
INTRODUCTION
Recently, we employed a label-free quantitative proteomics approach to discover new therapeutic targets in tumor-initiating cells (TICs) and/or cancer stem-like cells (CSCs) [1]. Briefly, ER-positive breast cancer lines were cultured under suspension conditions, allowing for the formation of mammospheres (a.k.a., tumor-spheres). Then, we compared the proteome of these tumor-spheres directly to attached monolayer cells grown in parallel. Using this approach, we identified several functional groups of proteins that were specifically up-regulated in tumor-spheres, relative to epithelial monolayers. More specifically, mitochondrial proteins were dramatically over-expressed, including key enzymes related to beta-oxidation and ketone metabolism, as well as proteins involved in mitochondrial biogenesis, and specific protein inhibitors of mitophagy [1]. Interestingly, our approach revealed > 60 mitochondrial-related target proteins that were significantly over-expressed in tumor-spheres. These mitochondrial proteins were also transcriptionally over-expressed in human breast cancer samples, highlighting their potential clinical importance. As such, we believe that increased mitochondrial biogenesis could provide a new mechanism for the accumulation of mitochondrial mass in CSCs.
We also utilized a pharmacological approach to functionally validate the importance of mitochondrial fuels, by employing a specific MCT1/2 inhibitor, which prevents the uptake of two mitochondrial fuels, ketone bodies and L-lactate. Notably, inhibition of MCT function significantly inhibited tumor-sphere formation in both ER-positive and ER-negative breast cancer cell lines, with an IC-50 of ~1–2 μM [1]. Oligomycin A, an inhibitor of Complex V, the mitochondrial ATP-synthase, showed very similar inhibitory activity. Taken together, these results provide suggestive evidence that the proliferative expansion of CSCs strictly depends upon oxidative mitochondrial metabolism [1]. These findings could have future clinical implications for eradicating CSCs, to minimize or prevent tumor recurrence, metastasis and drug resistance. Of course, this would depend on the clinical development of new non-toxic mitochondrial inhibitors that are safe for the treatment of breast cancer patients, which could require 10–15 years of intensive pharmaceutical development and investment.
In summary, using this unbiased proteomic approach, we identified a conserved dependence on mitochondrial biogenesis for the clonal expansion and survival of CSCs. Interestingly, it is well-known that certain FDA-approved antibiotics inhibit mitochondrial biogenesis as a mild, clinically manageably, side-effect [2–6]. Thus, we recently proposed that these antibiotics could be re-purposed as a relatively non-toxic approach to target CSCs. This approach would dramatically accelerate the potential use of mitochondrial inhibitors in cancer patients [7].
Using this strategy, we experimentally identified 5 different classes of mitochondrially-targeted antibiotics that successfully targeted CSCs [7]. Importantly, these FDA-approved drugs inhibited the growth of CSCs in 12 different cancer cell lines, across 8 different tumor types (breast, DCIS, ovarian, prostate, lung, pancreatic, melanoma, and glioblastoma (brain)). The use of generic antibiotics for cancer therapy could significantly reduce the costs of patient care, making treatment more accessible.
Doxycycline is one of the most promising drugs that emerged from these studies [7]. Since its FDA-approval in 1967, it has been successfully used as a broad-spectrum antibiotic for nearly 50 years, without any major side-effects. Currently, acne patients use doxycycline safely for chronic therapy, for up to 6 months at a time, at a standard dose of 200-mg per day [8, 9].
Mechanistically, in mammalian cells, doxycycline functions as an inhibitor of mitochondrial biogenesis, by binding to the small subunit of the mitochondrial ribosome [2–4, 10, 11]. Doxycycline targets the mitochondrial ribosome, as mitochondria evolved from bacteria over millions of years of evolution [12, 13]. Therefore, bacterial ribosomes and mitochondrial ribosomes show many conserved properties and protein homologies. Recent clinical trials with doxycycline (intended to target cancer-associated infections, but not cancer cells) have already shown positive therapeutic effects in lymphoma patients, although their ability to eradicate CSCs was not yet appreciated [14–15].
Here, to better understand the mechanism(s) of action of doxycycline on cancer cell metabolism, we used an unbiased proteomics approach to identify which proteins are effectively down-regulated by doxycycline. This led to identification of doxycycline as the first FDA-approved inhibitor of DNA-PK, which has broad clinical implications for the use of doxycycline as a radio-sensitizer, to overcome radio-resistance in CSCs.
Throughout this paper, we have used the term cancer stem-like cells (CSCs) or tumor-initiating cells (TICs). For a very insightful discussion of the emerging terminology surrounding the definition of CSCs or TICs, please see the following review (16).
RESULTS
Doxycycline inhibits mammosphere formation, as assessed using primary breast cancer samples derived from metastatic disease sites
Previously, we established that the antibiotic doxycycline effectively inhibits tumor-sphere formation in 12 different cell lines, across eight different cancer types, including breast cancer. More specifically, both ER-positive (MCF7/T47D) and ER-negative (MDA-MB-231) cell lines were all sensitive to doxycycline treatment [7]. Interestingly, all three of these cell lines were originally derived from the pleural effusions of breast cancer patients, with metastatic disease. Thus, we next tested if primary breast cancer samples derived from metastatic disease sites were also sensitive to doxycycline.
Importantly, Figure 1 shows that that doxycycline dose-dependently inhibits mammosphere formation in primary patient samples derived from metastatic disease sites (either pleural effusions or ascites fluid). Moreover, doxycycline appeared to work equally well in samples derived from either ER-positive or ER-negative patients (N = 4 patients in total) (See also Supplemental Figure 1). As such, we obtained quantitatively similar results with both well-established cell lines and primary breast cancer samples.
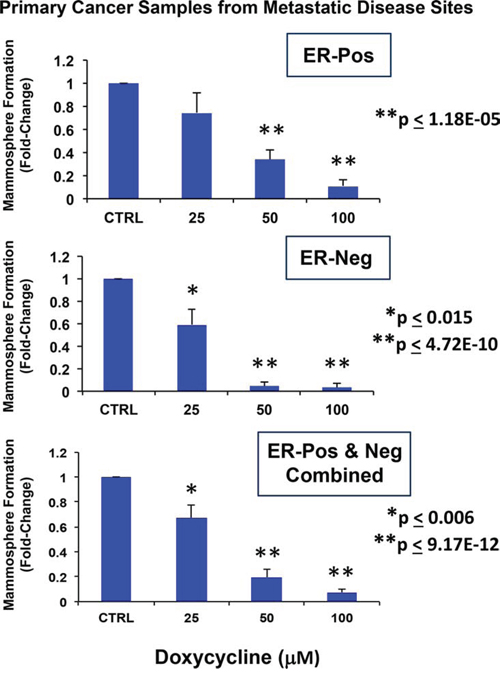
Figure 1: Doxycycline inhibits mammosphere formation, as assessed using primary breast cancer samples derived from metastatic disease sites. Upper panel: ER-positive (N = 2 patients); Middle panel: ER-negative (N = 2 patients); Lower panel: ER-positive and negative samples combined (N = 4 patients). Note that doxycycline dose-dependently inhibits mammosphere formation in primary patient’s samples derived from metastatic disease sites (either pleural effusions or ascites). Doxycycline appears to work equally well in samples derived from either ER-positive or ER-negative patients. All experiments were performed in triplicate.
These results are consistent with previous studies showing that doxycycline dramatically inhibits the in vivo growth of metastatic lesions (bone and soft tissue) in a mouse model of breast cancer, by up to 60-to-80% [17].
Doxycycline pre-treatment reduces the mammosphere forming capacity of MCF7 monolayer cells
To better understand how doxycycline inhibits the growth of CSCs, we used an unbiased proteomic approach to identify its potential molecular targets. For this purpose, we established conditions under which doxycycline selectively inhibits the proliferation of CSCs, but not “bulk” cancer cells.
First, MCF7 cells were pre-treated with doxycycline (50 μM) as monolayers for 7-days and then re-plated for the mammosphere assay, in the absence of doxycycline. Figure 2 shows that pre-treatment with doxycycline, under these conditions, is sufficient to significantly reduce mammosphere forming capacity. However, this 7-day treatment also significantly reduced proliferation in MCF7 cell monolayers to a similar extent, but did not affect the viability of the remaining cells.
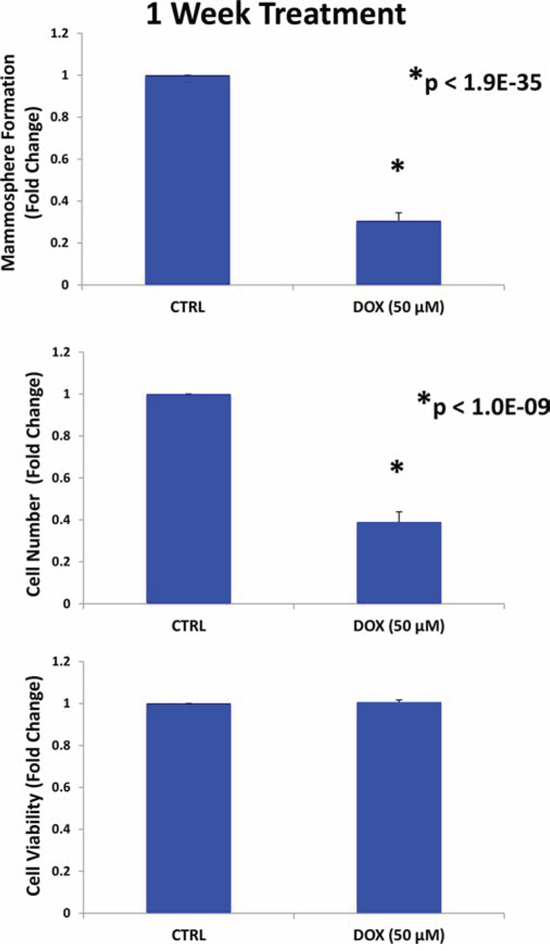
Figure 2: Doxycycline pre-treatment of MCF7 monolayers inhibits mammosphere formation: Effects at 7-days. MCF7 cells were pre-treated with doxycycline (50 μM) as monolayers for 7-days and then re-plated for the mammsphere assay, in the absence of doxycycline. Note that pre-treatment with doxycycline, under these conditions, is sufficient to significantly reduce mammosphere forming capacity. However, this 7-day treatment also reduced proliferation in MCF7 cell monolayers to a similar extent, but did not affect the viability of the remaining cells. Each data point in this figure is the average of 9 replicates.
