
Introduction
Despite being identified as particularly aggressive with high rates of relapse and poor survival over a decade ago, treatment and management of triple-negative breast cancer (TNBC) remains a significant clinical problem [1, 2]. Attacking these tumors with anthracycline-based chemotherapies does not improve outcomes [3]. By virtue of the fact that TNBCs are distinguished by the expression of the epidermal growth factor receptor (EGFR) reliance on the MAPK pathway may be their Achilles heel [4], a notion supported by small-molecule screens [5]. Unfortunately, EGFR inhibitors were met with repeated disappointment in clinical trials suggesting that other constituents of the MAPK pathway may be better targets. Large screening efforts using small interfering RNAs identified RPS6KA3 (encoding RSK2) as being absolutely required to sustain the growth of estrogen-receptor negative breast cancer suggesting it may be a relevant target [6, 7]. Soon following, RSK2 was validated as a target for TNBC in vitro and in vivo with the unique ability to eliminate the cancer stem cell (CSC) population which is believed to give rise to tumor recurrence [8]. In patients, RPS6KA3 mRNA is associated with TNBC [9] and poor outcome [8]. In the genesis of TNBC, RSK1 and RSK2 are upregulated as a consequence of expression of the Y-box binding protein-1 (YB-1) oncogene [10]. To date, there have been a small number of pan-RSK inhibitors reported, notably SL0101 [11], BI-D1870 [12], and luteolin [13]. However these small molecules are not highly specific for RSK and therefore more selective small molecules were recently developed, LJI308 and LJH685 [14]. Herein, we evaluated the potential of the new pan-RSK inhibitor LJI308 in inhibiting the growth and survival of TNBC. Further, we addressed its utility in preventing tumor recurrence by eliminating the CSC population, which we show is resistant to most traditional chemotherapies.
Results
Our group recently reported that induction of YB-1 could transform HMECs into carcinoma cells through a CSC-enriched intermediate (Fig. 1A), the phenotypes of which are summarized in Table 1 [10]. This process was driven by a self-activation loop between YB-1 and RSK (Fig. 1B) [10]. The tumorigenicity of these transformed cells was dependent on the level of YB-1 oncogene expression (Fig. 1C) and also correlated with RSK (Fig. 1D). Cultures established following long-term YB-1 over-expression (HTRY-LT1 and HTRY-LT2) formed tumors when transplanted into mice that were molecularly subtyped as TNBC [10]. The sensitivity of these cell lines to three commonly utilized therapeutic agents was measured at their approximate IC50 values reported in the established MDA-MB-231 TNBC breast cancer cell line: 50 µM 5-fluorouracil [15], 10 µM gefitinib [16], and 10 nM doxorubicin [17] (Fig. 1E). The HTRY-LT cell lines exhibited broad resistance to these therapies (Fig. 1E). We therefore questioned whether administration of RSK inhibitors could increase the efficacy of conventional chemotherapy. HTRY-LT cells were treated with a sub-lethal dose of RSK inhibitor, 1 µM BI-D1870 or 10 µM luteolin, alone or in combination with 5-fluorouracil, gefitinib, and doxorubicin. A sub-lethal dose of the RSK inhibitors were selected based on the respective IC50 values reported in other TNBC models [8, 13]. As expected, treatment with BI-D1870 and luteolin alone had little to no effect on cell viability; however, a synergistic combinatory effect was observed with 5-fluorouracil, gefitinib, and doxorubicin (Fig. 1F). Specifically, cell viability decreased by more than 80% with the combination treatments (Fig. 1F). These data suggest that a RSK inhibitor could be used in combination with traditional chemotherapy to increase drug efficacy.
Table 1: Phenotypic characterization of cell lines.
