
INTRODUCTION
Breast cancer is by far the most common non-cutaneous malignancy affecting women worldwide. Despite considerable progress in both diagnosis and treatment, breast cancer remains the second leading cause of cancer-related death in women [1, 2]. The importance of the human epidermal growth factor receptor (HER, also known as ErbB) family in the development and progression of various cancers is widely recognized [3]. Family members include four receptors: HER1 (EGFR/ErbB1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [4]. Each has been reported to be amplified or overexpressed in some forms of breast cancer, with HER2 and EGFR being the most extensively studied. Overexpression of HER2, found in more than 30% of breast cancer cases [5, 6], is associated with poor prognosis and considered as a predictive marker of chemoresistance. Thus, therapeutic strategies focusing on the inhibition of this oncogene are now being actively explored in breast cancer therapy.
Cancer metabolic reprogramming, such as enhanced glycolysis and lipid biosynthesis, has been recognized as one of the important characteristic features of cancer [7, 8]. Increased lipogenesis, found in approximately 20% to 90% of cancer cases, is a hallmark of aggressive cancers and is reflected in the up-regulation of key enzymes involved in this multiple step process [9]. These include fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC) and ATP-citrate lyase (ACLY). Several studies reported that these lipogenic enzymes are overexpressed in a number of human malignancies including breast cancer [9–14]. Interestingly, their inhibitions by specific inhibitors or siRNA caused significant growth suppression and induced apoptosis in different cancer models [15–20]. New and effective therapeutic agents and strategies targeting these key biosynthetic enzymes are now being actively explored in anticancer therapies [9, 15, 17, 21–24]. Importantly, recent investigations suggest a cross-talk between these metabolic enzymes and the human epidermal growth factor receptor family, especially HER2, via different signaling pathways [12, 25, 26].
Natural products derived from plants are major resources of prospective chemopreventive and chemotherapeutic candidates [27, 28]. Tanshinone IIA is one of the most active components found in the dried roots of Salvia miltiorrhiza Bunge (also known as Danshen). Previous studies indicated that tanshinone IIA possesses potent anti-inflammatory [29] and growth inhibitory properties against different cancer types [30–35]. Acetyltanshinone IIA (ATA) is a novel compound that was chemically modified from tanshinone IIA (Fig. 1A) and has been identified as a promising agent against breast cancer [36]. The current study show the potent anti-cancer activity of ATA on HER2-overexpressing breast cancer cells and explore its in vivo therapeutic potential in a mouse xenograft model of HER2-amplified breast cancer. Furthermore, mechanistic investigations described here reveal a novel anticancer mechanism of ATA via targeting different oncogenic signaling pathways.
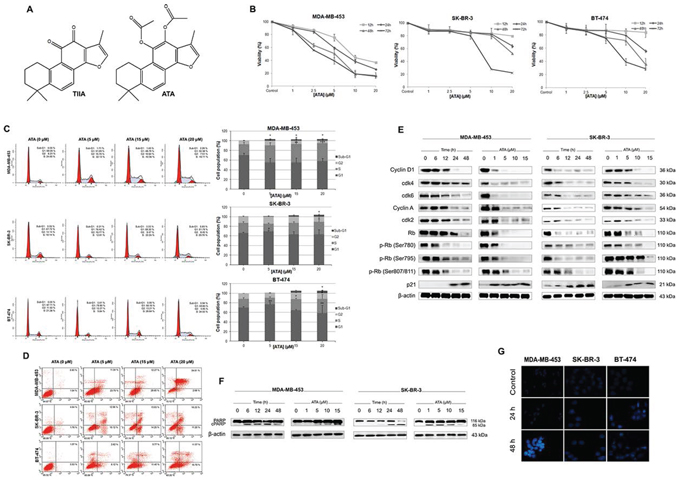
Figure 1: Induction of cell cycle arrest and apoptosis by ATA in HER2-positive breast cancer cells. A. Chemical structures of acetyltanshinone IIA (ATA) and tanshinone IIA (TIIA). B. Effect of ATA on the cell viability of MDA-MB-453, SK-BR-3, and BT-474. Cells were incubated with ATA at various concentrations (1 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM) and for different periods of time (12, 24, 48 and 72 h). Cell viability was measured by CCK-8 assay. C. ATA induced G1/S phase arrest. Following treatment with ATA (5 μM, 15 μM, and 20 μM) for 48 h, cells were analyzed for PI-stained DNA content by flow cytometry. Graphs represent different phases of the cell cycle with each bar representing the mean ± SD of data from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. D. ATA induced apoptosis in HER2-positive breast cancer cells. Treated and untreated cells were harvested, washed with PBS, stained with Annexin V-FITC and PI, and analyzed for Annexin V/PI positivity by flow cytometry. E. Cellular levels of key regulators of cell cycle: cyclin D1, cyclin A, cdk2, cdk4, cdk6, Rb, p-Rb (Ser 780), p-Rb (Ser 795), p-Rb (Ser 807/811), and p21 were analyzed by Western blotting. β-actin was used as loading control. F. Immunoblotting analysis of apoptosis-related protein, PARP. β-actin was used as loading control. G. Nuclear morphological changes induced by ATA in breast cancer cells. Following treatment with ATA (10 μM) for 24 h and 48 h, nuclei were stained with Hoechst 33258 (blue) and observed under fluorescence microscope (magnification, 200×).
RESULTS
ATA inhibited the viability of HER2-overexpressing human breast cancer cells
Initially we investigated the cytotoxic effect of ATA on the proliferation of a panel of HER2-overexpressing breast cancer cell lines including MDA-MB-453, SK-BR-3, and BT-474 cells. We found that ATA caused a significant time- and dose-dependent reduction in the viability of all tested cell lines (Fig. 1B) with a 50% inhibitory concentration (IC50) ranging between 2 μM and 9 μM as determined by CCK-8 assay (Table 1). To ascertain whether ATA had any selectivity for normal versus cancer cells, human normal breast epithelial cells MCF-10A was treated with ATA. We found that the IC50 value for this normal breast cell line is significantly higher than the IC50 values of ATA on breast cancer cells (Table 1). Thus, ATA was more able to selectively inhibit the growth of breast cancer cells than non-cancer ones.
