGG, PTEN and Akt signalings were significantly enriched. Mir122-KO or LKO mice provide us with a good example to study down-regulated microRNA in vivo.
A conditionally transgenic mouse model carrying miR-221 gene
Liver-specific miR-221 transgenic mouse is another mouse model targeting on the study of HCC related microRNA. In comparison with wild-type mice, these transgenic mice were characterized by steatohepatitic changes and alternative expression of genes connected to the modulation of the interferon-gamma pathway. After 9 months, about 50% male mice develop small liver tumors. Besides, they also exhibited an increased susceptibility to treatment with the carcinogen [104]. At 6 months, all male animals treated with diethylnitrosamine (DENA) showed evidence of multiple large tumors, whereas in females liver tumors were visible at 9 months. At the molecular level, miR-221 was up-regulated and the targets of miR-221 (p27, p57, and bmf) were down-regulated in the liver tissue. After in vivo delivery of anti-miR-221 oligonucleotides, number and size of tumors were reduced significantly accompanied by a persistent, significant decrease of miR-221 expression [104]. It was the first try to reveal the relationship of HCC and up-regulated miRNA in transgenic mice.
DIETHYLNITROSAMINE (DEN) INDUCED HEPATOCARCINOGENESIS
Chemically induced hepatocarcinogenesis begin with an irreversible process characterized by structural DNA changes [105]. Based on the carcinogens which increase the instability of DNA, chemically induced hepatocarcinogenesis can be categorized as DEN-induced carcinogenesis, aflatoxine-induced carcinogensis, carbon tetrachloride-induced carcinogenesis, dimethylnitrosamine-induced carcinogenesis and thioacetamide-induced carcinogenesis. Among them, DEN-induced hepatocarcinogenesis model is the most frequently used.
DEN is the hydroxylation of α-hydroxylnitrosamine. After a series of complicated chemical reactions in vivo, it turns into an electrophilic agent and reacts with DNA bases to form O6-ethylguanine. Then, to remove the adducts, base excision and the repair of interstrand crosslinking and strand breaks which are considered as major factors in determining DEN carcinogenicity occur [106]. DEN alone is effective to induce liver cancer in mice. In general, 15-days old male C57BL/6 × C3H F1 mice (B6C3F1 mice) are given a single intraperitoneal (i.p.) injection of DEN in a nontoxic dose ranging from 1.25 to 100 ug/g of body weight [107, 108]. Yet, Steffen et al. also induced tumors in 5-week-old male C3H mice by a single i.p. injection of DEN in a dose of 90 ug/g body weight. Tumors will form at least 6 months later. As time goes on, liver tumor incidence can reach 100% [108]. Both the frequency and the time of emergence of lesions is dose-dependent [109]. In addition, DEN-induced tumorigenesis varies with the age, mice strain and sex [110]. The enzymatic competence needed to hydroxylate DEN was already present in newborn animals. It increased and reached peak activity at between the 7th and 15th day of age, and then decreased with age [111]. For this reason, mice less than 15 days of age are more prone to DEN. As to strain differences, male C3H mice are much more sensitive to DEN than C57BL/6 mice. Yet, male C3H mice have the highest rate of spontaneous liver tumors (30–50%) [112], followed by B6C3F1 mice (20–30%) [112] and C57BL/6 mice (less than 2.5%) [113]. Thus, investigators may confuse DEN-induced liver tumors with spontaneous liver tumors. Gender disparity is another interesting phenomenon. HCC occurs mainly in men. Similar sex ratio is also seen in mice given DEN. The incidence of DEN-induced liver tumors is higher in male mice and can be reduced by orchiectomy or gonadotropin blockage; in females the incidence of tumors is increased by ovariectomy or testosterone treatment [114] Naugler et al. found that DEN exposure can promoted production of IL-6 in Kupffer cells (KCs), whereas estrogens can inhibit IL-6 promoter activity by decreasing the activity of the transcription factors nuclear factorkB (NF-kB). As what have mentioned before, IL-6 cause HCC in a manner dependent on signaling via STAT3. Thus, estrogen-mediated inhibition of IL-6 production reduces liver cancer risk in females. In addition, DEN-induced tumorigenesis is dose-dependent and even varies with the age, sex and mice strain [110]. Carcinogenicity of DEN is enhanced by phenobarbital (PB) [115] and by the expression of foxm1b [116], hepatocyte growth factor (HGF) [117], Interleukin-22 (IL-22) [118] or macrophage inflammatory protein-1 and its receptor (CCR1/CCL3) [119]. Meanwhile, curcumin [120], suppressor of cytokine signaling 3 (socs3) [121] and ptpn11 [122] are reported to act as tumor suppressors in the carcinogenicity of DEN.