HTRZ (non-tumorigenic) |
HTRY (CSC-enriched) |
HTRY-LT (TNBC) |
|
Spheroid formation |
+ |
++ |
++ |
Invasion |
- |
+ |
++ |
Soft agar growth |
- |
- |
+ |
Tumor-initiation in vivo |
- |
- |
+ |
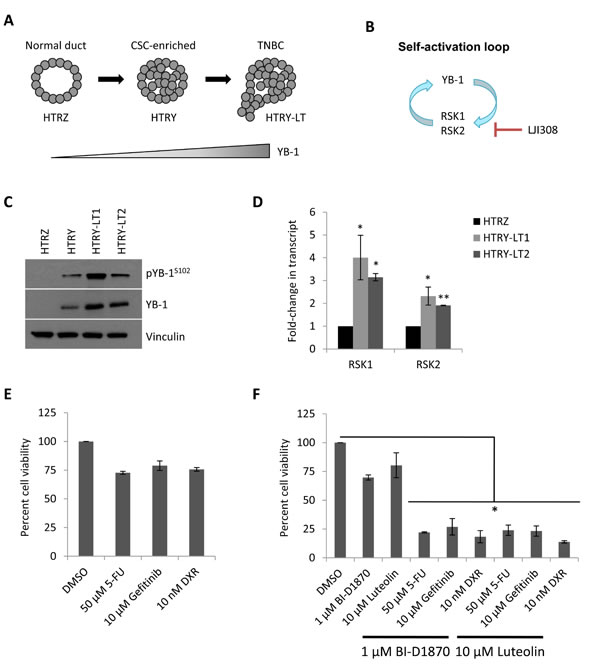
Figure 1: RSK is over-expressed in TNBC and confers multi-drug resistance. (A) Schematic representation of YB-1-driven breast cancer progression. Long-term YB-1 induction (30 day) transformed HMECs into cancerous cells of the TNBC subtype. (B) Schematic diagram depicts a RSK/YB-1 self-reinforcing loop. RSK induces ongoing YB-1 phosphorylation as well as YB-1-dependent RSK expression. (C) Immunoblot of YB-1 in HTRZ, HTRY (96-hour induction), and HTRY-LT cell lines. RSK expression had previously been characterized in these lysates (see [10]). Vinculin was used as a loading control in both instances. (D) Quantitative RT-PCR of RPS6KA1 (RSK1) and RPS6KA3 (RSK2) transcript in HTRY-LT cell lines relative to HTRZ cells. *, P < 0.05; **, P < 0.01. (E) Viability of HTRY-LT1 cells treated with 5-fluorouracil, gefitinib, or doxorubicin for 96 hours was assessed by Cellomics ArrayScan. Data is presented relative to DMSO control. 5-FU, 5-fluorouracil; DXR, doxorubicin. (F) Viability of HTRY-LT1 cells treated with BI-D1870 and luteolin alone or in combination with 5-fluorouracil, gefitinib, and doxorubicin for 96 hours. Data is presented relative to DMSO control. *, P < 0.05. 5-FU, 5-fluroruracil; DXR, doxorubicin.
A hurdle in translating current generation RSK inhibitors into the clinic has been their lack of specificity, for example, BI-D1870 also reportedly targets Aurora B and PLK1 as well as other kinases at high doses [12, 14]. Therefore, there has been a concerted effort to develop highly specific and potent RSK inhibitors, the most recent being LJI308 [14]. Treating HTRY-LT cell lines with increasing doses of LJI308 (1-10 µM) decreased cell viability by up to 90%; however, little to no effect was observed in the non-tumorigenic HTRZ control cells after 4 or 8 days (Fig. 2A, 2B). The cell lines proliferate at a similar rate [10] and thus the decrease in cell growth and viability can be directly attributed to LJI308 treatment. Notably, LJI308 also suppressed the growth of HTRY-LT cells in 3-dimensional soft agar cultures (Fig. 2C). A similar result was observed with the TNBC cell lines MDA-MB-231 and SUM149 (Fig. 2D). The decrease in cell growth was attributed to apoptosis based on an increase in annexin V-positive apoptotic cells following LJI308 treatment (Fig. 3A). These effects were mediated, at least in part, by the ability of LJI308 to inhibit the activation (phosphorylation) of the oncogenic YB-1 transcription factor (Fig. 3B).
