
BCR-ABL in Chronic Myeloid Leukemia and its targeting by TKIs
Chronic Myeloid leukemia (CML) is defined by the formation and presence of the Philadelphia (Ph) chromosome, which results from the reciprocal chromosomal translocation t(9;22) (q34;q11) [1, 2]. The protein product of the abnormal Bcr-Abl fusion gene is the oncoprotein BCR-ABL, which is expressed in CML and Ph+ ALL [2, 3]. BCR-ABL retains the tyrosine kinase ability of c-ABL but, contrary to c-ABL it is constitutively localized in the cytoplasm, resulting in the engagement and activation of multiple pro-proliferative and anti-apoptotic cascades in transformed cells [2, 3]. Among the cellular cascades activated by BCR-ABL there are mitogen activated protein kinase (MAPK) and phosphatidyl inositol 3’ kinase/AKT/mammalian target of rapamycin (PI3’K/AKT/ mTOR) pathways [4-7].
The identification of imatinib mesylate (STI-571; Gleevec) as a small molecule ATP-pocket inhibitor of BCR-ABL dramatically re-defined the treatment of CML and had a major impact in the survival of patients with CML and Ph+ ALL [8-13]. As this agent targets directly the ABL kinase domain, its introduction in clinical oncology provided a model for potential selective and specific therapeutic interventions in other malignancies with well-defined targets [11, 12]. Imatinib mesylate, along with second-generation tyrosine kinase inhibitors (TKI) such as nilotinib and dasatinib [14-22], have changed the natural history of CML and have provided important treatment options for this leukemia that in the past was uniformly fatal.
Despite such advances in the field, mutations rendering CML and Ph+ ALL patients non-responsive to TKI’s have been identified, including the threonine 315 to isoleucine (T315I) mutation and several others, which differentially prevent binding of different TKIs to the active site of the ABL kinase domain, thereby evading inhibition [23]. Beyond mutations in the kinase domain of BCR-ABL, additional mechanisms of resistance exist [23, 24], further complicating the management of such patients. Although identification of new molecular markers may facilitate response prediction to TKIs and allow optimization of their clinical use [25], there is a need for the development of agents that overcome BCR-ABL TKI resistance. This has led to the ongoing clinical development of new TKIs such as bosutinib that has activity against several imatinib-resistant BCR-ABL mutants with the exception of T315I-BCR-ABL [26] and ponatinib that has activity against T315I-BCR-ABL [28]. Beyond efforts to develop inhibitors that can overcome resistance to first and second generation TKIs, another approach of high potential value is the selective targeting and inhibition of cellular effectors downstream of BCR-ABL. As discussed below, the PI 3’K/mTOR cascade is a prime target for such purpose and has been the focus of extensive investigations.
Hyperactivation of the PI3’K/AKT/mTOR signaling pathway by BCR-ABL
Among the multiple cellular cascades that are activated by BCR-ABL, the PI3’K/AKT/mTOR pathway [5, 28-30] is of particular interest and has been the subject of extensive efforts by many groups in the CML and Ph+ ALL research fields. One mechanism by which the PI 3’K/mTOR pathway is engaged involves increased production of reactive oxygen species (ROS) by BCR-ABL, leading to inhibition of the serine/threonine phosphatase PP1α, a negative regulator of PI3’K/AKT, ultimately resulting in hyper-activation of the pathway [31, 32]. Blocking the PI3’K/AKT signaling in BCR-ABL cells with the pharmacological inhibitor LY294002 results in increased expression of the cell cycle regulator p27Kip1 [33] and decreased expression of VEGF and HIF1α [34]. Furthermore, combining pharmacological inhibition of the PI3’ kinase with BCR-ABL kinase inhibitors such imatinib mesylate results in enhanced anti-leukemic effects in Ph+ cells in vitro [35].