Table 1: Antiproliferative activity data of ATA against human breast cancer and non-cancer cell lines
Cell lines |
Origin |
Immunoprofile |
ATA IC50 (μM)a |
MDA-MB-453 |
Breast carcinoma |
ER -, PR -, HER2 + |
1.97 ± 1.16 |
SK-BR-3 |
Breast carcinoma |
ER -, PR -, HER2 + |
9.17 ± 0.42 |
BT-474 |
Breast carcinoma |
ER +, PR +/-, HER2 + |
6.79 ± 1.40 |
MCF-10A |
Non-cancer breast cell line |
ER -, PR -, HER2 - |
31.74 ± 4.24 |
aValues, expressed as IC50 (50% growth inhibition), are given in micromolar (μM) and are mean ± SD of three independent experiments.
ATA mediated inhibition of cancer cell growth by inducing cell cycle arrest and apoptosis
To determine the cause and the nature of cell death by which ATA mediated its cytotoxic effects on HER2-positive breast cancer cells, cell cycle progression and apoptosis were examined by flow cytometry. Data revealed that treatment with ATA caused S phase cell cycle arrest in MDA-MB-453 cells whereas SK-BR-3 and BT-474 cells were arrested at G1 phase when treated with the low dose of 5 μM and S phase when treated with the high dose of 20 μM. The representative FACS histograms are shown in Fig. 1C. Additionally, a dose-dependent increase in the sub-G1 and Annexin V-positive cell populations observed in ATA-treated groups indicate an induction of apoptosis (Fig. 1C and 1D).
To examine the molecular alterations associated with these events, expression levels of several cell cycle and apoptosis-related proteins were determined by Western blot analysis. As expected, cellular levels of G1-S phases proteins: Cyclin D1, cdk4 and 6 (G0/G1 phase); and Cyclin A, cdk2 (S phase) were markedly down-regulated following ATA treatment (Fig. 1E). Moreover, our data indicate that ATA resulted in an increase of cyclin-dependent kinase inhibitor p21 and a decrease of retinoblastoma (Rb) phosphorylation at serine 780, serine 795, and serine 807/811 (Fig. 1E). Poly (ADP-ribose) polymerase (PARP), an early marker of apoptosis, was found to be cleaved dose- and time-dependently following ATA exposure (Fig. 1F). Additionally, morphological changes demonstrated by Hoechst 33258 staining confirmed the induction of apoptosis (Fig. 1G). Together, our results clearly indicate that the anticancer activity of ATA on breast cancer cells is associated with induction of cell cycle arrest and apoptosis and that this action is not only time- and dose-dependent but also cell line-specific.
ATA depleted EGFR/HER2 levels and down-regulated multiple intracellular signaling pathways
EGFR and HER2 expression levels were evaluated in MDA-MB-453 and SK-BR-3 treated-cells. Results indicate that ATA induced a down-regulation of the cellular levels of HER2 and its dimerization partner EGFR (Fig. 2A). The phosphorylation/activation status of these oncoproteins was also strongly reduced in a dose- and time-dependent manner (Fig. 2A). Next, we analyzed the effect of ATA on signaling pathways usually activated by these receptor tyrosine kinases (RTKs). Western blotting data revealed a down-regulation of the phosphorylation status of Akt, Stat3, and NFκB p65 (Fig. 2A, 2B, and 2C). In some instances, the reduced phosphorylation was accompanied by concomitant reduction of total protein amounts (Fig. 2A, 2B, and 2C).
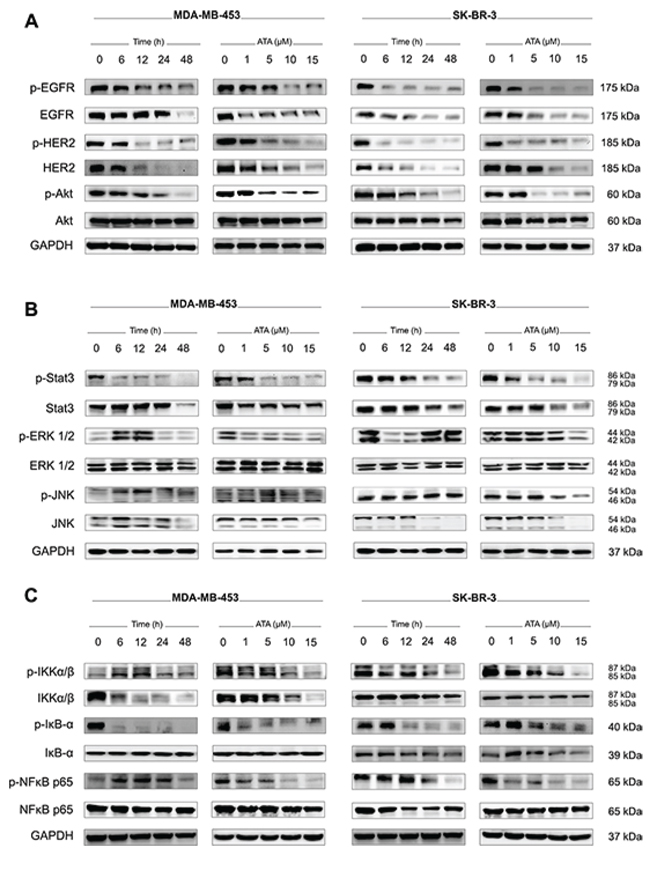
Figure 2: ATA down-regulates EGFR/HER2 cellular levels and inhibits downstream pro-survival signaling pathways. A. Representative Western blot analysis of p-EGFR, total EGFR, p-HER2, total HER2, p-Akt, and total Akt expressed in MDA-MB-453 and SK-BR-3 cells treated with ATA. GAPDH was used as loading control. B. Representative Western blot analysis of p-Stat3, total Stat3, p-ERK1/2, total ERK1/2, p-JNK, and total JNK expressed in MDA-MB-453 and SK-BR-3 cells treated with ATA. GAPDH was used as loading control. C. Representative Western blot analysis of p-IKKα/β, total IKKα/β, p-IκB-α, total IκB-α, p-NFκB p65, and total NFκB p65 expressed in MDA-MB-453 and SK-BR-3 cells treated with ATA. GAPDH was used as loading control.