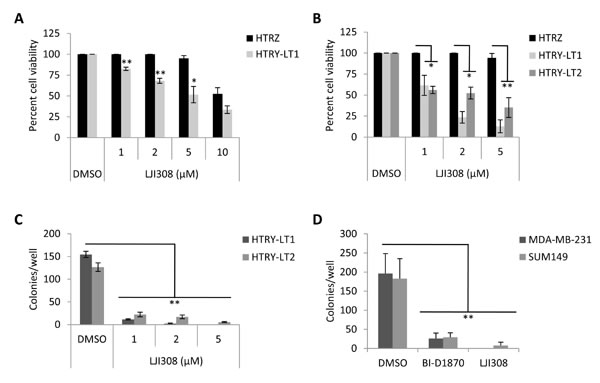
Figure 2: Inhibiting RSK suppresses the growth of TNBC cell lines. (A) Viability of HTRZ and HTRY-LT1 cell lines treated with increasing doses of LJI308 (1 - 10 µM) for 96 hours. DMSO treated cells served as a control. *, P < 0.05; **, P < 0.01. (B) HTRZ, HTRY-LT1, and HTRY-LT2 cell lines were treated with increasing doses of LJI308 (1 - 5 µM) for 8 days, with repeat dosing at 96 hours. DMSO treated cells served as a control. *, P < 0.05; **, P < 0.01. (C) Soft agar colony formation of HTRY-LT1 and HTRY-LT2 cells grown in the presence of LJI308 or DMSO control. Drug was replenished weekly and colonies quantified at 28 days. **, P < 0.01. (D) Soft agar colony formation of MDA-MB-231 and SUM149 cells grown in the presence of BI-D1870 (10 µM) or LJI308 (10 µM) assessed after 21 days. **, P < 0.01.
Due to the proposed role of CSCs in mediating tumor recurrence [18, 19], we wanted to evaluate whether LJI308 could not only eradicate bulk tumor cells but also this highly tumorigenic subpopulation. HTRY-LT cells were grown in spheroid cultures to enrich for CSCs [20], which could be repressed by treatment with LJI308 (Fig. 4A). This effect was mirrored using siRNA against RSK1 and RSK2 or alternatively by exposing cells to BI-D1870 (Fig. 4B). We subsequently isolated the CD44+/CD49f+ CSC population from the HTRY-LT cell line by FACS (Fig. 4C). As expected the CSCs over-expressed CD44 (Fig. 4D) and were found to be largely resistant to paclitaxel, 5-fluorouracil, gefitinib, and doxorubicin chemotherapy relative to the CD44-/CD49f- non-CSC population. We therefore questioned whether CSCs could be eradicated by a lethal IC90 dose of RSK inhibitor: 2 µM BI-D1870 [8], 5 µM LJI308, or 50 µM luteolin [13]. We observed a significant decrease in both CSC and non-CSC cell viability, with LJI308 demonstrating the greatest efficacy (Fig. 4E). These data strongly suggest that CSCs are refractory to traditional chemotherapy, but sensitive to RSK inhibitors.
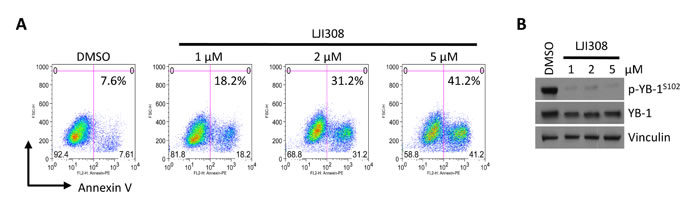
Figure 3: LJI308 kills TNBC correlative with YB-1 inhibition. (A) Annexin V staining of HTRY-LT1 cells treated with LJI308 analyzed by flow cytometry at 6 days post-treatment. DMSO treated cells served as a control. (B) Immunoblot of total and phosphorylated YB-1 in HTRY-LT1 cells treated with increasing dose of LJI308 for 72 hours. Vinculin was used as a loading control.