Regardless of the dose, sequential histochemical changes, from basophilic foci to hyperplastic nodules, then followed by hepatocellular adenomas, and finally hepatocellular carcinomas, will occurred during the hepatocarcinogenesis process induce by DEN [110]. In this process, the levels of fat and glycogen and the activities of several enzymes involved in cell membrane function, glycogen metabolism, the oxidative pentose phosphate pathway, and glycolysis [107]. All the changes are closely related to the neoplastic conversion of hepatocytes [123]. Comparative functional genomics showed that the gene expression patterns in HCCs in diethylnitrosamine-induced mouse liver cancers were most similar to those of the poorer survival group of human HCCs [124]. However, there are some problems concerning on this model. Though employing a various kind of methods to accelerate hepatocarcinogenesis, the average time for forming a tumor is still quite long. Besides, the temporal and spatial heterogeneity of tumor formation also bother the investigators. These shortcomings limit its usage severely.
SPONTANEOUS ANIMAL MODELS OF HCC
Due to different genetic background and numerous other factors, mouse strains differ in susceptibility to spontaneous hepatocellular neoplasia. In general, C3H mice and CBA mice are recognized as being prone to spontaneous liver tumors, while LP, 129, DBA/2, BALB/c, and C57BL are mouse strains with low incidence of spontaneous hepatocellular neoplasia. However, the specific incidence rate for each strain, which has been documented in the published literature since the late 1930’s and early 1940’s, could hardly be captured now [125]. The fatty liver Shionogi (FLS) mouse is an inbred strain that develops spontaneous fatty liver (hepatic steatosis) chronically without obesity. Masahiko et al. reported that the mice develop inflammatory, steatohepatitis and spontaneous hepatocellular tumors. An incidence of HCC was 40% in males at 15–16 months of age, while it was 0% at 13–16 months and 9.5% at 20–24 months in females [126]. Yet, with an extremely low incidence rate, the awfully long time needed to establish the model, and the heterogeneity of tumorigenesis, spontaneous HCC models are considered as an uncontrollable and unpredicted system, and therefore, are scarcely used nowadays.
CONCLUSION
An ideal animal model should faithfully reproduce the key biological behaviors of liver cancer, adequately mimic the human tumor microenvironment, be affordable and easy to manipulate [44]. However, ideal animal models for liver cancer research are unavailable to date. Fortunately, all of the existing liver cancer models can be regarded as valuable and predictive tools if used appropriately and taking the limits of these models into account. Usually, GEM are excellent models for studies concerning on the molecular mechanism of liver cancer development; with a relatively higher metastasis rate, orthotopic HCC models are appropriate for HCC metastasis investigations; both subcutaneous and orthotopic models could be applied to test new therapeutic strategies. With the development of genome editing techniques and the spread of valuable instruments, mice models of high quality will be more accessible to investigators and, doubtlessly, improve our understanding of liver carcinogenesis and the design of comprehensive treatment strategies.
ACKNOWLEDGMENTS
This work was financially supported by the National Natural Science Foundation of China (Nos. 81472832, 81372663).
CONFLICTS OF INTEREST
None declared.