ATA induced activation of AMPK and altered the expression levels of key enzymes involved in lipid and protein biosyntheses
Recent studies reported that inhibition of fatty acid synthesis can suppress tumor cell growth and induce apoptosis [15, 17, 18, 21, 37–42]. This inhibition can be achieved through impedance of a variety of enzymatic steps in the pathway. In the current study, expression levels of three key lipid biosynthetic enzymes (ACLY, FASN, ACC) were verified by Western blot. As shown in Fig. 3A, treatment with ATA resulted in a decrease of the protein levels of FASN and p-ACLY compared to those in DMSO-treated control cells. No change in total ACLY was observed. Simultaneously, cellular levels of p-ACC significantly increased following ATA exposure (Fig. 3A). Moreover, evaluation of the cellular level of AMPK, a metabolic sensor of cellular energy state, indicated that ATA induced the activation of AMPK with gradually increased levels of p-AMPK (Fig. 3A) whereas the cellular levels of p-mTOR and its downstream targets p-p70S6K and p-4E-BP1, key enzymes involved in protein synthesis, decreased (Fig. 3B). It seems that ATA inactivates mTOR via concomitant activation of AMPK and inhibition of Akt, thereby causing cell cycle arrest and apoptosis.
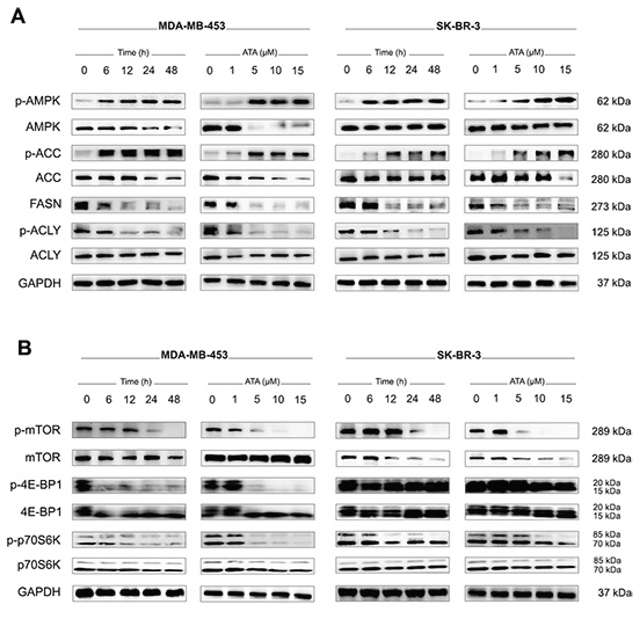
Figure 3: Western blotting for the effect of ATA on the signals associated with AMPK/ACC/FASN/ACLY and mTOR/4E-BP1/p70S6K pathways. A. Whole cell lysates were prepared and Western blot analysis was conducted using anti-p-AMPK, -AMPK, -p-ACC, -ACC, -FASN, -p-ACLY, and -ACLY antibodies. GAPDH was used as loading control. B. Representative Western blot analysis of p-mTOR, total mTOR, p-4E-BP1, total 4E-BP1, p-p70S6K, and total p70S6K expressed in MDA-MB-453 and SK-BR-3 cells treated with ATA. GAPDH was used as loading control.
ATA triggered ER and oxidative stresses in HER2-positive breast cancer cells
In eukaryotic cells, endoplasmic reticulum (ER) is an important player in regulating protein synthesis and lipid metabolism [43]. Perturbation of ER homeostasis is known as ER stress [44]. To assess whether ATA can cause ER stress in MDA-MB-453 and SK-BR-3 cells, we examined by Western blot analysis the phosphorylation status of the ER stress sensor, PERK. We found that ATA induced phosphorylation/activation of PERK which, in turn, phosphorylated/activated the translation initiation factor eIF2α (Fig. 4A). Transcription factor ATF4, a downstream signal of PERK-eIF2α pathway, was also found to be up-regulated. In addition, expression of CHOP, a hallmark of ER stress-mediated cell cycle arrest and apoptosis [45], was increased (Fig. 4A). Coupled with the phosphorylation of PERK, increases in steady-state levels of the pro-apoptotic proteins ATF-4 and CHOP with simultaneous decrease in general protein synthesis strongly indicate that the induction of ER stress might be involved in ATA-mediated cell death in HER2-positive breast cancer cells.