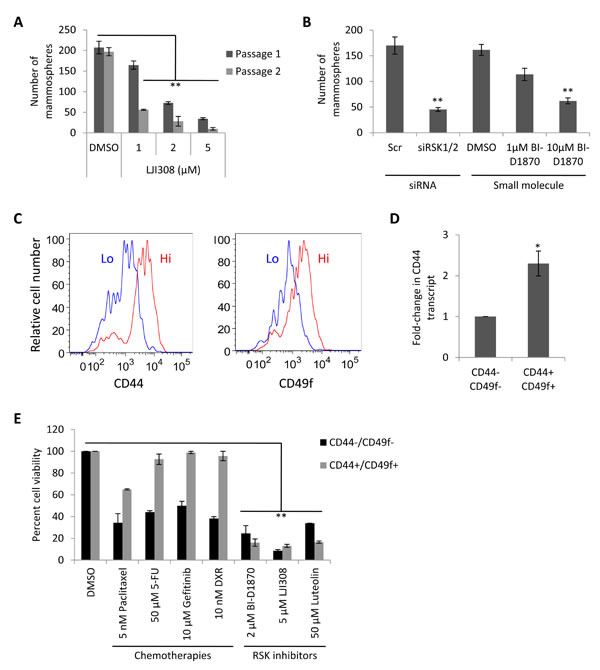
Figure 4: CSCs are sensitive to RSK inhibition. (A) HTRY-LT1 cells were propagated as mammospheres in the presence of LJI308 (passage 1). After 7 days, the spheroids were dissociated and serially passaged into secondary cultures absent of drug (passage 2). **, P < 0.01. (B) HTRY-LT1 cells were pre-treated with siRNA targeting RSK1 and RSK2 (20 nM) or the small molecule RSK inhibitor BI-D1870 for 48 hours prior to establishing mammosphere cultures. BI-D1870 pre-treated cells were plated in cultures supplemented with BI-D1870. The number of spheroids was quantified after 7 days. **, P < 0.01. Scr, scrambled control. (C) Flow cytometric analysis of CD44+/CD49f+ and CD44-/CD49f- populations in HTRY-LT1 cells. (D) Quantitative RT-PCR of CD44 transcript in CD44+/CD49f+ CSC and CD44-/CD49f- cell populations fractioned from the HTRY-LT1 cell line by FACS. *, P < 0.05. (E) Viability of CD44+/CD49f+ CSC and CD44-/CD49f- populations isolated from the HTRY-LT1 cell line treated with paclitaxel, 5-fluorouracil, gefitinib, doxorubicin, BI-D1870, LJI308, and luteolin for 96 hours. DMSO treated cells served as a control. **, P < 0.01. 5-FU, 5-fluorouracil; DXR, doxorubicin.
Discussion
Currently, there are no targeted therapies for TNBC, the most lethal form of breast cancer. While conventional chemotherapy is widely used and initially highly effective, the effect is short-lived; these tumors rapidly re-emerge and disseminate within one year of treatment [21]. Therefore, there is a strong clinical rationale that underlies the search for new targeted therapies. RSK represents a promising targeting point as it phosphorylates numerous downstream substrates associated with cell proliferation and motility [22]. We report the RSK inhibitor LJI308 as one of the first potent and effective targeted therapies for TNBC that, unlike currently utilized drugs, exhibits efficacy in eliminating the CSC population.
CSCs are widely accepted to be strongly therapy resistant and drive tumor dissemination and poor outcome in many cancer types, including breast [23]. Because these cells are enriched in residual breast cancer tumors following chemotherapy they may be responsible for driving relapse [23]. Accordingly, targeting CSCs could represent a promising therapeutic strategy for TNBC, a notion supported by the fact that these tumors have the highest percentage of CD44-positive CSCs compared to the other breast cancer subtypes [24]. Our group has previously reported that TNBC CSCs are dependent on RSK for growth and survival [8]. We have now further validated RSK as a bona fide target in new models of TNBC and identified a specific inhibitor LJI308 that is active in TNBC. Treatment with LJI308 induced apoptosis to eradicate both the CSC and non-CSC populations, suggesting it could be a “silver bullet” that not only debulks the tumor but also targets the “seeds” for cancer regrowth. This is particularly relevant in light of recent studies suggesting that non-CSCs are plastic and can acquire a CSC phenotype [25, 26]. Thus, eradication of existing CSCs may not be sufficient and RSK inhibitors that also target the non-CSCs may be mandatory.