Abbreviation
HCC, hepatocellular carcinoma; GEM, genetically engineered mouse models; miR, microRNA; CSCs, cancer stem cells; DEN, diethylnitrosamine; HBV, hepatitis B virus; HCV, hepatitis C virus; SCID, severe combined immune deficient; NOD/SCID, nonobese diabetic-severe combined immunodeficiency disease mice; TACE, transcatheter arterial chemoembolization; Chol-miR-375, cholesterol-conjugated 2’-O-methyl-modified microRNA-375 mimics; AEG-1, astrocyte elevated gene-1; FoxC1, forkhead box c1; BLI, bioluminescence molecular imaging; AFP, α-fetoprotein; MRI, magnetic resonance imaging; PET, positron emission tomography; FLI, fluorescence imaging; SV40 T-Ag, Simian Virus 40 T-antigen; STAT3, signal transducer and activator of transcription 3; HSCs, hepatic stellate cells; Tregs, regulatory T cells; MDSCs, myeloid-derived suppressor cells; PTEN, phosphatase and tensin homolog; 5-FU, fluorouracil; ROS, reactive oxygen species; HBx, hepatitis B virus X protein; HBsAg, hepatitis B virus surface antigen; HCV, hepatitis C virus; LAP, liver-activating protein; TGF-α, transforming growth factor α; Alb, albumin; E2F1, E2 promoter-binding factor 1; EGF, epidermal growth factor; tTA, tetracycline controlled transactivator; rtTA, reverse tetracycline controlled transactivator; Tet, tetracycline; Ptet, the promoter of Tet; Dox, doxycycline; tet-o-Myc, tetracycline-responsive minimal promoter; Cre-ER, Cre-estrogen receptor fusion protein; Apc, adenomatous polyposis coli; AdCre, adenovirus encoding Cre recombinase; TG, triglyceride; KO, knockout; LKO, liver-specific knockout; PB, phenobarbital; IL-22, Interleukin-22; HGF, hepatocyte growth factor; FLS, fatty liver Shionogi.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Blum HE. Hepatocellular carcinoma: therapy and prevention. World J Gastroenterol. 2005; 11:7391–7400.
3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003; 362:1907–1917.
4. Okuda K. Hepatocellular carcinoma. J Hepatol. 2000; 32:225–237.
5. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004; 127:S35–50.
6. Lai H, Mo X, Yang Y, He K, Xiao J, Liu C, Chen J, Lin Y. Association between aflatoxin B1 occupational airway exposure and risk of hepatocellular carcinoma: a case-control study. Tumour Biol. 2014; 35:9577–9584.
7. Avella DM, Li G, Schell TD, Liu D, Zhang SS, Lou X, Berg A, Kimchi ET, Tagaram HR, Yang Q, Shereef S, Garcia LS, Kester M. Regression of established hepatocellular carcinoma is induced by chemoimmunotherapy in an orthotopic murine model. Hepatology. 2012; 55:141–152.
8. Lu D, Han C, Wu T. 15-PGDH inhibits hepatocellular carcinoma growth through 15-keto-PGE2/PPARγ-mediated activation of p21WAF1/Cip1. Oncogene. 2014; 33:1101–1112.
9. Zheng F, Liao YJ, Cai MY, Liu TH, Chen SP, Wu PH, Wu L, Bian XW, Guan XY, Zeng YX, Yuan YF, Kung HF, Xie D. Systemic delivery of microRNA-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet. 2015; 11:e1004873.
10. He X, Liu F, Yan J, Zhang Y, Shang H, Dou Q, Zhao Q, Song Y. Trans-splicing repair of mutant p53 suppresses the growth of hepatocellular carcinoma cells in vitro and in vivo. Sci Rep. 2015; 5:8705.
11. Zhou HC, Fang JH, Luo X, Zhang L, Yang J, Zhang C, Zhuang SM. Downregulation of microRNA-100 enhances the ICMT-Rac1 signaling and promotes metastasis of hepatocellular carcinoma cells. Oncotarget. 2014; 5:12177–12188.
12. Heo MJ, Kim YM, Koo JH, Yang YM, An J, Lee SK, Lee SJ, Kim KM, Park JW, Kim SG. microRNA-148a dysregulation discriminates poor prognosis of hepatocellular carcinoma in association with USP4 overexpression. Oncotarget. 2014; 5:2792–2806.
13. Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta Pathol Microbiol Scand. 1969; 77:758–760.
14. He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F, Li PY, Song YH, Lin JS. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012; 31:3357–3369.
15. He XX, Guo AY, Xu CR, Chang Y, Xiang GY, Gong J, Dan ZL, Tian DA, Liao JZ, Lin JS. Bioinformatics analysis identifies miR-221 as a core regulator in hepatocellular carcinoma and its silencing suppresses tumor properties. Oncol Rep. 2014; 32:1200–1210.
16. Hu H, Qiu Y, Guo M, Huang Y, Fang L, Peng Z, Ji W, Xu Y, Shen S, Yan Y, Huang X, Zheng J, Su C. Targeted Hsp70 expression combined with CIK-activated immune reconstruction synergistically exerts antitumor efficacy in patient-derived hepatocellular carcinoma xenograft mouse models. Oncotarget. 2015; 6:1079–1089.