Therefore, we next shortened the pre-treatment period to 3-days. Importantly, under these new conditions, doxycycline (50 μM) reduced the mammosphere forming capacity of MCF7 cells by ~ 50%, without affecting the proliferation of the bulk monolayer cells (Figure 3). Thus, doxycycline can be used to selectively reduce “stemness” in MCF7 monolayers.
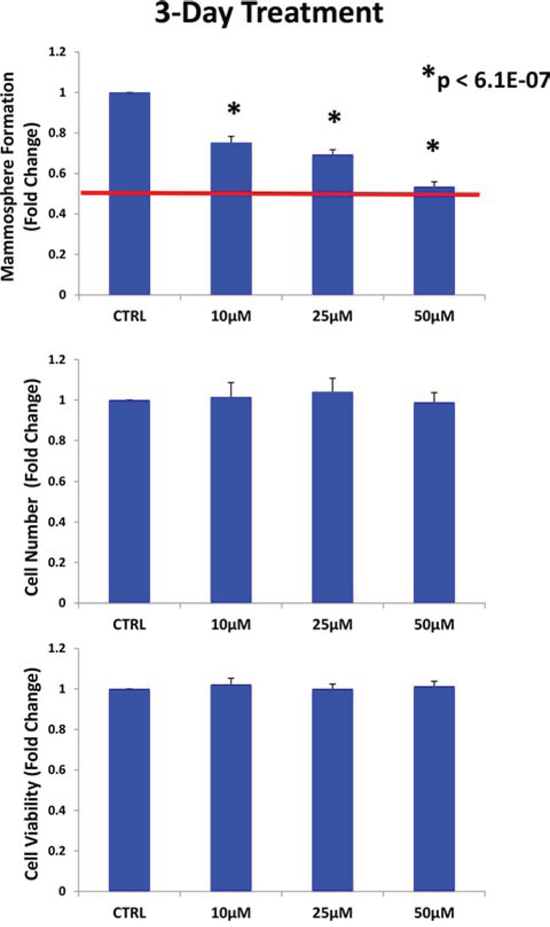
Figure 3: Doxycycline pre-treatment of MCF7 monolayers inhibits mammosphere formation: Effects at 3-days. MCF7 cells were pre-treated with doxycycline (50 μM) as monolayers for 3-days and then re-plated for the mammosphere assay, in the absence of doxycycline. Under these conditions, doxycycline (50 μM) reduced the mammosphere forming capacity of MCF7 cells by ~ 50%, without affecting the proliferation of the bulk monolayer cells As such, doxycycline can be used to selectively reduce “stemness” in MCF7 monolayers. Each data point in this figure is the average of 9 replicates.
Identification of the molecular targets of doxycycline, using unbiased label-free proteomics analysis: DNA-PK emerges as a new target
Next, to begin to understand the molecular basis of this selectively, we used these culture conditions to perform unbiased proteomics analysis on MCF7 monolayers (treated with doxycycline for 3-days). The results of this analysis are summarized in Table 1. Only proteins reduced by ≥ 1.5-fold (p < 0.05) were considered in the analysis. Importantly, Table 1 clearly highlights that doxycycline pre-treatment of MCF7 cell monolayers significantly reduced the expression of many key protein targets functionally associated with mitochondrial metabolism, glycolysis, the EMT, protein synthesis and the DNA damage response, as well as inflammation and protein degradation, in human breast cancer cells.
Table 1: MCF7 cell proteins down-regulated in response to doxycycline treatment of monolayer cultures (3-days at 50 μM)
Symbol |
Gene Description |
Down-regulation (fold-change) |
ANOVA |
Mitochondrial-related Proteins |
|||
PRKDC |
DNA-dependent protein kinase catalytic subunit (maintains mt-DNA integrity & copy number) |
14.71 |
0.02 |
GPD2 |
Glycerol-3-phosphate dehydrogenase, mitochondrial |
5.29 |
1.55E-06 |
MDH2 |
Malate dehydrogenase 2, mitochondrial |
3.08 |
4.01E-06 |
ECI1 |
Enoyl-CoA delta isomerase 1, mitochondrial |
2.95 |
0.018 |
VDAC1 |
Voltage-dependent anion-selective channel protein 1 |
2.71 |
9.65E-05 |
HSPD1 |
60 kDa heat shock protein, mitochondrial |
2.24 |
0.004 |
DECR1 |
2,4-dienoyl CoA reductase 1, mitochondrial |
1.94 |
0.02 |
UQCRC2 |
Cytochrome b-c1 complex subunit 2, mitochondrial |
1.91 |
0.002 |
SDHA |
Succinate dehydrogenase complex subunit A |
1.88 |
0.0001 |
ALDH18A1 |
Delta-1-pyrroline-5-carboxylate synthase, mitochondrial |
1.88 |
0.006 |
COX6A |
Cytochrome c oxidase subunit 6A, mitochondrial |
1.84 |
0.008 |
FASN |
Fatty acid synthase |
1.83 |
0.02 |
LRPPRC |
Leucine-rich PPR-motif containing (inhibitor of mitophagy) |
1.72 |
0.002 |
ORP150 |
150 kDa oxygen-regulated protein |
1.70 |
0.02 |
NDUFA4 |
NADH dehydrogenase (Ubiquinone) 1 alpha-subcomplex, 4 |
1.68 |
0.0007 |
COX5B |
Cytochrome c oxidase subunit 5B, mitochondrial |
1.66 |
0.0008 |
ATP5F1 |
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit b |
1.57 |
0.02 |
Glycolytic Enzymes |
|||
GPI |
Glucose-6-phosphate isomerase |
3.82 |
8.06E-05 |
LDHA |
L-lactate dehydrogenase A |
2.26 |
0.02 |
TPI1 |
Triosephosphate isomerase 1 |
2.25 |
0.004 |
ENO1 |
Enolase 1 |
1.71 |
0.005 |
ALDOA |
Fructose-bisphosphate aldolase A |
1.57 |
0.007 |
PGK1 |
Phosphoglycerate kinase 1 |
1.57 |
0.037 |
LDHB |
L-lactate dehydrogenase B |
1.53 |
0.03 |
PKM1/2 |
Pyruvate kinase |
1.45 |
0.03 |
EMT-markers/Muscle-related proteins (cytoskeletal)/Extracellular Matrix/Angiogenesis |
|||
SMOC2 |
SPARC-related modular calcium-binding protein 2 |
7.01 |
5.17E-06 |
MYO18B |
Unconventional myosin-XVIIIb |
6.48 |
0.0002 |
TUBB1 |
Tubulin beta-1 chain |
3.72 |
3.09E-05 |
ADAM22 |
A disintegrin and metalloproteinase domain 22 |
3.49 |
8.96E-05 |
PLEC1 |
Plectin 1, intermediate filament binding protein 500kDa |
2.86 |
0.0001 |
ATP2A2 |
Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 |
2.37 |
0.035 |
ACTBL2 |
Beta-actin-like protein 2 |
2.28 |
0.008 |
CTNNB1 |
Catenin (Cadherin-associated protein), beta 1, 88kDa |
2.07 |
0.02 |
CFL2 |
Cofilin-2 |
2.11 |
0.0004 |
CFL1 |
Cofilin-1 (Non-muscle), isoform |
2.01 |
0.04 |
DYNC1H1 |
Cytoplasmic dynein 1 heavy chain 1 |
1.91 |
0.04 |
SRRM2 |
Serine/arginine repetitive matrix protein 2 |
1.85 |
0.006 |
AMOT |
Angiomotin |
1.79 |
0.0005 |
MYH15 |
Myosin-15 |
1.76 |
0.0003 |
MYH10 |
Myosin-10 |
1.73 |
0.02 |
TUBA4B |
Putative tubulin-like protein alpha-4B |
1.67 |
0.003 |
USMG5 |
Up-regulated during skeletal muscle growth protein 5 |
1.58 |
0.02 |
KIF5C |
Kinesin heavy chain isoform 5C |
1.58 |
0.009 |
TUBA1A |
Tubulin alpha-1A chain |
1.50 |
0.01 |
Protein Synthesis and Transport, Glycosylation and Glycosaminoglycan Synthesis (GAGs) |
|||
EIF3C |
Eukaryotic translation initiation factor 3 subunit C |
7.24 |
0.0002 |
VCP |
Valosin-containing protein |
4.95 |
7.38E-06 |
RPL9 |
60S ribosomal protein L9 |
4.94 |
6.46E-05 |
SARS |
Seryl (serine)-tRNA synthetase |
4.85 |
0.0002 |
PMM2 |
Phosphomannomutase 2 |
4.45 |
1.43E-05 |
SURF4 |
Surfeit locus protein 4 |
3.53 |
5.63E-05 |