While a major concern in the development of CSC directed therapies is that signaling pathways active in CSCs may also be crucial for normal stem cell survival it has been reported that RSK2 knockout mice develop a normal haematopoietic stem cell compartment [27]. Moreover, our group has shown that treating hematopoietic stem cells with BI-D1870 has little effect at doses toxic to CSCs [8]. Together, this suggests RSK is uniquely linked to the CSC population and, as such, targeting it with inhibitors such as LJI308 has important implications in overcoming drug resistance and treating TNBC.
While our present research has focused on TNBC, RSK is also expressed in other subtypes of breast cancer. In HER2 amplified breast cancer, for example, activation of RSK has been directly linked to trastuzumab resistance [28]. The impact of this study likely transcends breast cancer as RSK has also been linked to drug resistance in other cancer types including prostate cancer [29], melanoma [30], and pediatric medulloblastoma [31].
In conclusion, we propose that the use of RSK inhibitors, such as LJI308, in combination with conventional chemotherapy could be used to overcome drug resistance and improve survival in patients with TNBC. The unique ability of these drugs to target the CSC population might provide sustained remission.
Materials and Methods
Cell lines and treatments
H16N2 HMECs expressing YB-1 (HTRY) or LacZ (HTRZ) under the control of a tetracycline-inducible promoter were cultured in F-12 media (Gibco, Burlington, Canada) containing 10% fetal bovine serum (FBS), as previously described [10, 32]. MDA-MB-231 (ATCC, Manassas, VA, USA) and SUM149 (Asterand, Detroit, MI, USA) cells were cultured as recommended. Cells were treated with the following drugs: 5-fluorouracil (Sigma-Aldrich, Oakville, Canada), BI-D1870 (Centre for Drug Research and Development, Vancouver, Canada), doxorubicin hydrochloride (Sigma-Aldrich), gefitinib (AstraZeneca, Mississauga, Canada), LJI308 (Novartis Pharma AG, Basel, Switzerland), luteolin (Sigma-Aldrich), and paclitaxel (Sigma-Aldrich).
Quantitative PCR
RNA was isolated using an RNeasy mini kit (Qiagen, Mississauga, Canada) and reverse transcribed using SuperScript III (Invitrogen, Burlington, Canada). Pre-designed Taqman Gene Expression Assays labelled with a FAM reporter (Applied Biosystems, Streetsville, Canada) were used to detect transcript level.
Immunoblotting
Proteins were harvested in erythrocyte lysis buffer (ELB) supplemented with protease and phosphatase inhibitors (Roche, Mississauga, Canada), run on a 12% SDS polyacrylamide gel, and transferred to a nitrocellulose membrane. The following antibodies were used: YB-1 (clone EP2708Y; Epitomics, Burlingame, CA, USA), p-YB-1Ser102 (Cell Signaling, Danvers, MA, USA), and vinculin (clone V284; Millipore, Etobicoke, Canada).
Cell viability assay
Cells were plated at a density of 5000 cells/well into 96-well plates. At the endpoint, cells were fixed with 2% paraformaldehyde/PBS and stained with Hoechst 33342 (1 µg/ml; Sigma-Aldrich) for 30 minutes at room temperature. Plate analysis was performed using an ArrayScan VTI high content screening instrument (Thermo Scientific, Waltham, MA, USA). Hoechst 33342-stained nuclei were used as a measure of cellularity.
Apoptosis analysis
Apoptosis analysis was performed with flow cytometry using the Annexin V:PE Apoptosis Detection Kit (BD Biosciences, Mississauga, Canada). Cells were stained with annexin V (1:20) on ice for 20 minutes prior to analysis.
Soft agar assay
Cells were embedded in soft agar medium (0.6% agarose) at a density of 5 x 103 per well in a 24-well plate. Drug treatments were replenished weekly. Colonies were quantified at 21-28 days, depending on the cell line.