17. Ge YY, Shi Q, Zheng ZY, Gong J, Zeng C, Yang J, Zhuang SM. MicroRNA-100 promotes the autophagy of hepatocellular carcinoma cells by inhibiting the expression of mTOR and IGF-1R. Oncotarget. 2014; 5:6218–6228.
18. Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998; 17:279–284.
19. Fan ZC, Yan J, Liu GD, Tan XY, Weng XF, Wu WZ, Zhou J, Wei XB. Real-time monitoring of rare circulating hepatocellular carcinoma cells in an orthotopic model by in vivo flow cytometry assesses resection on metastasis. Cancer Res. 2012; 72:2683–2691.
20. Lu YS, Kashida Y, Kulp SK, Wang YC, Wang D, Hung JH, Tang M, Lin ZZ, Chen TJ, Cheng AL, Chen CS. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology. 2007; 46:1119–1130.
21. Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye QH, Wang L, Zhou J, Qiu SJ, Li Y, Ji XN, Liu H, Xia JL, et al. A decade’s studies on metastasis of hepatocellular carcinoma. Journal of cancer research and clinical oncology. 2004; 130:187–196.
22. Xia L, Huang W, Tian D, Zhu H, Qi X, Chen Z, Zhang Y, Hu H, Fan D, Nie Y, Wu K. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology. 2013; 57:610–624.
23. Yao X, Hu JF, Daniels M, Yien H, Lu H, Sharan H, Zhou X, Zeng Z, Li T, Yang Y, Hoffman AR. A novel orthotopic tumor model to study growth factors and oncogenes in hepatocarcinogenesis. Clin Cancer Res. 2003; 9:2719–2726.
24. Kim SM, Im GH, Lee DG, Lee JH, Lee WJ, Lee IS. Mn(2+)-doped silica nanoparticles for hepatocyte-targeted detection of liver cancer in T1-weighted, MRI. Biomaterials. 2013; 34:8941–8948.
25. Baek S, Mueller A, Lim YS, Lee HC, Lee YJ, Gong G, Kim JS, Ryu JS, Oh SJ, Lee SJ, Bacher-Stier C, Fels L, Koglin N. (4S)-4-(3-18F-fluoropropyl)-L-glutamate for imaging of xC transporter activity in hepatocellular carcinoma using PET: preclinical and exploratory clinical studies. J Nucl Med. 2013; 54:117–123.
26. Zheng T, Yin D, Lu Z, Wang J, Li Y, Chen X, Liang Y, Song X, Qi S, Sun B, Xie C, Meng X, Pan S. Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in Hepatocellular Carcinoma. Mol Cancer. 2014; 13:133.
27. Gheysens O, Mottaghy FM. Method of bioluminescence imaging for molecular imaging of physiological and pathological processes. Methods. 2009; 48:139–145.
28. Cany J, Tran L, Gauttier V, Judor JP, Vassaux G, Ferry N, Conchon S. Immunotherapy of hepatocellular carcinoma: is there a place for regulatory T-lymphocyte depletion? Immunotherapy. 2011; 3:32–34.
29. Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer. 2014; 135:2857–2867.
30. McIlroy D, Barteau B, Cany J, Richard P, Gourden C, Conchon S, Pitard B. DNA/amphiphilic block copolymer nanospheres promote low-dose DNA vaccination. Mol Ther. 2009; 17:1473–1481.
31. Zhao W, Zhang L, Xu Y, Zhang Z, Ren G, Tang K, Kuang P, Zhao B, Yin Z, Wang X. Hepatic stellate cells promote tumor progression by enhancement of immunosuppressive cells in an orthotopic liver tumor mouse model. Lab Invest. 2014; 94:182–191.
32. Kornek M, Raskopf E, Guetgemann I, Ocker M, Gerceker S, Gonzalez-Carmona MA, Rabe C, Sauerbruch T, Schmitz V. Combination of systemic thioacetamide (TAA) injections and ethanol feeding accelerates hepatic fibrosis in C3H/He mice and is associated with intrahepatic up regulation of MMP-2, VEGF and ICAM-1. J Hepatol. 2006; 45:370–376.
33. Kornek M, Lukacs-Kornek V, Limmer A, Raskopf E, Becker U, Klöckner M, Sauerbruch T, Schmitz V. 1, 2-dioleoyl-3-trimethylammonium-propane (DOTAP)-formulated, immune-stimulatory vascular endothelial growth factor a small interfering RNA (siRNA) increases antitumoral efficacy in murine orthotopic hepatocellular carcinoma with liver fibrosis. Mol Med. 2008; 14:365–373.