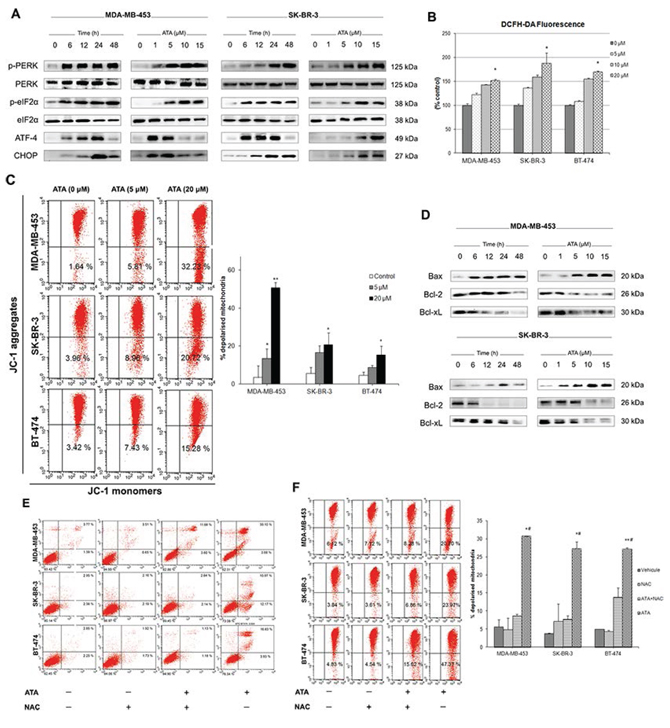
Figure 4: ATA triggers ER and oxidative stresses and induces loss of mitochondrial membrane potential. A. Representative Western blot analysis of p-PERK, total PERK, p-eIF2α, total eIF2α, ATF-4, and CHOP expressed in MDA-MB-453 and SK-BR-3 cells following ATA treatment. B. Effect of ATA on ROS generation in breast cancer cells. Cultured cells labeled with DCFH-DA were incubated in the presence or absence of ATA (5 μM, 10 μM, and 20 μM). Fluorescence signal was monitored time-dependently using a fluorescent plate reader. Each bar represents mean ± SD and is plotted for sextuplicate from each condition (*p < 0.05 versus control). C. ATA induced mitochondrial membrane depolarization. Graphs represent percentages of breast cancer cells with depolarized mitochondria as determined by flow cytometry. Data are presented as mean ± SD from three independent experiments (*p < 0.05, **p < 0.01 versus control). D. Representative Western blot analysis of Bax, Bcl-2, and Bcl-xL expressed in MDA-MB-453 and SK-BR-3 cells treated with ATA. E and F. NAC prevented ATA-induced apoptosis and loss of MMP. Breast cancer cells were pre-incubated for 2 h in the presence or absence of NAC, and then treated with ATA for 48 h. (E) Induction of apoptosis was determined by flow cytometry. (F) Loss of MPP was assessed using JC-1 assay kit. Percentages of breast cancer cells with depolarized mitochondria are plotted as mean ± SD (n = 3). *p < 0.05, **p < 0.01 versus control, #p < 0.05 compared to ATA+NAC group.
Accumulation of reactive oxygen species (ROS) in the cytoplasm has been shown to trigger ER stress and the unfolded protein response [46] leading to CHOP expression [47], cleavage of PARP, and apoptosis [48]. We used the fluorescent probe DCFH-DA to monitor intracellular ROS levels in the presence and absence of ATA. We found that ATA caused an early and slight, but statistically significant, increase in ROS accumulation (Fig. 4B). Furthermore, FACS analysis using JC-1 dye indicated a significant increase (p < 0.05) in the percentage of cells with depolarized mitochondria in ATA-treated cells compared to DMSO-treated control (Fig. 4C). Additionally, ATA notably increased the levels of proapoptotic protein Bax and concomitantly decreased the expression of antiapoptotic proteins Bcl-2 and Bcl-xL (Fig. 4D).
To determine whether increased production of ROS may have a role in ATA-induced apoptosis, loss of mitochondrial membrane potential (MMP), or cell cycle arrest; HER2-positive breast cancer cells were pretreated with the antioxidant N-acetyl-L-cysteine (NAC) two hours before adding ATA for a further 48 h. As shown in Fig. 4E and 4F, pretreatment with NAC conferred significant protection against ATA-induced apoptosis and loss of MMP. However, the same treatment did not prevent cell cycle arrest (data not shown). Together, these data indicate that ROS generation may be one of the important events in the ATA-activated apoptotic signaling cascade.
ATA inhibited tumor growth in vivo
We used a xenograft model of HER2-positive MDA-MB-453 breast cancer in nude mice to explore the in vivo therapeutic potential of ATA. We found that 35 mg/kg of ATA administered three times per week significantly reduced tumor volume (Fig. 5B) and tumor weight (Fig. 5C) but had no effect on mice body weight (Fig. 5A). In this tumor inhibition model, the average tumor volume in the negative control group increased from 95.79 ± 12.02 mm3 to 285.27 ± 25.24 mm3 at the end of the experiment, whereas the average tumor volume in the ATA-treated group decreased from 99.55 ± 11.13 mm3 to 46.49 ± 10.20 mm3. Tumor regression occurred in ATA-treated group by day 14 (~2%) after starting treatment and reached ~50% on day 33 (end-point of the experiment). Additionally, the average tumor weight in the negative control group was 96.33 ± 13.67 mg, whereas the average tumor weight in the ATA-treated group was only 26.5 ± 9.42 mg (Fig. 5C) suggesting that ATA strongly inhibited tumor growth in vivo.
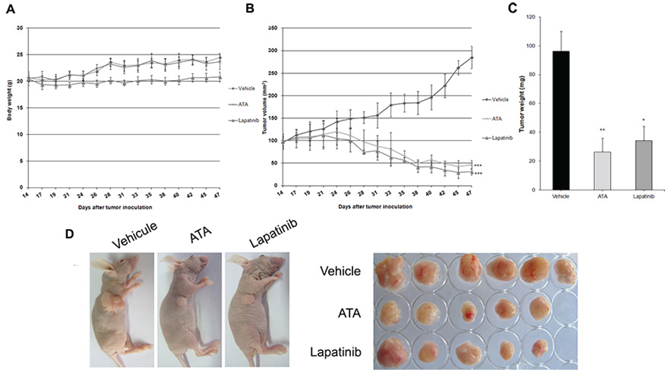
Figure 5: Effect of ATA on MDA-MB-453 xenograft tumor growth. MDA-MB-453 cells (2 × 107 per mouse) were implanted subcutaneously into the right flank of BALB/c nude mice. After solid tumors grew, mice were randomly divided into three groups (n = 6) and treated intraperitoneally with vehicle (negative control group), or ATA (35 mg/kg). Positive control group received lapatinib (80 mg/kg) via gastric gavage twice daily. ATA inhibited tumor growth as measured by tumor volume B. and tumor weight C. without side effects as shown by mice body weight A. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 versus control. D. At the end of the treatment-period, mice were sacrificed and tumors were excised.