Downstream of the PI3’K/AKT pathway, the mTOR signaling cascade is also hyperactive in CML [5, 7, 36-38]. mTOR is a central regulatory element in the control of mRNA translation in mammalian cells and functions as the catalytic subunit/kinase for two distinct protein complexes, TORC1 and TORC2 [39-45]. These complexes differ by in the mTOR-binding partners that they include, with TORC1 containing Raptor and TORC2 containing Rictor and mSin1 [39-45]. These complexes regulate distinct cellular processes, with TORC1 being the mediator of signals for initiation of mRNA translation and protein synthesis and TORC2 promoting survival pathways and cytoskeletal reorganization [39-45]. Previous studies have established that TORC1 and TORC2 play critical roles in growth and survival of BCR-ABL transformed cells, including myeloid (CML) and lymphoid (Ph+ ALL) cells [36-38, 46-49], underscoring the importance and relevance of the mTOR pathway in the pathogenesis and pathophysiology of Ph+ malignancies. Notably, the ATP-competitive dual mTORC1/2 inhibitors PP242 and OSI-027 have shown potent growth inhibitory and pro-apoptotic effects in a number of BCR-ABL cell lines and primary patient samples [47, 48] and in a mouse Ph+ ALL mouse model [48], suggesting that these or other similar agents with dual targeting capacities against TORC1 and TORC2 may provide a new alternative approach for the treatment of CML resistant to TKIs.
Effects of AMPK activation on BCR-ABL-transformed cells
Beyond agents that directly target and in inhibit TORCs, indirect suppression of mTOR function by modulation of the AMP-activated Protein Kinase (AMPK) pathway may provide an important alternative therapeutic approach [50]. AMPK regulates mTOR signaling both directly and indirectly. This heterotrimeric protein kinase is activated by means of phosphorylation on the Thr172 site of the α-subunit due to an increased AMP:ATP ratio [51]. Once active, AMPK phosphorylates and activates the TSC2 subunit of the TSC1/2 complex, which in turn suppresses Rheb activity, a small G-protein with regulatory functions on mTOR activation [52-54]. In addition, AMPK has been found to directly phosphorylate the Raptor subunit on Ser792, resulting in inactivation of the TORC1 complex [55].
In one of the initial studies in which the effects of modulation of AMPK in ALL cells were assessed, the AMP-analog AMPK-activator compound 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) was shown to exert antiproliferative effects on childhood ALL cells, including the SupB15 cell line which expresses the p185 BCR-ABL fusion protein [56]. In that study AICAR was found to induce phosphorylation of AMPKα on Thr172, resulting in inhibition of proliferation and cell-cycle arrest at the G1 phase, by increasing the levels of the cell cycle regulators p53, p27 and p21 [56]. The effects of AICAR were enhanced by combination with the mTOR inhibitor rapamycin in all different ALL cell lines tested in that study [56], while the growth inhibitory responses were mediated in part by engagement of the p38 MAP kinase pathway. Another study from the same group [57] established that phosphorylation of AKT on Thr308 and Ser473 increases following treatment of ALL cells with AICAR and demonstrated that AKT phosphorylation on Thr 308 is mediated by AMPK-induced IGF-1R activation and phosphorylation of IRS-1. In follow-up experiments, the authors of that work were able to demonstrate that concomitant inhibition of IGF-1R activity using a tyrosine kinase inhibitor and AMPK activation using AICAR resulted in substantially enhanced antileukemic effects [57].
Resveratrol, a naturally occurring substance found in grapes, has been shown to modulate AMPK in BCR-ABL transformed cells and to exhibit antileukemic effects [57-58]. Treatment of either imatinib mesylate-sensitive or imatinib mesylate-resistant CML cells with resveratrol resulted in cell cycle arrest and apoptotic cell death [57]. Notably, among the resistant cells that were sensitive to resveratrol, there were also cells expressing the T315I BCR-ABL mutant [57]. Subsequent studies demonstrated that resveratrol induces autophagic cell death in CML cells by a dual mechanism involving both engagement of AMPK and JNK-mediated overexpression of p62/SQSTM1 [58].