34. Kornek M, Lukacs-Kornek V, Limmer A, Raskopf E, Becker U, Klockner M, Sauerbruch T, Schmitz V. 1, 2-dioleoyl-3-trimethylammonium-propane (DOTAP)-formulated, immune-stimulatory vascular endothelial growth factor a small interfering RNA (siRNA) increases antitumoral efficacy in murine orthotopic hepatocellular carcinoma with liver fibrosis. Mol Med. 2008; 14:365–373.
35. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, Reid LM, Minato H, Honda M. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009; 136:1012–1024.
36. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008; 68:4311–4320.
37. Damia G, D’Incalci M. Contemporary pre-clinical development of anticancer agents—what are the optimal preclinical models? Eur J Cancer. 2009; 45:2768–2781.
38. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. Lupeol targets liver tumor-initiating cells through phosphatase and tensin homolog modulation. Hepatology. 2011; 53:160–170.
39. Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007; 132:2542–2556.
40. Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009; 7:e1000121.
41. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009; 4:226–235.
42. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, Barnard GF, Doki Y, Mori M. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010; 120:3326–3339.
43. Yang ZF, Ngai P, Ho DW, Yu WC, Ng MN, Lau CK, Li ML, Tam KH, Lam CT, Poon RT, Fan ST. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology. 2008; 47:919–928.
44. Li Y, Tang ZY, Hou JX. Hepatocellular carcinoma: insight from animal models. Nat Rev Gastroenterol Hepatol. 2012; 9:32–43.
45. Koike K. Hepatocarcinogenesis in hepatitis viral infection: lessons from transgenic mouse studies. J Gastroenterol. 2002; 37:55–64.
46. Feitelson MA, Larkin JD. New animal models of hepatitis B and C. ILAR J. 2001; 42:127–138.
47. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991; 351:317–320.
48. Kalra N, Kumar V. c-Fos is a mediator of the c-myc-induced apoptotic signaling in serum-deprived hepatoma cells via the p38 mitogen-activated protein kinase pathway. J Biol Chem. 2004; 279:25313–25319.
49. Singh M, Kumar V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev Med Virol. 2003; 13:243–253.
50. Robinson WS. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu Rev Med. 1994; 45:297–323.
51. Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, Geller DA, Will H, Harris CC. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A. 1997; 94:14707–14712.
52. Su Q, Liu YF, Zhang JF, Zhang SX, Li DF, Yang JJ. Expression of insulin-like growth factor II in hepatitis B, cirrhosis and hepatocellular carcinoma: its relationship with hepatitis B virus antigen expression. Hepatology. 1994; 20:788–799.
53. Kim SO, Park JG, Lee YI. Increased expression of the insulin-like growth factor I (IGF-I) receptor gene in hepatocellular carcinoma cell lines: implications of IGF-I receptor gene activation by hepatitis B virus X gene product. Cancer Res. 1996; 56:3831–3836.
54. Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Kurokawa K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994; 19:810–819.
55. Wang Y, Cui F, Lv Y, Li C, Xu X, Deng C, Wang D, Sun Y, Hu G, Lang Z, Huang C, Yang X. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology. 2004; 39:318–324.
56. Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res. 1990; 50:3400–3407.
57. Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989; 59:1145–1156.
58. Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, Chang WT, Huang W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis. 2004; 25:2023–2032.
59. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998; 4:1065–1067.
60. Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997; 78:1527–1531.
61. Caporaso N, Romano M, Marmo R, de Sio I, Morisco F, Minerva A, Coltorti M. Hepatitis C virus infection is an additive risk factor for development of hepatocellular carcinoma in patients with cirrhosis. J Hepatol. 1991; 12:367–371.
62. Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, Imai K, Todoroki T, Kimura S. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001; 61:4365–4370.
63. Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, Weinman SA. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005; 280:37481–37488.
64. Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002; 122:366–375.
65. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992; 359:288–294.
66. Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992; 359:328–330.
67. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992; 359:295–300.
68. Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, Lemon SM. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002; 122:352–365.
69. Kamegaya Y, Hiasa Y, Zukerberg L, Fowler N, Blackard JT, Lin W, Choe WH, Schmidt EV, Chung RT. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology. 2005; 41:660–667.
70. Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990; 61:1137–1146.
71. Lou DQ, Molina T, Bennoun M, Porteu A, Briand P, Joulin V, Vasseur-Cognet M, Cavard C. Conditional hepatocarcinogenesis in mice expressing SV 40 early sequences. Cancer Lett. 2005; 229:107–114.
72. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004; 431:1112–1117.
73. Sandgren EP, Quaife CJ, Pinkert CA, Palmiter RD, Brinster RL. Oncogene-induced liver neoplasia in transgenic mice. Oncogene. 1989; 4:715–724.
74. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998; 95:8847–8851.
75. Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993; 53:1719–1723.
76. Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005; 33:181–184.
77. Factor V, Oliver AL, Panta GR, Thorgeirsson SS, Sonenshein GE, Arsura M. Roles of Akt/PKB and IKK complex in constitutive induction of NF-kappaB in hepatocellular carcinomas of transforming growth factor alpha/c-myc transgenic mice. Hepatology. 2001; 34:32–41.
78. Santoni-Rugiu E, Jensen MR, Thorgeirsson SS. Disruption of the pRb/E2F pathway and inhibition of apoptosis are major oncogenic events in liver constitutively expressing c-myc and transforming growth factor alpha. Cancer Res. 1998; 58:123–134.
79. Factor VM, Kiss A, Woitach JT, Wirth PJ, Thorgeirsson SS. Disruption of redox homeostasis in the transforming growth factor-alpha/c-myc transgenic mouse model of accelerated hepatocarcinogenesis. J Biol Chem. 1998; 273:15846–15853.
80. Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004; 126:1374–1386.
81. Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res. 2001; 61:2085–2091.
82. Conner EA, Lemmer ER, Sanchez A, Factor VM, Thorgeirsson SS. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models. Biochem Biophys Res Commun. 2003; 302:114–120.
83. Tonjes RR, Lohler J, O’Sullivan JF, Kay GF, Schmidt GH, Dalemans W, Pavirani A, Paul D. Autocrine mitogen IgEGF cooperates with c-myc or with the Hcs locus during hepatocarcinogenesis in transgenic mice. Oncogene. 1995; 10:765–768.
84. Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009; 137:1005–1017.
85. Wang B, Hsu SH, Wang X, Kutay H, Bid HK, Yu J, Ganju RK, Jacob ST, Yuneva M, Ghoshal K. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: role of E2F1 and transcription factor dimerization partner 2. Hepatology. 2014; 59:555–566.
86. Gossen M, Bujard H. Studying gene function in eukaryotes by conditional gene inactivation. Annu Rev Genet. 2002; 36:153–173.
87. Hardouin SN, Nagy A. Mouse models for human disease. Clin Genet. 2000; 57:237–244.
88. Tumurbaatar B, Sun Y, Chan T, Sun J. Cre-estrogen receptor-mediated hepatitis C virus structural protein expression in mice. J Virol Methods. 2007; 146:5–13.
89. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Developmental biology. 2002; 244:305–318.
90. Mehasseb MK, Bell SC, Habiba MA. The effects of tamoxifen and estradiol on myometrial differentiation and organization during early uterine development in the CD1 mouse. Reproduction. 2009; 138:341–350.
91. Higashi AY, Ikawa T, Muramatsu M, Economides AN, Niwa A, Okuda T, Murphy AJ, Rojas J, Heike T, Nakahata T, Kawamoto H, Kita T, Yanagita M. Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J Immunol. 2009; 182:5633–5640.
92. Hsieh YJ, Liu RS, Hwu L, Ke CC, Wang FH, Wang HE, Chen FD. Cre/loxP system controlled by specific promoter for radiation-mediated gene therapy of hepatoma. Anticancer Res. 2007; 27:1571–1579.
93. Wang Y, Krushel LA, Edelman GM. Targeted DNA recombination in vivo using an adenovirus carrying the cre recombinase gene. Proc Natl Acad Sci U S A. 1996; 93:3932–3936.
94. Vetter D, Cohen-Naftaly M, Villanueva A, Lee YA, Kocabayoglu P, Hannivoort R, Narla GM, Llovet J, Thung SN, Friedman SL. Enhanced hepatocarcinogenesis in mouse models and human hepatocellular carcinoma by coordinate KLF6 depletion and increased messenger RNA splicing. Hepatology. 2012; 56:1361–1370.
95. Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA. 2004; 101:17216–17221.
96. Harada N, Miyoshi H, Murai N, Oshima H, Tamai Y, Oshima M, Taketo MM. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002; 62:1971–1977.
97. Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006; 25:2537–2545.
98. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002; 12:735–739.
99. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012; 122:2884–2897.
100. Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, Mao H, Wei M, Clark KR. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012; 122:2871–2883.
101. Hsu SH, Wang B, Kutay H, Bid H, Shreve J, Zhang X, Costinean S, Bratasz A, Houghton P, Ghoshal K. Hepatic loss of miR-122 predisposes mice to hepatobiliary cyst and hepatocellular carcinoma upon diethylnitrosamine exposure. Am J Pathol. 2013; 183:1719–1730.
102. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010; 140:197–208.
103. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011; 70:i104–108.
104. Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, Fornari F, Giacomelli L, D’Abundo L, Ferracin M, Bassi C, Zagatti B, Corrà F, Miotto E, et al. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012; 56:1025–1033.
105. Pitot HC, Dragan YP. Facts and theories concerning the mechanisms of carcinogenesis. FASEB J. 1991; 5:2280–2286.
106. Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). 2007; 6:1079–1099.
107. Hacker HJ, Mtiro H, Bannasch P, Vesselinovitch SD. Histochemical profile of mouse hepatocellular adenomas and carcinomas induced by a single dose of diethylnitrosamine. Cancer Res. 1991; 51:1952–1958.
108. Park TJ, Kim JY, Oh SP, Kang SY, Kim BW, Wang HJ, Song KY, Kim HC, Lim IK. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology. 2008; 47:1533–1543.
109. Goldfarb S, Pugh TD, Kosciuk S, Chover J. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1985; 45:909–912.
110. Vesselinovitch SD, Mihailovich N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983; 43:4253–4259.
111. Rao KV, Vesselinovitch SD. Age- and sex-associated diethylnitrosamine dealkylation activity of the mouse liver and hepatocarcinogenesis. Cancer Res. 1973; 33:1625–1627.
112. Rao MS, Dwivedi RS, Subbarao V, Reddy JK. Induction of peroxisome proliferation and hepatic tumours in C57BL/6N mice by ciprofibrate, a hypolipidaemic compound. Br J Cancer. 1988; 58:46–51.
113. Fausto N, Campbell JS. Mouse models of hepatocellular carcinoma. Semin Liver Dis. 2010; 30:87–98.
114. Nakatani T, Roy G, Fujimoto N, Asahara T, Ito A. Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn J Cancer Res. 2001; 92:249–256.
115. Tamano S, Merlino GT, Ward JM. Rapid development of hepatic tumors in transforming growth factor alpha transgenic mice associated with increased cell proliferation in precancerous hepatocellular lesions initiated by N-nitrosodiethylamine and promoted by phenobarbital. Carcinogenesis. 1994; 15:1791–1798.
116. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, Costa RH. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004; 18:830–850.
117. Horiguchi N, Takayama H, Toyoda M, Otsuka T, Fukusato T, Merlino G, Takagi H, Mori M. Hepatocyte growth factor promotes hepatocarcinogenesis through c-Met autocrine activation and enhanced angiogenesis in transgenic mice treated with diethylnitrosamine. Oncogene. 2002; 21:1791–1799.
118. Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011; 54:900–909.
119. Yang X, Lu P, Fujii C, Nakamoto Y, Gao JL, Kaneko S, Murphy PM, Mukaida N. Essential contribution of a chemokine, CCL3, and its receptor, CCR1, to hepatocellular carcinoma progression. Int J Cancer. 2006; 118:1869–1876.
120. Chuang SE, Kuo ML, Hsu CH, Chen CR, Lin JK, Lai GM, Hsieh CY, Cheng AL. Curcumin-containing diet inhibits diethylnitrosamine-induced murine hepatocarcinogenesis. Carcinogenesis. 2000; 21:331–335.
121. Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med. 2008; 205:91–103.
122. Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, Varki NM, Wang H, Feng GS. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011; 19:629–639.
123. Bannasch P, Enzmann H, Klimek F, Weber E, Zerban H. Significance of sequential cellular changes inside and outside foci of altered hepatocytes during hepatocarcinogenesis. Toxicol Pathol. 1989; 17:617–628. discussion 629.
124. Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004; 36:1306–1311.