ATA suppressed the motility of HER2-overexpressing breast cancer cells and inhibited angiogenesis in vitro
It is a well-known fact that cell migration and invasion plays a crucial role in tumor metastasis. In the present study, wound-closure and transwell migration assays were employed to assess the effects of ATA on the motility of HER2-positive breast cancer cells. Results shown in Fig. 6A and 6B indicate an apparent antimobility effect of ATA as illustrated with MDA-MB-453 and SK-BR-3 cells compared to their respective negative controls.
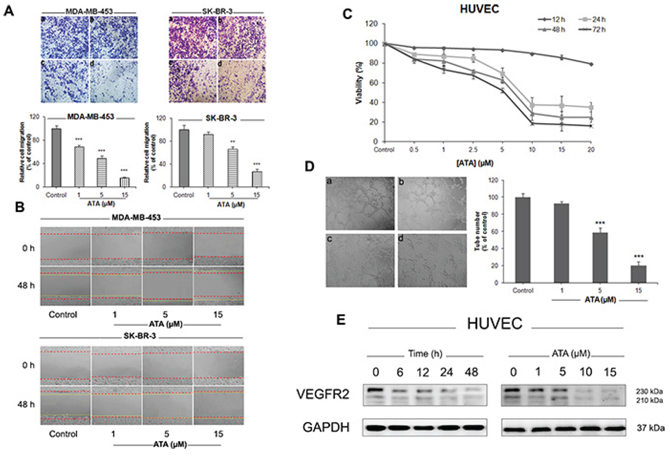
Figure 6: ATA supresses the motility of HER2-positive breast cancer cells and inhibits HUVECs proliferation and tubular formation. A. ATA inhibited MDA-MB-453 and SK-BR-3 cells migration in a transwell assay. Representative images and summary data (bar graph on bottom side) indicate the inhibition of migration. Cells were treated for 48 h with vehicle (a), 1 μM ATA (b), 5 μM ATA (c), or 15 μM ATA (d). Magnification: 100×. The results are expressed as mean ± SD (n = 3); **p < 0.01, ***p < 0.001 versus control. B. ATA treatment inhibited cell motility in a wound-healing test. Representative images before and after incubation with vehicle or ATA (1 μM, 5 μM, and 15 μM) (red lines indicate time 0 h and yellow lines indicate time 48 h). Magnification: 100×. C. Effect of ATA on the viability of HUVECs. Cells were exposed to different concentrations of ATA as indicated for 12, 24, 48 and 72 h. At the end of each time-period, cell viability was measured by CCK-8 assay. D. ATA inhibited HUVECs tube formation. Representative images and summary data (bar graph on right hand side) indicate the inhibition of tube formation. HUVECs were treated with vehicle (a), 1 μM ATA (b), 5 μM ATA (c), or 15 μM ATA (d) for 6 h. Magnification: 100×. The results are expressed as mean ± SD (n = 3); ***p < 0.001 versus control. E. Representative Western blot analysis of VEGFR-2 expressed in HUVECs treated with ATA. GAPDH was used as loading control.
We further investigated the effect of ATA on the proliferation, migration, invasion, and tube formation of human umbilical vascular endothelial cells (HUVECs). As depicted in Fig. 6C, ATA resulted in a dramatic inhibition of HUVE cell proliferation. Capillary-like tube formation assay indicates that ATA exposure markedly and dose-dependently suppressed the formation of functional tube-like networks (Fig. 6D). Furthermore, ATA inhibited the chemotactic motility and invasion of HUVECs as revealed by wound-healing and transwell techniques (data not shown). To investigate the molecular mechanism underlying this anti-angiogenic effect, we evaluated the cellular levels of vascular endothelial growth factor receptor-2 (VEGFR2) by Western blot analysis. As shown in Fig. 6E, ATA time- and dose-dependently down-regulated the protein expression of VEGFR2 in HUVECs. Cumulatively, these data suggest that ATA may inhibit angiogenesis in vitro.
DISCUSSION
The development of innovative ways to prevent and/or to treat resistant subtypes of breast cancer is being actively explored in cancer chemotherapy. We focused our investigation on acetyltanshinone IIA (ATA) which is described as a promising cytotoxic agent from herbal origin with potent in vitro and in vivo anticancer properties [36]. We explored for the first time the molecular mechanisms by which ATA inhibits angiogenesis, metastasis, and induces cell death in HER2-overexpressing breast cancer cells. We further examined the intricate relationship between ATA-induced apoptosis and inhibition of key metabolic enzymes involved in lipid and protein biogenesis.
Data reported here show that ATA inhibited dose- and time-dependently the proliferation of HER2-positive MDA-MB-453, SK-BR-3, and BT-474 cell lines. More importantly, when we investigated whether ATA has a selective activity on cancer cells, we found that its action is relatively less potent on MCF-10A, a non-tumorigenic breast cell line (Table 1). These results enhance the desirability of ATA as an anticancer agent because they suggest that it selectively kills breast cancer cells while minimizing damage to normal breast tissue.
FACS and Western blot analyses revealed that the anticancer activity of ATA is associated with cell cycle arrest and apoptosis. Perturbations of cell cycle were mainly found in the increase of G1 and S phases cell populations. Further investigations on the status of key factors known to regulate cell cycle progression indicated that treatment with ATA promoted the levels of cyclin-dependent kinase inhibitor p21 and down-regulated the protein expressions of cyclin D1, cyclin A, cdk2, cdk4, and cdk6. This action prevented cyclin D/cdk4/cdk6 and cyclin A/cdk2 complexes from phosphorylating Rb protein at different serine positions thereby arresting cell growth. Moreover, observations that ATA induced S phase arrest in MDA-MB-453 and G1 and S phases arrest in SK-BR-3 and BT-474 depending on the dose used for treatment suggest that ATA-mediated growth inhibition may be cell specific.