EEF1G |
Elongation factor 1-gamma |
3.18 |
7.42E-05 |
EIF3A |
Eukaryotic translation initiation factor 3 subunit A |
3.04 |
0.0007 |
RPB4 |
60S ribosomal protein L4 |
2.81 |
0.02 |
EIF3G |
Eukaryotic translation initiation factor 3 subunit G |
2.77 |
7.33E-06 |
RAB2 |
RAB2, member RAS oncogene family |
2.59 |
0.03 |
COPG1 |
Coatomer subunit gamma-1 |
2.34 |
0.003 |
HSP90AB1 |
Heat shock protein 90kDa alpha (Cytosolic), class B member 1 |
2.29 |
0.02 |
UGDH |
UDP-glucose 6-dehydrogenase |
2.28 |
0.03 |
EEF1A1 |
Eukaryotic translation elongation factor 1 alpha 1 |
2.21 |
0.004 |
RRBP1 |
p180/ribosome receptor |
2.19 |
0.04 |
AP1G1 |
AP-1 complex subunit gamma-1 |
1.98 |
0.002 |
RPS18 |
40S ribosomal protein S18 |
1.97 |
0.007 |
RAB21 |
RAB21, member RAS oncogene family |
1.88 |
0.02 |
RPL15 |
60S ribosomal protein L15 |
1.82 |
0.026 |
RPL7 |
60S ribosomal protein L7 |
1.75 |
0.03 |
STIP1 |
Stress-induced-phosphoprotein 1 (Hsp70/Hsp90-organizing protein) |
1.72 |
0.003 |
P4HB |
Protein disulfide-isomerase |
1.69 |
0.02 |
RPS16 |
40S ribosomal protein S16 |
1.59 |
0.015 |
RPS2 |
40S ribosomal protein S2 |
1.57 |
0.008 |
RPL35 |
60S ribosomal protein L35 |
1.55 |
0.02 |
SEC22B |
Vesicle-trafficking protein SEC22b |
1.50 |
0.037 |
DNA-binding, Nuclear-related, and Cell Cycle Control |
|||
PRKDC |
DNA-dependent protein kinase catalytic subunit (non-homologous end joining (NHEJ)) |
14.71 |
0.02 |
XRN2 |
5’-3’ exoribonuclease 2 |
5.53 |
0.0006 |
NAP1L4 |
Nucleosome assembly protein 1-like 4 |
4.46 |
0.002 |
ESF1 |
Nucleolar Pre-RRNA Processing Protein, Homolog |
4.30 |
2.00E-08 |
NASP |
Nuclear autoantigenic sperm protein |
4.12 |
0.009 |
HNRNPA1 |
Heterogeneous nuclear ribonucleoprotein A1 |
3.85 |
9.30E-05 |
FUBP1 |
Far upstream element-binding protein 1 |
3.21 |
1.44E-05 |
POLD3 |
DNA polymerase delta subunit 3 |
3.09 |
0.0007 |
RPA1 |
Replication protein A 70 kDa DNA-binding subunit |
2.80 |
0.02 |
CHD4 |
Chromodomain-helicase-DNA-binding protein 4 |
2.62 |
5.68E-05 |
CTPS1 |
CTP synthase |
2.37 |
0.0003 |
LMNA |
Prelamin-A/C |
2.22 |
0.01 |
CEP110 |
Centrosomal protein 110kDa |
2.02 |
0.03 |
MCM7 |
MCM7 minichromosome maintenance deficient 7 |
1.96 |
0.03 |
NAP1L1 |
Nucleosome assembly protein 1-like 1 |
1.86 |
0.015 |
HDAC1 |
Histone deacetylase 1 |
1.71 |
0.045 |
SP100 |
Nuclear autoantigen Sp-100 |
1.64 |
0.03 |
DHX9 |
ATP-dependent RNA helicase A |
1.47 |
0.02 |
Inflammation/Immune Function |
|||
LTA4H |
Leukotriene A(4) hydrolase |
3.52 |
1.16E-06 |
CCT4 |
T-complex protein 1 subunit delta |
2.13 |
0.0003 |
CCT2 |
T-complex protein 1 subunit beta |
1.53 |
0.04 |
Protein Degradation |
|||
UBR4 |
E3 ubiquitin-protein ligase UBR4 |
3.47 |
0.002 |
STUB1 |
E3 ubiquitin-protein ligase CHIP |
2.55 |
0.01 |
PSMC3 |
Proteasome (prosome, macropain) 26S subunit, ATPase 3 |
1.79 |
0.04 |
PSMB4 |
Proteasome subunit beta type-4 |
1.72 |
0.003 |
PSMD2 |
Proteasome 26S non-ATPase subunit 2 |
1.58 |
0.04 |
UBE1 |
Ubiquitin-activating enzyme E1 (A1S9T and BN75 temperature sensitivity complementing) |
1.55 |
0.003 |
UBE2V1 |
Ubiquitin-conjugating enzyme E2 variant 1 |
1.47 |
0.03 |
USP14 |
Ubiquitin carboxyl-terminal hydrolase 14 |
1.47 |
0.007 |
Note that doxycycline targets mitochondrial metabolism, glycolysis, the EMT, protein synthesis and the DNA damage response, as well as inflammation and protein degradation, in human breast cancer cells.
Interestingly, using this approach, we identified DNA-PK as the protein target that was most dramatically down-regulated by doxycycline, by nearly 15-fold (> 90% reduction) (Table 1). DNA-PK (also known as PRKDC) is the catalytic subunit of the DNA-dependent protein kinase involved in DNA-repair. DNA-PK is required for DNA-repair using the mechanism of NHEJ (non-homologous end joining) [18] [19]. DNA-PK also functions to maintain the integrity and copy number of mt-DNA, so there is a clear link with mitochondrial metabolic function, as well [20].
Consistent with our current findings, we also observed that DNA-PK is significantly over-expressed in both MCF7 and T47D mammospheres (Table 2). Remarkably, DNA-PK is infinitely upregulated in MCF7 mammospheres and nearly 15-fold increased in T47D mammospheres, relative to monolayer cultures.
Table 2: DNA-PK is highly up-regulated in both MCF7 and T47D mammospheres, as compared with monolayer cultures
Cell Line |
Symbol |
Gene Description |
Up-regulation (fold-change) |
ANOVA |
MCF7 |
PRKDC |
DNA-dependent protein kinase, catalytic subunit |
Infinity |
1.13E-10 |
T47D |
PRKDC |
DNA-dependent protein kinase, catalytic subunit |
14.85 |
2.60E-05 |
Clinical relevance of “doxycycline reduced targets” in human breast cancers
To determine the potential clinical relevance of our proteomic findings, we next assessed whether the protein targets reduced by doxycycline were transcriptionally upregulated in human breast cancer cells in vivo.
For this purpose, we employed a published clinical data set of N = 28 breast cancer patients in which their tumor samples were subjected to laser-capture micro-dissection, to physically separate epithelial cancer cells from their adjacent tumor stroma [21]. Table 3 presents a summary of these findings. Importantly, most of the protein targets reduced by doxycycline were transcriptionally upregulated in human breast cancer cells in tumors excised from patients.