In a recent study [59], we assessed whether modulation of AMPK by AICAR and the FDA-approved anti-diabetic drug metformin [60-62], exert suppressive effects on BCR-ABL transformed cells. In that study [59] we found that both AICAR and metformin induce AMPK kinase activity, as reflected by phosphorylation of ACC and TSC2. The phosphorylation of various effectors downstream of mTOR was compromised upon treatment in all cell lines tested, including cells harboring the T315I mutation. These studies have indicated that the effects of mTOR inhibition by AICAR and metformin occur irrespectively of TKI-sensitivity [59] and further raised the prospect of using AMPK activators in the treatment of Ph+ leukemias refractory to TKIs, including CML and Ph+ ALL.
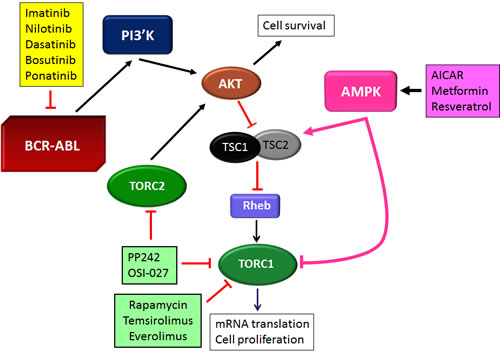
Figure 1: Different target points of AMPK and mTOR pathways in BCR-ABL transformed cells and known pharmacological agents that can be used to target them.
Conclusions and Future Directions
There have been remarkable advances in the treatment of CML over the last decade. With the introduction of first and second generation TKIs in the treatment of CML, the majority of patients achieve complete hematological, cytogenetic and molecular responses [4, 12]. Also, the introduction of imatinib mesylate, nilotinib and dasatinib in the treatment of patients with Ph+ ALL has dramatically improved their outcome. Nevertheless, large numbers of patients eventually become unresponsive to treatment due to mutations that occur either in the TKI-binding kinase domain of BCR-ABL or due to the emergence of other resistance mechanisms downstream of the kinase [24]. Approaches to target cellular effectors of BCR-ABL may provide a complementary/enhancing approach to the use of TKIs and/or help overcome resistance in cases of resistant CML or Ph+ ALL. Importantly, modulation of the AMPK pathway may allow targeting of mTOR in a distinct, non-overlapping way, from mTOR inhibitors and other suppressors of the PI3’K/AKT/mTOR cascade and AMPK activators could be potentially combined with PI 3’K/mTOR inhibitors in the treatment of refractory myeloid leukemias. Notably, the anti-tumor effects of metformin, a commonly used anti-diabetic drug, have been observed in a number of other hematologic malignancies, including acute myeloid leukemia (AML) [63, 64] and various solid tumors, including breast cancer, lung cancer and others [50, 65-77], suggesting that the clinical development of this drug in clinical oncology may offer advantages for a variety of malignancies. In fact, efforts to combine metformin with mTOR inhibitors for the treatment of solid tumors are already underway [78] and similar studies in Ph+ leukemias and other hematological malignancies are probably warranted from the emerging experimental evidence.
References
1. Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973; 243:290-93.
2. Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999; 340:1330-40.
3. Lugo TC, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990; 247:1079-82.
4. Platanias LC. Mechanisms of BCR-ABL leukemogenesis and novel targets for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2011; 52: 2-3.
5. Redig AJ, Vakana E, Platanias LC. Regulation of mammalian target of rapamycin and mitogen activated protein kinase pathways by BCR-ABL. Leuk Lymphoma. 2011; 52:45-53.
6. Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, Salgia R, Podar K, Griffin JD, Sattler M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005; 105:1717-23.