Cancers overexpressing receptor tyrosine kinases (RTKs) EGFR and/or HER2, particularly breast cancers [49], are known to be resistant to established chemotherapy [50]. Thus, dual kinase inhibition directed against EGFR and HER2 was and still is an attractive potential approach for breast cancer therapy. Our data showed a decrease in the phosphorylation/activation status of HER2 and EGFR oncoproteins following ATA exposure which consecutively inhibited the activation of downstream pro-survival growth pathways.
The survival of breast cancer cells with HER2 gene amplification has been reported to be heavily dependent on lipid metabolism [51]. Several studies reported a crosstalk between FASN and HER2 oncoprotein [52–55]. Indeed, HER2 overexpression was found to increase translation of FASN [12, 56], whereas FASN overexpression was found to markedly increase EGFR and HER2 signalings [25]. Furthermore, tumor cells are well-characterized by their rapid cell growth which requires active synthesis of proteins, ribosomal RNA, and lipids. As all of these functions are switched off by AMPK [57, 58], tumor cells developed mechanisms to down-regulate AMPK and thus escape from its restraining influences on growth and biosynthesis. Herein, we have shown that treatment with ATA induced phosphorylation/activation of AMPK and that this effect translated to downstream targets such as phosphorylation/inhibition of ACC, down-regulation of FASN and p-ACLY, and inhibition of mTOR/4EB-P1/p70S6K signaling pathway. Interestingly, we found that ATA triggered ER and oxidative stresses, induced loss of MMP, and altered protein expression of Bcl-2, Bcl-xL, and Bax in HER2-positive breast cancer cells. More important, employing the powerful antioxidant NAC [59], we demonstrated that ROS play a critical role in ATA-induced apoptosis and loss of MMP.
Overexpression of HER2 in human tumor cells is closely associated with increased angiogenesis and expression of vascular endothelial growth factor (VEGF) [60]. Proliferation, migration, and tube formation of endothelial cells represent the three primary steps of angiogenesis [61] and play a central role in tumor growth, metastasis, and survival. In the present study, we provided evidence that ATA significantly and dose-dependently inhibits the mobility of HER2-positive breast cancer cells. We further showed that endothelial cells (HUVECs) proliferation, migration, invasion, and tubule formation are markedly disrupted following ATA exposure. Consistent with these results, VEGFR2 protein expression was found to be down-regulated (Fig. 6E). As VEGFR2 is the main receptor involved in endothelial cell survival, proliferation, and vascular permeability; VEGFR2 suppression may represent a critical mechanism by which ATA mediates its anti-angiogenic effects.
To the best of our knowledge, this is the first study demonstrating the detailed molecular mechanism of ATA on HER2-overexpressing breast cancer cells. Altogether our findings described the ability of ATA to perturb four major cellular signaling and metabolic pathways: HER2/EGFR, ROS/ER stress, proteins and lipids biogenesis. On the basis of these promising results, the present study suggests that ATA is a potential option for the treatment of patients with HER2-driven breast cancer.
MATERIALS AND METHODS
Drugs and reagents
ATA (MW 380 g/mol) was provided by China Pharmaceutical University Research Institute of Pharmaceutical Chemistry. It was obtained as a white powder of high purity (> 97%). Stock solutions of 10 mM were dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich), stored at − 20°C, and diluted in fresh medium just before use. Lapatinib (MW 925.3 g/mol) was obtained from Jiangsu Kanion Pharmaceutical Co., Ltd. (China). Drugs dilutions were made in serum-free media with a final DMSO concentration less than 0.1%. Media and reagents for cell culture were acquired from Thermo Scientific HyClone (Beijing, China). N-acetyl-L-cysteine (NAC) was purchased from Sigma-Aldrich (China). Antibodies employed in this study were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and Cell Signaling Technology (Beverly, MA, USA). Hoechst 33258 was from Invitrogen (CA, US). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Annexin V-FITC Apoptosis Detection Kit was from BD Pharmingen™ (San Diego, CA, USA). Other kits were acquired from Beyotime Biotechnology Co., Ltd. (Nantong, China) and KeyGEN Biotechnology Co., Ltd. (Nanjing, China).
Cells and culture conditions
Three human breast cancer cell lines (MDA-MB-453, SK-BR-3, and BT-474) characterized by amplification and/or overexpression of the HER2 oncogene and a normal breast epithelial cell line (MCF-10A) were used in this study. Cells were obtained from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China) and were grown as monolayers in the following media: RPMI 1640 (BT-474 and MCF-10A); DMEM (SK-BR-3); and Leibovitz’s (MDA-MB-453). All media were supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotics. Cells were maintained at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air except MDA-MB-453 cells which were maintained in an atmosphere without CO2.
Cell viability assay
The effect of ATA on cell viability was determined by Cell Counting Kit-8 (CCK-8) assay. Briefly, exponentially growing cells were plated at appropriate seeding density in 96-well plates and allowed to attach overnight. Next day, cells were treated with media containing DMSO alone as control or with ATA (1 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM) for 12, 24, 48, and 72 h. Cell viability was determined at the end of each time-period using CCK-8 reagent. Absorbance was measured at 450 nm with a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
Flow cytometry
To examine the mitochondrial membrane potential (MMP) after ATA-treatment, JC-1 (5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazolylcarbocyanine iodide) dye was used [62] according to the manufacturer’s instructions. For apoptosis detection, Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences Pharmingen) was employed following the manufacturer’s protocol. For cell cycle analysis, treated and untreated cells were washed with phosphate buffered saline (PBS), fixed overnight in ice-cold 70% (v/v) ethanol, and stained with a staining buffer containing RNase and propidium iodide (PI). Data acquisition was done by flow cytometry (Becton Dickinson, San Jose, CA) using Cell Quest and ModFit LT softwares.