Table 3: Doxycycline-targets normally up-regulated in human breast cancer cells in vivo
Symbol |
Gene Description |
Up-regulation (fold-change) |
P-value |
Mitochondrial-related Proteins |
|||
ATP5F1 |
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit b |
5.39 |
7.83E-07 |
COX5B |
Cytochrome c oxidase subunit 5B, mitochondrial |
5.03 |
2.86E-06 |
UQCRC2 |
Cytochrome b-c1 complex subunit 2, mitochondrial |
4.84 |
5.73E-06 |
COX6A |
Cytochrome c oxidase subunit 6A, mitochondrial |
4.46 |
2.07E-05 |
LRPPRC |
Leucine-rich PPR-motif containing (inhibitor of mitophagy) |
4.34 |
3.15E-05 |
NDUFA4 |
NADH dehydrogenase (Ubiquinone) 1 alpha-subcomplex, 4 |
4.25 |
4.29E-05 |
MDH2 |
Malate dehydrogenase 2, mitochondrial |
4.18 |
5.32E-05 |
HSPD1 |
60 kDa heat shock protein, mitochondrial |
3.42 |
5.93E-04 |
DECR1 |
2,4-dienoyl CoA reductase 1, mitochondrial |
3.38 |
6.86E-04 |
VDAC1 |
Voltage-dependent anion-selective channel protein 1 |
2.64 |
5.35E-03 |
PRKDC |
DNA-dependent protein kinase catalytic subunit (maintains mt-DNA integrity & copy number) |
2.14 |
0.02 |
Glycolytic Enzymes |
|||
TPI1 |
Triosephosphate isomerase 1 |
4.21 |
4.88E-05 |
ALDOA |
Fructose-bisphosphate aldolase A |
3.60 |
3.45E-04 |
GPI |
Glucose-6-phosphate isomerase |
3.36 |
7.28E-04 |
PKM2 |
Pyruvate kinase |
3.26 |
9.79E-04 |
PGK1 |
Phosphoglycerate kinase 1 |
2.46 |
8.66E-03 |
LDHA |
L-lactate dehydrogenase A |
2.42 |
9.42E-03 |
ENO1 |
Enolase 1 |
1.96 |
0.03 |
EMT-markers/Muscle-related proteins (cytoskeletal) |
|||
CFL1 |
Cofilin-1 (Non-muscle), isoform |
2.39 |
0.01 |
TUBB1 |
Tubulin beta-1 chain |
2.32 |
0.01 |
TUBA1A |
Tubulin alpha-1A chain |
2.17 |
0.02 |
ATP2A2 |
Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 |
2.07 |
0.02 |
CTNNB1 |
Catenin (Cadherin-associated protein), beta 1, 88kDa |
2.05 |
0.02 |
MYH10 |
Myosin-10 |
1.82 |
0.037 |
KIF5C |
Kinesin heavy chain isoform 5C |
1.82 |
0.037 |
Protein Synthesis and Transport |
|||
RPL7 |
60S ribosomal protein L7 |
5.21 |
1.53E-06 |
RPS18 |
40S ribosomal protein S18 |
4.96 |
3.71E-06 |
HSP90AB1 |
Heat shock protein 90kDa alpha (Cytosolic), class B member 1 |
4.94 |
4.03E-06 |
RPS2 |
40S ribosomal protein S2 |
4.77 |
7.21E-06 |
RPL15 |
60S ribosomal protein L15 |
4.60 |
1.28E-05 |
EIF3C |
Eukaryotic translation initiation factor 3 subunit C |
4.48 |
1.94E-05 |
RPL35 |
60S ribosomal protein L35 |
4.10 |
7.05E-05 |
RAB2 |
RAB2, member RAS oncogene family |
3.73 |
2.29E-04 |
RPL9 |
60S ribosomal protein L9 |
3.71 |
2.49E-04 |
EEF1G |
Elongation factor 1-gamma |
3.71 |
2.44E-04 |
EEF1A1 |
Eukaryotic translation elongation factor 1 alpha 1 |
3.16 |
1.30E-03 |
RPS16 |
40S ribosomal protein S16 |
3.05 |
1.77E-03 |
RPL4 |
60S ribosomal protein L4 |
3.05 |
1.79E-03 |
SEC22B |
Vesicle-trafficking protein SEC22b |
3.04 |
1.81E-03 |
EIF3A |
Eukaryotic translation initiation factor 3 subunit A |
2.51 |
7.57E-03 |
SARS |
Seryl (serine)-tRNA synthetase |
2.15 |
0.02 |
P4HB |
Protein disulfide-isomerase |
2.15 |
0.02 |
EIF3G |
Eukaryotic translation initiation factor 3 subunit G |
1.92 |
0.03 |
DNA-binding, Nuclear-related, and Cell cycle control |
|||
HDAC1 |
Histone deacetylase 1 |
4.15 |
6.02E-05 |
NAP1L4 |
Nucleosome assembly protein 1-like 4 |
4.07 |
7.61E-05 |
HNRNPA1 |
Heterogeneous nuclear ribonucleoprotein A1 |
4.03 |
8.90E-05 |
RPA1 |
Replication protein A 70 kDa DNA-binding subunit |
2.52 |
7.30E-03 |
MCM7 |
MCM7 minichromosome maintenance deficient 7 |
2.23 |
0.015 |
NAP1L1 |
Nucleosome assembly protein 1-like 1 |
2.19 |
0.017 |
PRKDC |
DNA-dependent protein kinase catalytic subunit (non-homologous end joining (NHEJ)) |
2.14 |
0.02 |
CTPS1 |
CTP synthase |
2.09 |
0.02 |
CHD4 |
Chromodomain-helicase-DNA-binding protein 4 |
1.87 |
0.034 |
Inflammation/Immune Function |
|||
CCT4 |
T-complex protein 1 subunit delta |
3.25 |
1.00E-03 |
LTA4H |
Leukotriene A(4) hydrolase |
2.94 |
2.40E-03 |
CCT2 |
T-complex protein 1 subunit beta |
2.85 |
3.06E-03 |
Protein Degradation |
|||
PSMB4 |
Proteasome subunit beta type-4 |
4.16 |
5.70E-05 |
PSMD2 |
Proteasome 26S non-ATPase subunit 2 |
3.31 |
8.29E-04 |
USP14 |
Ubiquitin carboxyl-terminal hydrolase 14 |
3.23 |
1.05E-03 |
STUB1 |
E3 ubiquitin-protein ligase CHIP |
1.95 |
0.03 |
UBR4 |
E3 ubiquitin-protein ligase UBR4 |
1.77 |
0.04 |
In light of these findings, the new “doxycycline reduced targets” that we identified may be particularly relevant, for improving both the diagnosis and treatment of human breast cancers.
Validation of DNA-PK as a therapeutic target in CSCs
To validate the potential relevance of DNA-PK for maintaining the growth of CSCs, we next used an sh-RNA-approach. Briefly, MCF7 cells were transduced with lentiviral vectors harboring either control sh-RNA or sh-RNA species targeting the expression of DNA-PK. Figure 4A illustrates that MCF7 cells harboring the DNA-PK sh-RNA show a 50% reduction in mammosphere forming capacity, as predicted. Loss of DNA-PK expression was indeed confirmed by immunoblot analysis, using specific antibody probes (Figure 4A).
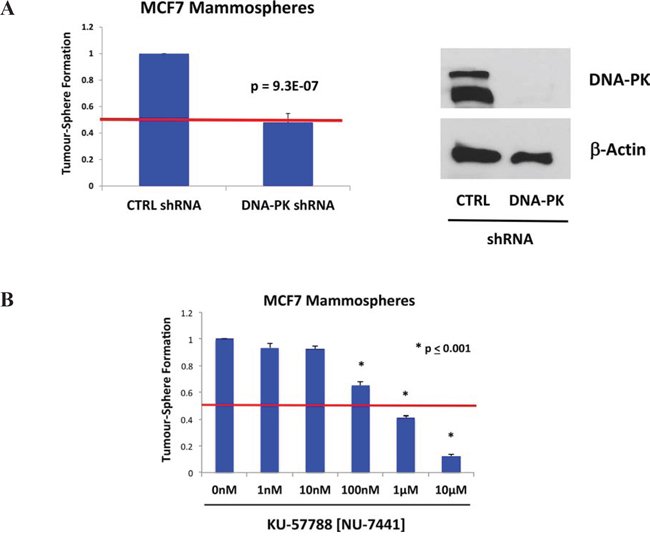
Figure 4: DNA-PK is required for mammosphere formation in MCF7 cells. A. Genetic approach. MCF7 cells were transduced with lentiviral vectors harboring either control sh-RNA or sh-RNA species targeting the expression of DNA-PK. Note that MCF7 cells harboring the DNA-PK sh-RNA show a 50% reduction in mammosphere forming capacity, as predicted. B. Pharmacological approach. Note that the well-established DNA-PK inhibitor, namely KU-57788 [NU-7441], dose-dependently inhibits MCF7 mammosphere formations, with an IC-50 between 100 nM and 1 μM. As such, DNA-PK activity is required for the efficient clonal expansion and anchorage-independent growth of CSCs, as observed using the mammosphere assay. Each data point in this figure is the average of 9 replicates.
Validation was also performed using a well-established DNA-PK inhibitor, namely KU-57788 [NU-7441] [22, 23]. Figure 4B directly shows that KU-57788 [NU-7441] dose-dependently inhibits MCF7 mammosphere formations, with an IC-50 between 100 nM and 1 μM.
Thus, DNA-PK activity appears to be required for the efficient clonal expansion and anchorage-independent growth of CSCs, as observed using the mammosphere assay.
Doxycycline pre-treatment sensitizes CSCs to radiation treatment
Since DNA-PK has been previously implicated in the radio-resistance of cancer cells [24, 25] and we show here that doxycycline functionally reduces the expression of DNA-PK, we would predict that doxycycline treatment should radio-sensitize CSCs.
To test this hypothesis directly, MCF7 cell monolayers were pre-treated with doxycycline (50 μM) for 3-days and then irradiated. After radiation treatment, monolayers were trypsinized and re-plated to evaluate mammosphere growth over a 5-day period. Figure 5 shows that radiation treatment significantly increases the growth of CSCs by up to 1.45-fold, as expected. In contrast, doxycycline pre-treatment increased the sensitivity of CSCs to radiation by up to 4.5-fold. However, under these conditions, doxycycline pre-treatment (with or without radiation) had little or no effect on the proliferation or viability of the “bulk” cancer cells (Figure 6A, 6B).
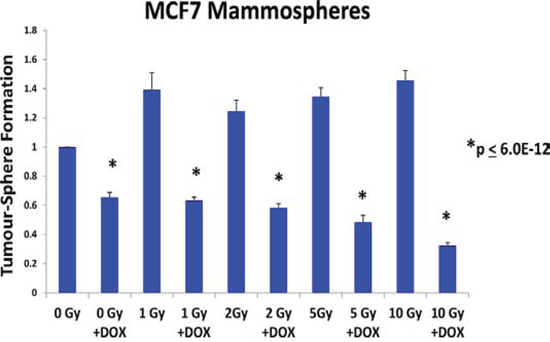
Figure 5: Doxycycline pre-treatment sensitizes cancer stem cells to radiation treatment. MCF7 cell monolayers were pre-treated with doxycycline (50 μM) for 3-days and then irradiated. After radiation treatment, monolayers were trypsinized and re-plated to evaluate mammosphere growth over a 5-day period. Note that radiation treatment significantly increases the growth of CSCs by up to 1.45-fold, as expected. In contrast, doxycycline pre-treatment increased the sensitivity of CSCs to radiation by up to 4.5-fold. In conclusion, doxycycline pre-treatment functionally sensitizes CSCs to radiation, as predicted based on its ability to reduce DNA-PK expression. Each data point in this experiment is the average of 18 replicates.