Mammosphere assay
Cells were washed through a 40 μm filter to obtain a single-cell suspension and seeded at 2 x 104 per well into ultra-low attachment 6-well culture plates (Corning, Corning, NY, USA) in MammoCult Basal media (StemCell Technologies, Vancouver, Canada). Spheres with a minimum diameter of 50 μm were counted at 7 days. For serial passaging, mammospheres were collected by centrifugation at 350g for 2 minutes, dissociated with 0.25% trypsin, and re-seeded.
Fluorescence activated cell sorting (FACS)
Cells were prepared according to standard protocols and suspended in 2% FBS/PBS on ice before FACS. Cells were sorted on a BD FACSCalibur using PE-conjugated CD44 (clone 515; BD Biosciences) and FITC-conjugated CD49f (clone GoH3; BD Biosciences).
Acknowledgments
We thank Dr. Isabelle Berquin (Wake Forest University School of Medicine) for providing the HMECs with Tet-On YB-1, Dr. Kristen Hurov and Dr. A.B. Jefferson (Novartis Institutes for BioMedical Research) for LJI308, and Lisa Xu at the Child and Family Research Institute FACS core for technical assistance.
Funding
This work was supported by the Canadian Institutes of Health Research (S. Dunn), with additional support from a Frederick Banting and Charles Best Doctoral Research Award (A. Davies).
Conflicts of Interest
The authors declare no conflict of interest.
References
1. Pal SK, Childs BH and Pegram M. Triple negative breast cancer: unmet medical needs. Breast cancer research and treatment. 2011; 125:627-636.
2. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:10869-10874.
3. Cheang MC, Voduc KD, Tu D, Jiang S, Leung S, Chia SK, Shepherd LE, Levine MN, Pritchard KI, Davies S, Stijleman IJ, Davis C, Ebbert MT, Parker JS, Ellis MJ, Bernard PS, et al. Responsiveness of intrinsic subtypes to adjuvant anthracycline substitution in the NCIC.CTG MA.5 randomized trial. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18:2402-2412.
4. Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM and Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14:1368-1376.
5. Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, Sartor CI, Rieger-House T, Bernard PS, Carey LA and Perou CM. EGFR associated expression profiles vary with breast tumor subtype. BMC genomics. 2007; 8:258.
6. Speers C, Tsimelzon A, Sexton K, Herrick AM, Gutierrez C, Culhane A, Quackenbush J, Hilsenbeck S, Chang J and Brown P. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15:6327-6340.
7. Brough R, Frankum JR, Sims D, Mackay A, Mendes-Pereira AM, Bajrami I, Costa-Cabral S, Rafiq R, Ahmad AS, Cerone MA, Natrajan R, Sharpe R, Shiu KK, Wetterskog D, Dedes KJ, Lambros MB, et al. Functional viability profiles of breast cancer. Cancer discovery. 2011; 1:260-273.
8. Stratford AL, Reipas K, Hu K, Fotovati A, Brough R, Frankum J, Takhar M, Watson P, Ashworth A, Lord CJ, Lasham A, Print CG and Dunn SE. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem cells. 2012; 30:1338-1348.
9. Hsu YH, Yao J, Chan LC, Wu TJ, Hsu JL, Fang YF, Wei Y, Wu Y, Huang WC, Liu CL, Chang YC, Wang MY, Li CW, Shen J, Chen MK, Sahin AA, et al. Definition of PKC-alpha, CDK6, and MET as Therapeutic Targets in Triple-Negative Breast Cancer. Cancer research. 2014; 74:4822-4835.
10. Davies AH, Reipas KM, Pambid MR, Berns R, Stratford AL, Fotovati A, Firmino N, Astanehe A, Hu K, Maxwell C, Mills GB and Dunn SE. YB-1 transforms human mammary epithelial cells through chromatin remodeling leading to the development of basal-like breast cancer. Stem cells. 2014; 32:1437-1450.
11. Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM and Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer research. 2005; 65:1027-1034.
12. Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P and Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. The Biochemical journal. 2007; 401:29-38.