7. Parmar S, Smith J, Sassano A, Uddin S, Katsoulidis E, Majchrzak B, Kambhampati S, Eklund EA, Tallman MS, Fish EN, Platanias LC. Differential regulation of the p70 S6 kinase pathway by interferon alpha and imatinib mesylate (STI571) in chronic myelogenous leukemia cells. Blood 2005; 106:2436-43.
8. Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355:2408-17.
9. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fischer T, O’Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002; 99:3530-39.
10. Druker BJ. Imatinib as a paradigm of targeted therapies. Adv Cancer Res. 2004; 91:1-30.
11. Lydon NB, Druker BJ. Lessons learned from the development of imatinib. Leuk Res. 2004; 28:S29-38.
12. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008; 112:4808-17.
13. Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest. 2007; 117:2067-74.
14. Cortes J, Hochhaus A, Hughes T, Kantarjian H. Front-line and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. J Clin Oncol. 2011; 29:524-31.
15. Fullmer A, Kantarjian H, Cortes J, Jabbour E. New developments in the treatment of chronic myeloid leukemia and Philadelphia-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2011;52:81-91.
16. Tauchi T, Ohyashiki K. The second generation of BCR-ABL tyrosine kinase inhibitors. Int J Hematol. 2006; 83:294-00.
17. Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006; 354:2542-51.
18. Shah NP, Tran C, Lee Fy, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 305:399-01.
19. Guilhot F, Apperley J, Kim DW, Bullorsky EO, Baccarani M, Roboz GJ, Amadori S, de Souza CA, Lipton JH, Hochhaus A, Heim D, Larson RA, Branford S, Muller MC, Agarwal P, Gollerkeri A. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood. 2007; 109:4143-50.
20. Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, Facon T, Goldberg SL, Cervantes F, Niederwieser D, Silver RT, Stone RM, Hughes TP, Muller MC, Ezzeddine R, Countouriotis AM. Dasatinib induces notable hematologic and cytogenetic responses in chronic phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007; 109:2303-09.
21. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354:2531-41.
22. Kantarjian H, Jabbour E, Grimley J, Kirkpatrick P. Dasatinib. Nat Rev Drug Discov 2006; 5:717-18.
23. Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology Am Soc Hematol Educ Program. 2009; 461-476.
24. Diamond JM, Melo JV. Mechanisms of resistance to BCR-ABL kinase inhibitors. Leuk Lymphoma. 2011; 52:12-22.
25. Mitchell B, Deininger M. Techniques for risk stratification of newly diagnosed patients with chronic myeloid leukemia. Leuk Lymphoma. 2011; 52: 4-11.
26. Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011; 118:4567-76.
27. O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009; 16:401-12.
28. Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004; 18:189-18.
29. McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008; 22:708-22.
30. Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005; 65:2047-53.
31. Naughton R, Quiney C, Turner SD, Cotter TG. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia. 2009; 23:1432-40.
32. Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, Greenfield EA, Salgia R, Griffin JD. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000; 275:24273-278.
33. Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem. 2000; 275:39223-230.
34. Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002; 100:3767-75.
35. Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene 2002; 21:5868-76.
36. Mohi MG, Boulton C, Gu TL, Sternberg DW, Neuberg D, Griffin JD, Gilliland DG, Neel BG. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004; 101:3130–35.
37. Ly C, Arechiga AF, Melo JV, Walsh CM, Ong ST. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res 2003; 63:5716-22.
38. Mayerhofer M, Aichberger KJ, Florian S, Krauth MT, Hauswirth AW, Derdak S, Sperr WR, Esterbauer H, Wagner O, Marosi C, Pickl WF, Deininger M, Weisberg E, Druker BJ, Griffin JD, Sillaber C, et al. Identification of mTOR as a novel bifunctional target in chronic myeloid leukemia: dissection of growth-inhibitory and VEGF-suppressive effects of rapamycin in leukemic cells. Faseb J 2005; 19:960-62.
39. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006; 6:729- 34.
40. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007; 12:9-22.
41. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007; 12:487-02.
42. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat Rev Immunol. 2005; 5:375-86.
43. Altman JK, Sassano A, Platanias LC. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget. 2011 Jun;2(6):510-7.
44. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004; 14:1296-02.
45. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004; 6:1122-28.
46. Carayol N, Katsoulidis E, Sassano A, Altman JK, Druker BJ, Platanias LC. Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J Biol Chem 2008; 283:8601-10.
47. Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, Platanias LC. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A 2010; 107:12469-474.
48. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 2010; 16:205-13.
49. Cilloni D, Saglio G. Molecular Pathways: BCR-ABL. Clin Cancer Res. 2011 Dec 8. [Epub ahead of print].
50. Vakana E, Altman JK, Platanias LC. Targeting AMPK in the treatment of malignancies. J Cell Biochem. 2011 Sep 16. doi: 10.1002/jcb.23369. [Epub ahead of print]
51. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996; 271:27879-887.
52. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829-34.
53. Inoki K, Li Y, Zhu T, Wu J, Guan KL: TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648-57.
54. Inoki K, Zhu T, Guan KL: TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-590.
55. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ: AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-226.
56. Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer 2007; 6:46.
57. Puissant A, Grosso S, Jacquel A, Belhacene N, Colosetti P, Cassuto JP, Auberger P. Imatinib mesylate-resistant human chronic myelogenous leukemia cell lines exhibit high sensitivity to the phytoalexin resveratrol. Faseb J 2008; 22:1894-04.
58. Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P: Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Research 2010; 70:1042-52.
59. Vakana E, Altman JK, Glaser H, Donato NJ, Platanias LC. Antileukemic effects of AMPK activators on BCR-ABL expressing cells. Blood. 2011 118:6399-402.
60. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001; 108:1167-1174.
61. Li D. Metformin as an antitumor agent in cancer prevention and treatment. J Diabetes. 2011; 3 :320-7.
62. Aljada A, Mousa SA. Metformin and neoplasia: Implications and indications. Pharmacol Ther. 2012;133:108-15.
63. Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle 2011; 10:2115-20.
64. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, Boyer O, Bardet V, Park S, Foretz M, Viollet B, Ifrah N, Dreyfus F, Hermine O, Moura IC, Lacombe C,: The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010; 116:4262-73.
65. Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008; 67:1-7.
66. Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, Jellema G, Deharo S, Hardie DG, Pusztai L, Moulder-Thompson S, Dewar JA, Thompson AM. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat 2011;128:783-94.
67. Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res. 2010; 3:1066-76.
68. Woodard J, Joshi S, Viollet B, Hay N, Platanias LC. AMPK as a therapeutic target in renal cell carcinoma. Cancer Biol Ther. 2010;10:1168-77.
69. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA.Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010; 9 :4461-8.
70. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010; 9: 3807-14.
71. Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9: 1057-64.
72. Jiralerspong S, Gonzalez-Angulo AM, Hung MC. Expanding the arsenal: metformin for the treatment of triple-negative breast cancer? Cell Cycle. 2009; 8 :2681.
73. Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009; 8: 2031-40.
74. Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, Zabezhinski MA, Anikin IV, Karkach AS, Romanyukha AA. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010; 9: 188-97.
75. Anisimov VN, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Egormin PA, Yurova MV, Rosenfeld SV, Semenchenko AV, Kovalenko IG, Poroshina TE, Berstein LM.Gender differences in metformin effect on aging, life span and spontaneous tumorigenesis in 129/Sv mice. Aging (Albany NY). 2010; 2: 945-58.
76. Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY). 2010; 2: 760-74.
77. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009; 8: 88-96.
78. Mackenzie MJ, Ernst S, Johnson C, Winquist E. A phase I study of temsirolimus and metformin in advanced solid tumours. Invest New Drugs. 2010 Oct 27. [Epub ahead of print]