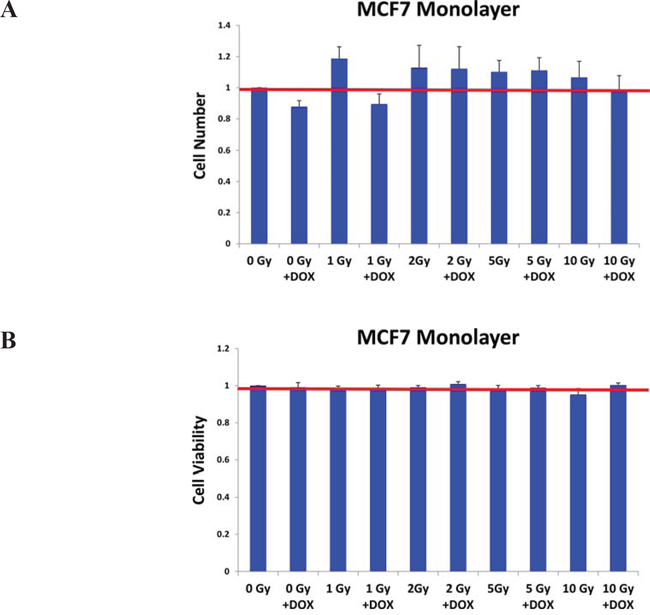
Figure 6: Doxycycline pre-treatment does not inhibit the growth and viability of MCF7 cell monolayers. As in Figure 4, MCF7 cell monolayers were pre-treated with doxycycline (50 μM) for 3-days and then irradiated. However, note that under these conditions doxycycline pre-treatment (with or without radiation), had little or no effect on the proliferation or viability of the “bulk” cancer cells. Each data point in this experiment is the average of 18 replicates.
Thus, doxycycline pre-treatment functionally sensitizes CSCs to radiation, as predicted based on its ability to reduce DNA-PK expression.
Validation of the metabolic phenotype induced by doxycycline pre-treatment
Based on our proteomics analysis presented in Table 1, both the levels of key mitochondrial proteins and glycolytic enzymes were significantly reduced by doxycycline pre-treatment. Thus, these results suggest that doxycycline should reduce overall metabolic activity in cancer cells.
To test this hypothesis further, we next examined the metabolic profile of MCF7 cell monolayers pre-treated with doxycycline (50 μM) for 2-days. Interestingly, Figure 7 shows that the rates of both oxidative mitochondrial metabolism and glycolysis were dramatically reduced by doxycycline pre-treatment, as measured using the Seahorse XFe96 analyzer to measure metabolic flux. This resulted in significant reductions in respiration (basal and maximal), as well as reduced ATP levels (Figure 8). Finally, as seen in Figure 9, MCF7 cancer cells were shifted from a highly energetic to a metabolically quiescent state.
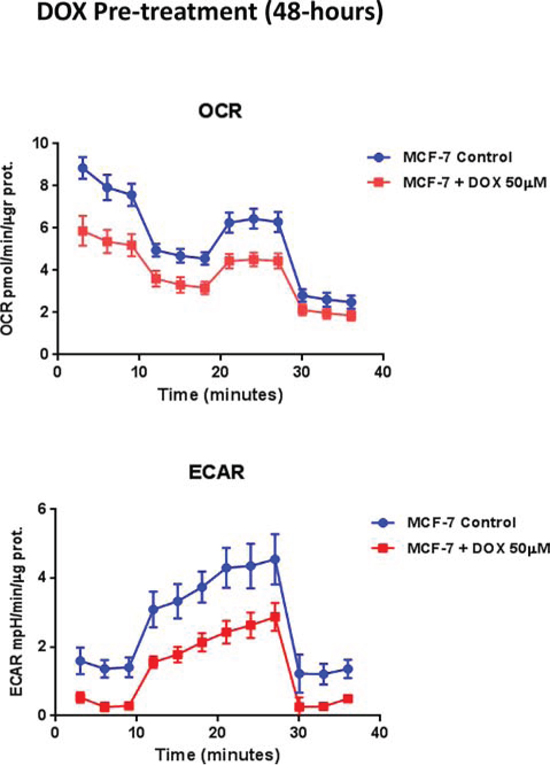
Figure 7: Doxycycline treatment reduces the rates of both oxidative mitochondrial metabolism and glycolysis. We examined the metabolic profile of MCF7 cell monolayers pre-treated with doxycycline (50 μM) for 2-days. Note that the rates of both oxidative mitochondrial metabolism and glycolysis were significantly reduced by doxycycline pre-treatment, as measured using the Seahorse XFe96 analyzer to measure metabolic flux.
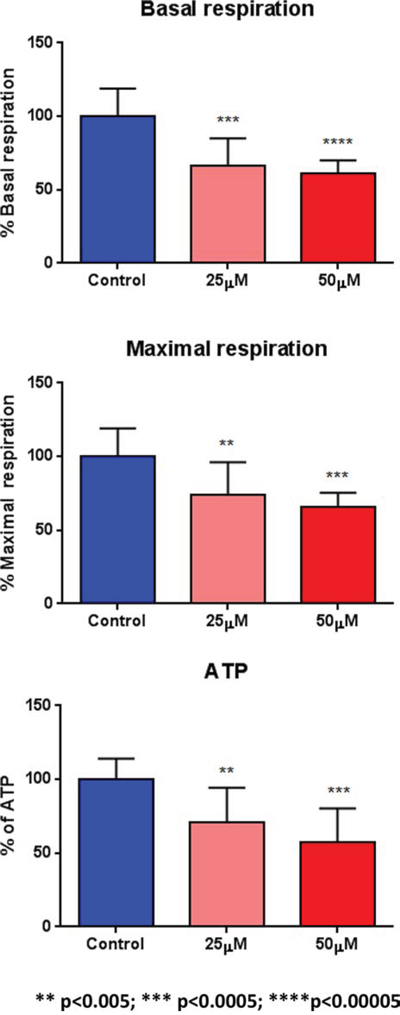
Figure 8: Doxycycline quantitatively reduces respiration (basal and maximal) and ATP levels. We examined the metabolic profile of MCF7 cell monolayers pre-treated with doxycycline (50 μM) for 2-days, using the Seahorse XFe96 analyzer to measure metabolic flux. Note that significant reductions in respiration (basal and maximal), as well as reduced ATP levels, were observed experimentally. Each data point in this figure is the average of 9 replicates.
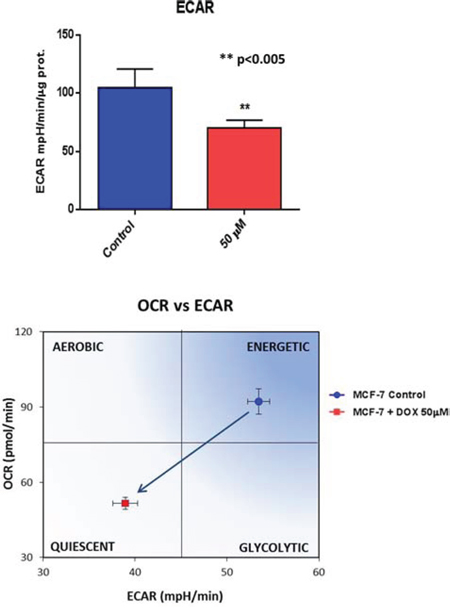
Figure 9: Doxycycline shifts MCF7 cancer cells from a highly energetic to a metabolically quiescent state. We examined the metabolic profile of MCF7 cell monolayers pre-treated with doxycycline (50 μM) for 2-days, using the Seahorse XFe96 analyzer to measure metabolic flux. Note that MCF7 cancer cells were shifted towards a metabolically quiescent state. Each data point in this figure is the average of 9 replicates.
Doxycycline reduces the anoikis-resistance of MCF7 cells, prior to mammosphere formation
Anoikis is a specific type of programmed cell death (apoptosis) that is induced in certain cell types, under anchorage-independent growth conditions [26, 27]. Interestingly, epithelial CSCs show anoikis-resistance, and are able to undergo mammosphere formation under these anchorage-independent conditions [28].
Next, to determine the possible effects on anoikis-resistance, MCF7 cells were pre-treated with doxycycline (at 25 or 50 μM) as monolayers for 2-days and then re-plated on low-attachment plates, for either the anoikis assay or the mammosphere assay, in the absence of doxycycline.
Importantly, Figure 10 shows that doxycycline pre-treatment dose-dependently reduced the number of live cells remaining after 10 hours of seeding on low-attachment plates. Under these conditions, doxycycline pre-treatment also dose-dependently reduced the mammosphere forming capacity of MCF7 cells, by up to ~ 50%. Thus, doxycycline may exert its inhibitory effects on mammosphere formation, in part, by effectively reducing anoikis-resistance, during the initial time spent under low-attachment conditions.