13. Reipas KM, Law JH, Couto N, Islam S, Li Y, Li H, Cherkasov A, Jung K, Cheema AS, Jones SJ, Hassell JA and Dunn SE. Luteolin is a novel p90 ribosomal S6 kinase (RSK) inhibitor that suppresses Notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget. 2013; 4:329-345.
14. Aronchik I, Appleton BA, Basham SE, Crawford K, Del Rosario M, Doyle LV, Estacio WF, Lan J, Lindvall MK, Luu CA, Ornelas E, Venetsanakos E, Shafer CM and Jefferson AB. Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Molecular cancer research : MCR. 2014; 12:803-812.
15. Takahashi K, Tanaka M, Inagaki A, Wanibuchi H, Izumi Y, Miura K, Nagayama K, Shiota M and Iwao H. Establishment of a 5-fluorouracil-resistant triple-negative breast cancer cell line. International journal of oncology. 2013; 43:1985-1991.
16. Corkery B, Crown J, Clynes M and O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2009; 20:862-867.
17. Smith L, Watson MB, O’Kane SL, Drew PJ, Lind MJ and Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Molecular cancer therapeutics. 2006; 5:2115-2120.
18. Visvader JE and Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell stem cell. 2012; 10:717-728.
19. Dean M, Fojo T and Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005; 5:275-284.
20. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA and Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer research. 2005; 65:5506-5511.
21. Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U and Harbeck N. Triple-negative breast cancer--current status and future directions. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2009; 20:1913-1927.
22. Stratford AL and Dunn SE. The promise and challenges of targeting RSK for the treatment of cancer. Expert opinion on therapeutic targets. 2011; 15:1-4.
23. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J and Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008; 100:672-679.
24. Park SY, Lee HE, Li H, Shipitsin M, Gelman R and Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16:876-887.
25. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C and Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011; 146:633-644.
26. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN and Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature neuroscience. 2013; 16:1373-1382.
27. Kang S, Elf S, Dong S, Hitosugi T, Lythgoe K, Guo A, Ruan H, Lonial S, Khoury HJ, Williams IR, Lee BH, Roesel JL, Karsenty G, Hanauer A, Taunton J, Boggon TJ, et al. Fibroblast growth factor receptor 3 associates with and tyrosine phosphorylates p90 RSK2, leading to RSK2 activation that mediates hematopoietic transformation. Molecular and cellular biology. 2009; 29:2105-2117.
28. Astanehe A, Finkbeiner MR, Krzywinski M, Fotovati A, Dhillon J, Berquin IM, Mills GB, Marra MA and Dunn SE. MKNK1 is a YB-1 target gene responsible for imparting trastuzumab resistance and can be blocked by RSK inhibition. Oncogene. 2012; 31:4434-4446.
29. Shiota M, Itsumi M, Yokomizo A, Takeuchi A, Imada K, Kashiwagi E, Inokuchi J, Tatsugami K, Uchiumi T and Naito S. Targeting ribosomal S6 kinases/Y-box binding protein-1 signaling improves cellular sensitivity to taxane in prostate cancer. The Prostate. 2014; 74:829-838.
30. Ray-David H, Romeo Y, Lavoie G, Deleris P, Tcherkezian J, Galan JA and Roux PP. RSK promotes G2 DNA damage checkpoint silencing and participates in melanoma chemoresistance. Oncogene. 2013; 32:4480-4489.
31. Pambid MR, Berns R, Adomat HH, Hu K, Triscott J, Maurer N, Zisman N, Ramaswamy V, Hawkins CE, Taylor MD, Dunham C, Guns E and Dunn SE. Overcoming resistance to Sonic Hedgehog inhibition by targeting p90 ribosomal S6 kinase in pediatric medulloblastoma. Pediatric blood & cancer. 2014; 61:107-115.
32. Berquin IM, Pang B, Dziubinski ML, Scott LM, Chen YQ, Nolan GP and Ethier SP. Y-box-binding protein 1 confers EGF independence to human mammary epithelial cells. Oncogene. 2005; 24:3177-3186.