125. Maronpot RR. Biological Basis of Differential Susceptibility to Hepatocarcinogenesis among Mouse Strains. J Toxicol Pathol. 2009; 22:11–33.
126. Soga M, Kishimoto Y, Kawamura Y, Inagaki S, Makino S, Saibara T. Spontaneous development of hepatocellular carcinomas in the FLS mice with hereditary fatty liver. Cancer Lett. 2003; 196:43–48.
127. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006; 66:11851–11858.
128. Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, Tanabe KK. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008; 68:2391–2399.
129. Li Y, Yu DC, Chen Y, Amin P, Zhang H, Nguyen N, Henderson DR. A hepatocellular carcinoma-specific adenovirus variant, CV890, eliminates distant human liver tumors in combination with doxorubicin. Cancer Res. 2001; 61:6428–6436.
130. Pei Z, Chu L, Zou W, Zhang Z, Qiu S, Qi R, Gu J, Qian C, Liu X. An oncolytic adenoviral vector of Smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology. 2004; 39:1371–1381.
131. Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, Mills AS, Waxman S, Fisher RA, Llovet JM, Fisher PB, Sarkar D. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. The Journal of clinical investigation. 2009; 119:465–477.
132. Chen LD, Liu J, Yu XF, He M, Pei XF, Tang ZY, Wang QQ, Pang DW, Li Y. The biocompatibility of quantum dot probes used for the targeted imaging of hepatocellular carcinoma metastasis. Biomaterials. 2008; 29:4170–4176.
133. Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL. Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2008; 68:6698–6707.
134. Hu J, Dong A, Fernandez-Ruiz V, Shan J, Kawa M, Martínez-Ansó E, Prieto J, Qian C. Blockade of Wnt signaling inhibits angiogenesis and tumor growth in hepatocellular carcinoma. Cancer Res. 2009; 69:6951–6959.
135. Zhu LM, Shi DM, Dai Q, Cheng XJ, Yao WY, Sun PH, Ding Y, Qiao MM, Wu YL, Jiang SH, Tu SP. Tumor suppressor XAF1 induces apoptosis, inhibits angiogenesis and inhibits tumor growth in hepatocellular carcinoma. Oncotarget. 2014; 5:5403–5415.
136. Peng H, Dara L, Li TW, Zheng Y, Yang H, Tomasi ML, Tomasi I, Giordano P, Mato JM, Lu SC. MAT2B-GIT1 interplay activates MEK1/ERK 1 and 2 to induce growth in human liver and colon cancer. Hepatology. 2013; 57:2299–2313.
137. Yang XZ, Dou S, Sun TM, Mao CQ, Wang HX, Wang J. Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J Control Release. 2011; 156:203–211.
138. Wang R, Zhao N, Li S, Fang JH, Chen MX, Yang J, Jia WH, Yuan Y, Zhuang SM. MicroRNA-195 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting the expression of VEGF, VAV2, and CDC42. Hepatology. 2013; 58:642–653.
139. Yan XL, Jia YL, Chen L, Zeng Q, Zhou JN, Fu CJ, Chen HX, Yuan HF, Li ZW, Shi L, Xu YC, Wang JX, Zhang XM. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology. 2013; 57:2274–2286.
140. Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, Han YM, Lee CS, Park JS, Lee CH, Hyun BH, Murakami S, Lee KK. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol. 1999; 31:123–132.
141. Santoni-Rugiu E, Nagy P, Jensen MR, Factor VM, Thorgeirsson SS. Evolution of neoplastic development in the liver of transgenic mice co-expressing c-myc and transforming growth factor-alpha. Am J Pathol. 1996; 149:407–428.
142. Calvisi DF, Conner EA, Ladu S, Lemmer ER, Factor VM, Thorgeirsson SS. Activation of the canonical Wnt/beta-catenin pathway confers growth advantages in c-Myc/E2F1 transgenic mouse model of liver cancer. J Hepatol. 2005; 42:842–849.
143. Lee GH, Merlino G, Fausto N. Development of liver tumors in transforming growth factor alpha transgenic mice. Cancer Res. 1992; 52:5162–5170.
144. Dubois N, Bennoun M, Allemand I, Molina T, Grimber G, Daudet-Monsac M, Abelanet R, Briand P. Time-course development of differentiated hepatocarcinoma and lung metastasis in transgenic mice. J Hepatol. 1991; 13:227–239.