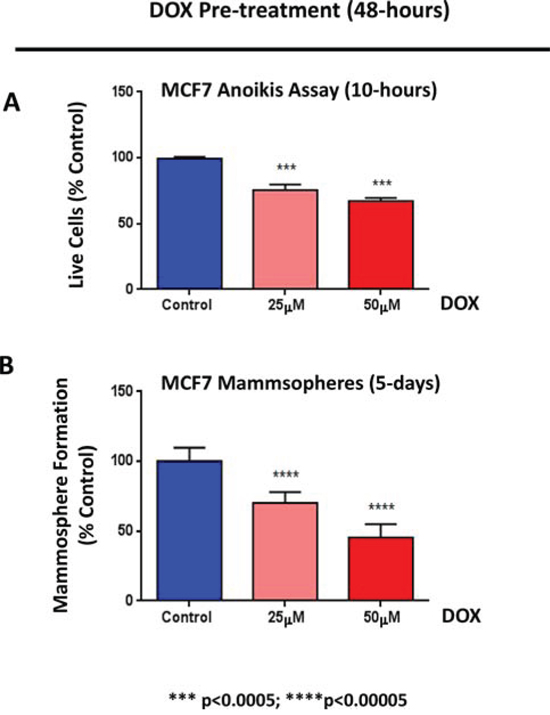
Figure 10: Doxycycline reduces the anoikis-resistance of MCF7 cells, prior to mammosphere formation. MCF7 cells were pre-treated with doxycycline (at 25 or 50 μM) as monolayers for 2-days and then re-plated on low-attachment plates, for the anoikis assay or the mammosphere assay, in the absence of doxycycline. A. Anoikis Assay. Note that doxycycline pre-treatment dose-dependently reduced the number of live cells remaining after 10 hours of seeding on low-attachment plates. B. Mammosphere formation. Note that, under these conditions, doxycycline pre-treatment dose-dependently reduced the mammosphere forming capacity of MCF7 cells, by up to ~ 50%. Each data point in this figure is the average of 9 replicates.
Doxycycline simultaneously inhibits the functional activity of multiple stem-cell associated signal transduction pathways
To better understand which signaling pathways are affected by doxycycline treatment, we used a panel of eight MCF7-GFP cell lines engineered to express different luciferase-based transcriptional reporters. The functional activation state of these reporters was then normalized by GFP expression, to account for cell number.
Interestingly, Figure 11 directly shows that doxycycline treatment of MCF7 monolayer cultures inhibits both the anti-oxidant response (NRF1/2) and STAT1/3 signaling, especially at 72 and 96 hours. Similarly, Figure 12 illustrates that doxycycline also dampens signaling along four other stem-cell associated pathways, including Sonic Hedgehog, Notch, WNT and TGF-beta signaling, again most prominently at 72 and 96 hours. Interestingly, in several cases, a bi-phasic response was noted, with activation of signaling at 24 hours, and progressive inhibition from 48-to-96 hours.
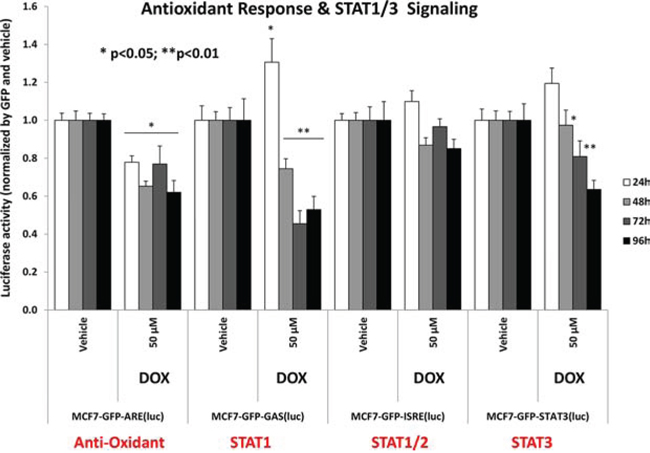
Figure 11: Doxycycline treatment of MCF7 monolayers inhibits the anti-oxidant response (NRF1/2) and STAT1/3 signaling. Note that doxycycline is especially effective at 72 and 96 hours. Each data point in this figure is the average of at least 8 replicates.
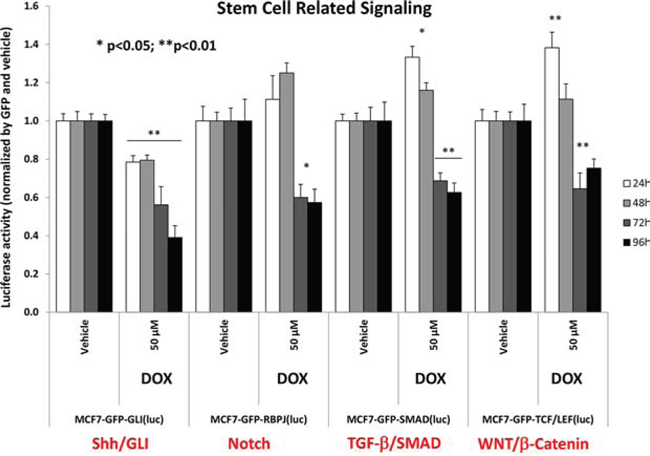
Figure 12: Doxycycline dampens signaling along four major stem-cell associated pathways in MCF7 cells, namely Sonic Hedgehog, Notch, WNT and TGF-beta signaling. Note that the functional effects of doxycycline were most prominent at 72 and 96 hours. In several cases, a bi-phasic response was noted, with activation of signaling at 24 hours, and progressive inhibition from 48-to-96 hours. Each data point in this figure is the average of at least 8 replicates.
These results directly support our observation that doxycycline potently inhibits mammosphere formation, by blocking the clonal expansion of CSCs and reduces anoikis-resistance. Indeed, it has been reported that most of these signaling pathways such as Notch, Hedgehog, WNT, TGFβ, STAT3 or NRF1/2 are able to confer anoikis-resistance, and inhibition of these pathways sensitizes CSCs to anoikis.
DISCUSSION
Recently, we proposed that FDA-approved antibiotics that adversely effect mitochondria could be used to effectively target the cancer stem cell population, by inhibiting mitochondrial biogenesis in tumor-initiating cells (TICs) [7]. One of these promising antibiotics is doxycycline, a member of the tetracycline class, with excellent pharmacokinetics.
To better understand how doxycycline exerts its therapeutic effects, here we used a chemical proteomics approach [29, 30] to more effectively interrogate the target-phenotype relationship. Our results directly show that doxycycline treatment significantly reduced the expression of many key protein targets functionally associated with mitochondrial metabolism, glycolysis, the EMT, protein synthesis and the DNA damage response, as well as inflammation and protein degradation, in human breast cancer cells. We further validated that doxycycline suppresses both oxidative mitochondrial metabolism and glycolysis, using metabolic flux analysis. Doxycycline also inhibited signal transduction along multiple stem-cell associated communication pathways, namely Sonic Hedgehog, Notch, WNT and TGF-beta signaling.
Interestingly, using this approach, we also identified DNA-PK as the protein target that was most dramatically down-regulated by doxycycline, by nearly 15-fold (> 90% reduction) (Figure 13). DNA-PK is required for effective cellular DNA-repair and also maintains the integrity and copy number of mitochondrial DNA [20, 31, 32]. DNA-PK is thought to confer radiation resistance in cancer cells [33–35], but has never been previously implicated in the growth of CSCs.
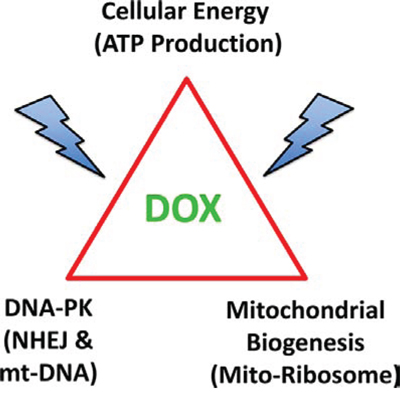
Figure 13: Doxycycline targets mitochondrial biogenesis and DNA-repair, ultimately converging on ATP production and energy metabolism in cancer cells. DNA-PK activity is normally required for repairing mt-DNA and maintaining mt-DNA copy number. Mitochondrial biogenesis is dependent on mitochondrial protein synthesis, which is carried out, in part, by mitochondrial ribosomes, which share significant homology with bacterial ribosomes. Doxycycline targets both mitochondrial ribosomes and DNA-PK, thereby reducing ATP levels, as observed experimentally.
Additional validation experiments, using both genetic (DNA-PK sh-RNA) and pharmacological (DNA-PK inhibitor: NU-7441) approaches directly showed that DNA-PK expression or activity is required for MCF7 cells to efficiently form mammospheres. This is the first demonstration that DNA-PK is required for the clonal expansion and survival of CSCs. Most importantly, however, pre-treatment of MCF7 cell monolayers with doxycycline was sufficient to increase the sensitivity of CSCs to radiation treatment, by up to 4.5-fold.
Unfortunately, no FDA-approved DNA-PK inhibitors have emerged, despite many years of drug discovery and lead optimization. This is largely because existing DNA-PK inhibitors suffer from poor pharmacokinetics. They are not well absorbed and/or are unstable, with a short plasma half-life [36–38]. In contrast, doxycycline has excellent pharmacokinetics, with nearly 100% oral absorption and a long serum half-life (18–22 hours), at a standard dose of 200-mg per day [39, 40].
Thus, we propose that the efficacy of doxycycline as a DNA-PK inhibitor should be tested in Phase-II clinical trials, in combination with radio-therapy. In further support of this idea, we show that doxycycline effectively inhibits the mammosphere-forming activity of primary breast cancer samples, derived from metastatic disease sites (pleural effusions or ascites fluid).