Immunoblotting
Following ATA exposure, whole-cell lysates were isolated using KeyGEN Total Protein Extraction Kit following the manufacturer’s instructions. Protein concentrations were determined by the BCA method and protein extracts were stored at -80°C until use. For Western blot analysis, equal amounts of proteins were electrophoresed on sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes (0.45 μm, Bio-Rad Laboratories, Hercules, CA, USA) by electroblotting. Proteins were then detected with specific antibodies. For each test, an anti-β-actin or anti-GAPDH antibody was used as loading control. Protein signals on nitrocellulose membranes were assessed with ChemiDoc XRS imaging densitometer (Bio-Rad) using Quantity One software program (Bio-Rad Laboratories, CA, USA).
Measurement of ROS production
The intracellular ROS levels were determined spectroflurometrically using the membrane-permeable fluorescent probe 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime). Briefly, cells (2.5 × 104 per well) were seeded in 96-well black wall/clear bottom microplates. After allowing cells to attach overnight, 100 μL of DCFH-DA staining solution (5 μM) was added to each well and cells were incubated in the dark at 37°C for 1 h. At the end of the incubation time, cells were washed twice with serum-free media and 100 μL of ATA dilutions (5 μM, 10 μM, and 20 μM) were added to each well (six wells per concentration). Background wells (untreated or ATA treated cells) as well as blank wells (media only) were included in each test. Fluorescence signal was then monitored time-dependently using a fluorescent plate reader (Tecan Austria Gmbh) with Magellan software. Blank readings were subtracted from all measurements and relative ROS production was expressed as fold change in fluorescence compared with the fluorescence of the corresponding control.
Transwell migration and wound healing assays
Transwell motility assays were performed as previously described [63] with minor modifications. The chemoattractant used for these tests was complete media. Assays were performed using 8-μm transwell inserts (Millipore). Migrated cells were fixed with 4% paraformaldehyde and stained with Crystal Violet. For wound healing tests, confluent monolayers of cells were scratched with a micro pipette tip. All assays were monitored over a 48 h-period. Pictures of five random fields were captured with an Olympus IX71 microscope. All experiments were performed in triplicate.
Capillary-like tube formation (HUVEC) assay
Growth factor-depleted Matrigel from BD Pharmingen (San Jose, CA, USA) was applied to a 96-well tissue culture plate (50 μl per well) [64]. Following polymerization, human umbilical vein endothelial cells (HUVECs) were seeded at a density of 2 × 104 cells per well (final volume 180 μL) on the polymerized Matrigel in the presence or absence of ATA (1 μM, 5 μM, and 15 μM). Plate was incubated at 37°C, 5% CO2 for 6 h. Photographs of five random fields were taken under an inverted microscope and tube formation was quantified using Adobe Photoshop software. The inhibition of tube formation was expressed relative to untreated control wells which were set at 100%.
In vivo study
5–6 weeks old female BALB/c nude mice weighing ~20 g each were purchased from Shanghai Rubicam Laboratory Animal Ltd (Shanghai, China) and housed in a controlled environment at 22°C with a 12 h light/dark cycle. MDA-MB-453 cells with ~92% viability were subcutaneously injected into the right flank of each mouse (2 × 107 per mouse). After tumors grew to ~90 mm3, mice were randomly selected (6 mice/group) for the intraperitoneal administration of either ATA at a dose of 35 mg/kg or vehicle three times per week. The positive control group was treated by gastric gavage with lapatinib (80 mg/kg) twice daily. The body weight of each mouse was recorded three times a week (Monday, Wednesday, and Friday). At the same time, solid tumor volume (in cubic millimeters) was determined using digital Vernier caliper measurements and the formula of a × b2 × 0.5, where a is the longest diameter of the tumor and b is the shortest diameter of the tumor. At the end of the treatment period, mice were sacrificed and tumors were excised. All animal procedures were approved by the Animal Ethics Committee of China Pharmaceutical University, Nanjing University, and Laboratory Animal Management Committee of Jiangsu Province.
Statistical analysis
Student’s two-tailed t test was used to determine the statistical significance of difference in the measured variables between control and treated groups. For all analyses, a p value less than 0.05 was considered statistically significant.
GRANT SUPPORT
This study was partially supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Fundamental Research Funds for the Central Universities (YD2014SK0002), the 111 Project (111-2-07), 333 high level project of Jiangsu Province (BRA2014245), and the National 12th Five-year Plan “Major Scientific and Technological Special Project for Significant New Drugs Creation” project (NO.2012ZX09504001-001).
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
REFERENCES
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127:2893–2917.
2. Li Y, Luo Q, Yuan L, Miao C, Mu X, Xiao W, Li J, Sun T, Ma E. JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol Appl Pharmacol. 2012; 263:21–31.
3. Kolibaba KS, Druker BJ. Protein tyrosine kinases and cancer. Biochim Biophys Acta. 1997; 1333:F217–248.
4. Shah S, Chen B. Testing for HER2 in Breast Cancer: A Continuing Evolution. Patholog Res Int. 2011; 2011:903202.
5. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989; 244:707–712.
6. Isola J, Chu L, DeVries S, Matsumura K, Chew K, Ljung BM, Waldman FM. Genetic alterations in ERBB2-amplified breast carcinomas. Clin Cancer Res. 1999; 5:4140–4145.
7. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. 2010; 1805:141–152.
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
9. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007; 7:763–777.
10. Szutowicz A, Kwiatkowski J, Angielski S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br J Cancer. 1979; 39:681–687.
11. Yancy HF, Mason JA, Peters S, Thompson CE, 3rd, Littleton GK, Jett M, Day AA. Metastatic progression and gene expression between breast cancer cell lines from African American and Caucasian women. J Carcinog. 2007; 6:8.
12. Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, Park BW, Kim KS. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem. 2007; 282:26122–26131.
13. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012; 279:2610–2623.
14. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 2012; 72:3709–3714.
15. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005; 8:311–321.
16. Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005; 24:6314–6322.
17. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008; 68:8547–8554.
18. Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther. 2012; 11:1925–1935.
19. Chypre M, Zaidi N, Smans K. ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun. 2012; 422:1–4.
20. Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, Sukhatme VP. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol. 2012; 227:1709–1720.
21. Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009; 100:1369–1372.
22. Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007; 67:8180–8187.
23. Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005; 65:6719–6725.
24. De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003; 63:3799–3804.
25. Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008; 41:59–85.
26. Mukherjee A, Wu J, Barbour S, Fang X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J Biol Chem. 2012; 287:24990–25000.
27. Lo C, Lai TY, Yang JS, Yang JH, Ma YS, Weng SW, Lin HY, Chen HY, Lin JG, Chung JG. Gallic acid inhibits the migration and invasion of A375.S2 human melanoma cells through the inhibition of matrix metalloproteinase-2 and Ras. Melanoma Res. 2011; 21:267–273.
28. Bishayee A, Ahmed S, Brankov N, Perloff M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front Biosci (Landmark Ed). 2011; 16:980–996.
29. Jang SI, Jeong SI, Kim KJ, Kim HJ, Yu HH, Park R, Kim HM, You YO. Tanshinone IIA from Salvia miltiorrhiza inhibits inducible nitric oxide synthase expression and production of TNF-alpha, IL-1beta and IL-6 in activated RAW 264.7 cells. Planta Med. 2003; 69:1057–1059.
30. Zhou Z, Zheng J, Xu W. [Study on the effect of ofloxacin and tanshinone II A on human leukocyte chemotactic migration in vitro]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao, 1997; 19:232–235.
31. Sung HJ, Choi SM, Yoon Y, An KS. Tanshinone IIA, an ingredient of Salvia miltiorrhiza BUNGE, induces apoptosis in human leukemia cell lines through the activation of caspase-3. Exp Mol Med. 1999; 31:174–178.
32. Yoon Y, Kim YO, Jeon WK, Park HJ, Sung HJ. Tanshinone IIA isolated from Salvia miltiorrhiza BUNGE induced apoptosis in HL60 human premyelocytic leukemia cell line. J Ethnopharmacol. 1999; 68:121–127.
33. Tang Z, Tang Y, Fu L. Growth inhibition and apoptosis induction in human hepatoma cells by tanshinone II A. J Huazhong Univ Sci Technolog Med Sci. 2003; 23:166–168, 172.
34. Yuan SL, Wei YQ, Wang XJ, Xiao F, Li SF, Zhang J. Growth inhibition and apoptosis induction of tanshinone II-A on human hepatocellular carcinoma cells. World J Gastroenterol. 2004; 10:2024–2028.
35. Wang X, Wei Y, Yuan S, Liu G, Lu Y, Zhang J, Wang W. Potential anticancer activity of tanshinone IIA against human breast cancer. Int J Cancer. 2005; 116:799–807.
36. Tian HL, Yu T, Xu NN, Feng C, Zhou LY, Luo HW, Chang DC, Le XF, Luo KQ. A novel compound modified from tanshinone inhibits tumor growth in vivo via activation of the intrinsic apoptotic pathway. Cancer Lett. 2010; 297:18–30.
37. Gao Y, Islam MS, Tian J, Lui VW, Xiao D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014; 349:15–25.
38. Zaytseva YY, Rychahou PG, Gulhati P, Elliott VA, Mustain WC, O'Connor K, Morris AJ, Sunkara M, Weiss HL, Lee EY, Evers BM. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012; 72:1504–1517.
39. Wang Y, Shen L, Pang Y, Qiao Z, Liu P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep. 2012; 27:1156–1162.
40. Hanai JI, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013; 4:e696.
41. Migita T, Okabe S, Ikeda K, Igarashi S, Sugawara S, Tomida A, Taguchi R, Soga T, Seimiya H. Inhibition of ATP citrate lyase induces an anticancer effect via reactive oxygen species: AMPK as a predictive biomarker for therapeutic impact. Am J Pathol. 2013; 182:1800–1810.
42. Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 2003; 63:7330–7337.
43. Borgese N, Francolini M, Snapp E. Endoplasmic reticulum architecture: structures in flux. Curr Opin Cell Biol. 2006; 18:358–364.
44. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014; 5:112.
45. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998; 12:982–995.
46. Guan L, Han B, Li Z, Hua F, Huang F, Wei W, Yang Y, Xu C. Sodium selenite induces apoptosis by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction in human acute promyelocytic leukemia NB4 cells. Apoptosis. 2009; 14:218–225.
47. Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005; 25:9554–9575.
48. Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998; 273:33533–33539.
49. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001; 2:127–137.
50. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004; 319:1–11.
51. Kourtidis A, Srinivasaiah R, Carkner RD, Brosnan MJ, Conklin DS. Peroxisome proliferator-activated receptor-gamma protects ERBB2-positive breast cancer cells from palmitate toxicity. Breast Cancer Res. 2009; 11:R16.
52. Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A. 2004; 101:10715–10720.
53. Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses. 2005; 64:997–1001.
54. Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep. 2007; 18:973–980.
55. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007; 31:769–776.
56. Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A. 1994; 91:6379–6383.
57. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011; 25:1895–1908.
58. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014; 15:156–169.
59. Spagnuolo G, D'Anto V, Cosentino C, Schmalz G, Schweikl H, Rengo S. Effect of N-acetyl-L-cysteine on ROS production and cell death caused by HEMA in human primary gingival fibroblasts. Biomaterials. 2006; 27:1803–1809.
60. Kumar R, Yarmand-Bagheri R. The role of HER2 in angiogenesis. Semin Oncol. 2001; 28:27–32.
61. Sharma PS, Sharma R, Tyagi T. VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present and future. Curr Cancer Drug Targets. 2011; 11:624–653.
62. Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997; 411:77–82.
63. Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999; 147:631–644.
64. Soeda S, Kozako T, Iwata K, Shimeno H. Oversulfated fucoidan inhibits the basic fibroblast growth factor-induced tube formation by human umbilical vein endothelial cells: its possible mechanism of action. Biochim Biophys Acta. 2000; 1497:127–134.