MATERIALS AND METHODS
Materials
Breast cancer cell lines (MCF7 and T47D) were purchased from the ATCC. Gibco-brand cell culture media (DMEM and DMEM/F12) was purchased from Life Technologies. Doxycycline was purchased from Sigma-Aldrich. Sh-RNA lentiviral particles targeting DNA-PK (sc-35200-V) were obtained commercially from Santa Cruz Biotech (USA), along with appropriate sh-RNA control particles (sc-108080). Antibodies directed against DNA-PK (MS-423-P0) for immunoblot analysis were obtained from Thermo Scientific. KU-57788 [NU-7441] was obtained from Selleckchem.
Description of primary tumor samples from metastatic disease sites
Ethical approval was granted by the Central Office for Research Ethics Committee (study numbers: 05/Q1402/25 and 05/Q1403/159) and patients gave written informed consent. Metastatic fluid samples were obtained from 4 patients undergoing palliative drainage of symptomatic ascites or pleural effusions at The Christie Hospital, Manchester, UK. Estrogen, progesterone, and HER2 receptor status of the primary tumors were reported by the Department of Pathology at The Christie, according to established criteria [41].
Isolation of breast cancer epithelial cells
Metastatic breast cancer cells were harvested as previously described, with minor modifications [42]. Invasive breast cancer tissue (1–2 cm3) was collected, dissected into 1–2 mm3 cubes, and digested in media comprised of Dulbecco’s Modified Eagle’s Media (DMEM), 15 mM HEPES, and 10% collagenase/hyaluronidase (Stem Cell Technologies) (supplemented with Pen-Strep) at 37oC for 16 hours. Digested tissue was filtered sequentially through 100, 70, and 40 μm sieves. Red blood cells were removed using Lymphoprep (Axis-Shield), and leukocytes were removed with CD45-negative magnetic sorting according to the manufacturer’s instructions (Miltenyi Biotech).
Pre-treatment of monolayers with doxycycline, with and without radiation treatment
MCF7 cells were plated in normal medium (DMEM, 10% FCS, L-glutamine, supplemented with Pen-Strep) for 24-hr. Cells were then treated, initially for 7 days with doxycycline, with media changed every 3 days. For subsequent experiments, cells were treated for 72-hr. For radiation experiments, monolayer cells pre-treated with doxycycline, were exposed to increasing levels of radiation, and the cells collected by trypsinization and centrifugation. To quantitatively determine cell growth, the number of cells remaining after treatment was counted using an automatic cell counter (Biorad), compared to untreated cells and expressed as fold-change. To assess cell viability, cells were incubated for 1 minute with Trypan Blue (Sigma, #T8145) using a 1:1 ratio. The number of Trypan Blue positive cells (non-viable) was measured using an automatic cell counter (Biorad) and compared to untreated controls. Cells were also plated into mammosphere cultures to assess stem cell-like activity with no further drug treatment. All experiments were performed in triplicate and repeated at least three times independently.
Irradiation of MCF7 monolayers using a cell x-ray unit
MCF7 cells were pre-treated at a monolayer for 3 days, with either doxycycline (50 μM) or with vehicle-alone, and then were irradiated with 0–10 Gy at room temperature, using an X-ray unit, at a dose-rate of 1.37 Gy/min. More specifically, irradiation was performed using a free-standing 320 kV x-ray system (Gulmay Medical Ltd, now Xstrahl Ltd, Camberley, UK). The machine was operated at 300 kV, 10 mA with the addition of a 0.75 mm Cu filter to give a beam quality with half-value layer (HVL) of 2.3 mm Cu. Samples were positioned at a distance of 500 mm from the x-ray focus and a backscatter block was utilised to ensure good dose uniformity. The dose rate to samples was determined to be 1.37 Gy/min [43]. This dose rate was checked regularly using the IPEM 2005 protocol and equipment whose calibration was traceable to UK national standards.
Lentiviral transduction
Lentiviral particles harboring human DNA-PK sh-RNA (#sc-35200-V, Santa Cruz) or control shRNA lentiviral Particles (#sc-108080, Santa Cruz), were used to stably transduce MCF7 cells, according to the manufacturer’s protocol (in the presence of 5 μg/ml polybrene). Twenty-four hours post-infection, media containing the virus was removed and replaced with standard media. Cells were then selected with 2 μg/ml puromycin, for up to 10 days.
Immunoblot analysis
MCF7 cells were seeded in 10 cm dishes for 72 hrs. Then, cells were lysed in RIPA buffer (Sigma), containing proteinase inhibitors (Roche) and kept at 4°C for 30 minutes. Lysates were collected by centrifugation for 10 minutes at 10,000 × g, and protein concentration were determined using the BCA protein assay kit (Pierce). Samples were diluted into SDS-PAGE sample buffer and were boiled for 5 minutes before being separated by SDS-PAGE, using a 4–15% gradient Mini-PROTEAN TGX Gel (Biorad). Samples were then transferred onto a nitrocellulose membrane (Biorad), blocked in 5% milk in TBS-Tween 20 (Sigma) and probed with antibodies directed against DNA-PK (ThermoScientific, #MS-423-P0) and β-actin (Santa Cruz Biotechnology, #sc-1616), using a secondary antibody at a dilution of 1 to 5000. Bound antibodies were detected using the Supersignal West Pico Chemiluminiscent substrate (ThermoScientific).
Mammosphere culture
A single cell suspension was prepared using enzymatic (1x Trypsin-EDTA, Sigma Aldrich, #T3924), and manual disaggregation (25 gauge needle) to create a single cell suspension [44]. Cells were plated at a density of 500 cells/cm2 in mammosphere medium (DMEM-F12/B27/20 ng/ml EGF/PenStrep) in non-adherent conditions, in culture dishes coated with (2-hydroxyethylmethacrylate) (poly-HEMA, Sigma, #P3932). Cells were grown for 5 days and maintained in a humidified incubator at 37°C at an atmospheric pressure in 5% (v/v) carbon dioxide/air. After 5 days for culture, spheres > 50 μm were counted using an eye piece graticule, and the percentage of cells plated which formed spheres was calculated and is referred to as percentage mammosphere formation, and was normalized to one (1 = 100% MSF). For proteomic analysis, mammospheres were collected by centrifugation at 800 rpm for 10 minutes and compared to monolayer cells grown for 5 days.
Label-free quantitative proteomics analysis
Cell lysates were prepared for trypsin digestion by sequential reduction of disulphide bonds with TCEP and alkylation with MMTS [45]. Then, the peptides were extracted and prepared for LC-MS/MS. All LC-MS/MS analyses were performed on an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA) coupled to an Ultimate 3000 RSLCnano system (Thermo Scientific, formerly Dionex, The Netherlands). Xcalibur raw data files acquired on the LTQ-Orbitrap XL were directly imported into Progenesis LCMS software (Waters Corp., Milford, MA, formerly Non-linear dynamics, Newcastle upon Tyne, UK) for peak detection and alignment. Data were analyzed using the Mascot search engine. Five replicates were analyzed for each sample type (N = 5). Statistical analyses were performed using ANOVA and only fold-changes in proteins with a p-value less than 0.05 were considered significant.
Data mining
To firmly establish the clinical relevance of our results from the quantitative proteomics analysis of mammosheres, we re-analyzed the transcriptional profiles of epithelial breast cancer cells and adjacent tumor stromal cells that were physically separated by laser-capture microdissection (from N = 28 human breast cancer patients) [21].
Seahorse XFe96 metabolic flux analysis
Extracellular acidification rates (ECAR) and real-time oxygen consumption rates (OCR) for MCF7 and MCF7 treated with doxycycline were determined using the Seahorse Extracellular Flux (XFe96) analyzer (Seahorse Bioscience, MA, USA). MCF7 cells were maintained in DMEM supplemented with 10% FBS (fetal bovine serum), 2 mM GlutaMAX, and 1% Pen-Strep. We seeded 7,000 cells per well into XFe96 well cell culture plates and they were incubated overnight at 37°C in a 5% CO2 humidified atmosphere. After 24 h, the cells were treated with DOX (25 μM and 50 μM) for 48 h in XFe96 cell culture plates. After 48 h of incubation in DMEM media, MCF-7 cells were washed in XF assay media (or for OCR measurement, XF assay media supplemented with 10 mM glucose, 1 mM Pyruvate, 2 mM L-glutamine and adjusted at 7.4 pH), which were pre-warmed to 37°C. MCF7 cells were then maintained in 175 μL/well of XF assay media at 37°C, in a non-CO2 incubator for 1 h. During the cell incubation time, we loaded 25 μL of 80 mM glucose, 9 μM oligomycin, 1M 2-deoxyglucose (for ECAR measurement) and 10 μM oligomycin, 9 μM FCCP, 10 μM rotenone, 10 μM antimycin A (for OCR measurement), in XF assay media into the injection ports in the XFe96 sensor cartridge. Measurements were normalized by protein content. Data set was analyzed by XFe96 software and GraphPad Prism software, using one-way ANOVA and Student’s t-test calculations. All experiments were performed in quintuplicate, three times independently, such that each data point represents the average of 15 replicates.
Anoikis assay
Following doxycycline treatment, the CSC population was enriched by seeding on low-attachment plates. Under these conditions, the non-CSCs undergo anoikis (a form of apoptosis induced by a lack of cell-substrate attachment) and CSCs are believed to survive. The surviving “CSC fraction” was analyzed by FACS analysis. Briefly, 1 × 104 MCF7 cells were treated with doxycycline (25 μM and 50 μM) for 48 h in 6-well plates, grown as a monolayer. Then, the monolayer cells were trypsinized and seeded in low-attachment plates in mammosphere media. After 10 h under low-attachment conditions, MCF7 cells were spun down and incubated with LIVE/DEAD dye (Fixable Dead Violet reactive dye; Invitrogen) for 20 minutes to distinguish between the live and dead populations of cells (cell viability), during anoikis. Samples were then analyzed by FACS (Fortessa, BD Bioscence) and the data were analysed using FlowJo software.
Monitoring cell signal transduction pathways
The Cignal Lenti luciferase reporter assay was used to monitor the activity of several signaling pathways in MCF7-GFP cells, essentially as previously described [46, 47]. Briefly, viral particles diluted 1:10 in complete media containing polybrene (sc-134220, Santa Cruz) were added to the cells. Puromycin treatment (P9620, Sigma) was added 48 h later in order to stably select infected cells. Luciferase assays (E1501, Promega) were performed according to manufacturer’s instructions. Approximately 6 × 103 MCF7 cells were seeded in black-walled 96 well plates. When cells were attached, drug treatments were added for 24, 48, 72 and 96 h. Four replicates were used for each condition. After treatment, the luciferase assay was performed according to manufacturer’s instructions and light signal was acquired in the Xenogen VivoVision IVIS Lumina. Results were normalized by GFP fluorescence.
ACKNOWLEDGMENTS
We thank the University of Manchester for providing start-up funds that contributed to the success of this study. The Sotgia and Lisanti Laboratories were supported, in part, by funding from the European Union (ERC Advanced Grant), Breakthrough Breast Cancer, and the Manchester Cancer Research Centre (MCRC). DLS was core-funded by CRUK.
REFERENCES
1. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014; 5:11029–11037.
2. McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrobial agents and chemotherapy. 2006; 50:2042–2049.
3. Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell reports. 2015; 10:1681–1691.
4. Riesbeck K, Bredberg A, Forsgren A. Ciprofloxacin does not inhibit mitochondrial functions but other antibiotics do. Antimicrobial agents and chemotherapy. 1990; 34:167–169.
5. Bottger EC, Springer B, Prammananan T, Kidan Y, Sander P. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO reports. 2001; 2:318–323.
6. Leach KL, Swaney SM, Colca JR, McDonald WG, Blinn JR, Thomasco LM, Gadwood RC, Shinabarger D, Xiong L, Mankin AS. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Molecular cell. 2007; 26:393–402.
7. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget. 2015.
8. Kircik LH. Doxycycline and minocycline for the management of acne: a review of efficacy and safety with emphasis on clinical implications. Journal of drugs in dermatology: JDD. 2010; 6:4569–4584.
9. Maibach H. Second-generation tetracyclines, a dermatologic overview: clinical uses and pharmacology. Cutis. 1991; 48:411–417.
10. Ahler E, Sullivan WJ, Cass A, Braas D, York AG, Bensinger SJ, Graeber TG, Christofk HR. Doxycycline alters metabolism and proliferation of human cell lines. PloS one. 2013; 8:e64561.
11. Zhang L, Ging NC, Komoda T, Hanada T, Suzuki T, Watanabe K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS letters. 2005; 579:6423–6427.
12. Zimorski V, Ku C, Martin WF, Gould SB. Endosymbiotic theory for organelle origins. Current opinion in microbiology. 2014; 22:38–48.
13. Degli Esposti M, Chouaia B, Comandatore F, Crotti E, Sassera D, Lievens PM, Daffonchio D, Bandi C. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PloS one. 2014; 9:e96566.
14. Ferreri AJ, Dolcetti R, Magnino S, Doglioni C, Cangi MG, Pecciarini L, Ghia P, Dagklis A, Pasini E, Vicari N, Dognini GP, Resti AG, Ponzoni M. A woman and her canary: a tale of chlamydiae and lymphomas. Journal of the National Cancer Institute. 2007; 99:1418–1419.
15. Ferreri AJ, Ponzoni M, Guidoboni M, Resti AG, Politi LS, Cortelazzo S, Demeter J, Zallio F, Palmas A, Muti G, Dognini GP, Pasini E, Lettini AA, Sacchetti F, De Conciliis C, Doglioni C, et al. Bacteria-eradicating therapy with doxycycline in ocular adnexal MALT lymphoma: a multicenter prospective trial. Journal of the National Cancer Institute. 2006; 98:1375–1382.
16. Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007; 6:1684–90.
17. Duivenvoorden WC, Popovic SV, Lhotak S, Seidlitz E, Hirte HW, Tozer RG, Singh G. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer research. 2002; 62:1588–1591.
18. Davis AJ, Lee KJ, Chen DJ. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. The Journal of biological chemistry. 2013; 288:7037–7046.
19. Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA repair. 2014; 17:21–29.
20. Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH, Reiser J, D’Agati VD, Gharavi AG. Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. The Journal of clinical investigation. 2010; 120:4055–4064.
21. Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Weaver D, Muss H, Plaut K. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast cancer research and treatment. 2009; 114:47–62.
22. Hardcastle IR, Cockcroft X, Curtin NJ, El-Murr MD, Leahy JJ, Stockley M, Golding BT, Rigoreau L, Richardson C, Smith GC, Griffin RJ. Discovery of potent chromen-4-one inhibitors of the DNA-dependent protein kinase (DNA-PK) using a small-molecule library approach. Journal of medicinal chemistry. 2005; 48:7829–7846.
23. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer research. 2006; 66:5354–5362.
24. Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast cancer research and treatment. 2014; 143:47–55.
25. Hsu FM, Zhang S, Chen BP. Role of DNA-dependent protein kinase catalytic subunit in cancer development and treatment. Translational cancer research. 2012; 1:22–34.
26. Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochemical pharmacology. 2008; 76:1352–1364.
27. Taddei ML, Giannoni E, Fiaschi T, Chiarugi P. Anoikis: an emerging hallmark in health and diseases. The Journal of pathology. 2012; 226:380–393.
28. Kwon OJ, Valdez JM, Zhang L, Zhang B, Wei X, Su Q, Ittmann MM, Creighton CJ, Xin L. Increased Notch signalling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nature communications. 2014; 5:4416.
29. Rix U and Superti-furga, G. Target profiling of small molecules by chemical proteomics. Nature chemical biology. 2009; 5:616–624.
30. Jeffery DA, Bogyo M. Chemical proteomics and its application to drug discovery. Current opinion in biotechnology. 2003; 14:87–95.
31. O’Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007; 26:7816–7824.
32. Kashishian A, Douangpanya H, Clark D, Schlachter ST, Eary CT, Schiro JG, Huang H, Burgess LE, Kesicki EA, Halbrook J. DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Molecular cancer therapeutics. 2003; 2:1257–1264.
33. An J, Xu QZ, Sui JL, Bai B, Zhou PK. Downregulation of c-myc protein by siRNA-mediated silencing of DNA-PKcs in HeLa cells. International journal of cancer Journal international du cancer. 2005; 117:531–537.
34. Peng Y, Zhang Q, Nagasawa H, Okayasu R, Liber HL, Bedford JS. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer research. 2002; 62:6400–6404.
35. Novotna E, Tichy A, Pejchal J, Lukasova E, Salovska B, Vavrova J. DNA-dependent protein kinase and its inhibition in support of radiotherapy. International journal of radiation biology. 2013; 89:416–423.
36. Nutley BP, Smith NF, Hayes A, Kelland LR, Brunton L, Golding BT, Smith GC, Martin NM, Workman P, Raynaud FI. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. British journal of cancer. 2005; 93:1011–1018.
37. Davidson D, Amrein L, Panasci L, Aloyz R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Frontiers in pharmacology. 2013; 4:5.
38. Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer discovery. 2014; 4:1126–1139.
39. Bahrami F, Morris DL, Pourgholami MH. Tetracyclines: drugs with huge therapeutic potential. Mini reviews in medicinal chemistry. 2012; 12:44–52.
40. Welling PG, Koch PA, Lau CC, Craig WA. Bioavailability of tetracycline and doxycycline in fasted and nonfasted subjects. Antimicrobial agents and chemotherapy. 1977; 11:462–469.
41. Singh JK, Farnie G, Bundred NJ, Simoes BM, Shergill A, Landberg G, Howell SJ, Clarke RB. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013; 19:643–656.
42. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010; 70:709–718.
43. Aukett RJ, Burns JE, Greener AG, Harrison RM, Moretti C, Nahum AE, Rosser KE, Party IW. Addendum to the IPEMB code of practice for the determination of absorbed dose for x-rays below 300 kV generating potential. 0.035 mm Al-4 mm Cu HVL. Physics in medicine and biology. 2005; 50:2739–2748.
44. Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G, Clarke RB. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of mammary gland biology and neoplasia. 2012; 17:111–117.
45. Holland M, Castro FV, Alexander S, Smith D, Liu J, Walker M, Bitton D, Mulryan K, Ashton G, Blaylock M, Bagley S, Connolly Y, Bridgeman J, Miller C, Krishnan S, Dempsey C, et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood. 2011; 118:638–649.
46. Peiris-Pagès M, Sotgia F, Lisanti MP. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signaling in breast cancer cells. Oncotarget. 2015; 6:10728–107245.
47. Fiorillo M, Verre AF, Iliut M, Peiris-Pagés M, Ozsvari B, Gandara R, Cappello AR, Sotgia F, Vijayaraghavan A, Lisanti MP. Graphene oxide selectively targets cancer stem cells, across multiple tumor types: Implications for non-toxic cancer treatment, via “differentiation-based nano-therapy”. Oncotarget. 2015; 6:3553–62.