
Introduction
Cutaneous T-Cell lymphoma (CTCL) is characterized by the proliferation of malignant T cells in a chronic inflammatory microenvironment. The etiology of CTCL remains only partially understood and, unfortunately, it has not been possible to identify one unifying genetic event or one particular oncogene of central importance for the malignant transformation and cancer progression. Instead, deregulation of signaling pathways including Signal Transducers and Activators of Transcription (STAT), src kinases, c-Myc, COX-2, NFκB, GATA3, TOX, and embryonic stem cell regulators appears to play an important role in the pathogenesis [1-11]. In particular, the Interleukin-2 receptor common gamma chain (IL-2Rgc), the associated Janus kinase-3 (Jak3), and the down-stream effectors (STAT3 and STAT5) have attracted substantial interest in this regard. Indeed, the IL-2Rgc-signaling cytokines IL-2, IL-4, IL-7, IL-15, and IL-21 are implicated in early pathogenesis whereas constitutive, interleukin-independent activation of the Jak3/STAT3 pathway is believed to play a key role in progressive and advanced disease [12-15]. The aberrant activation of the Jak3/STAT pathway and interleukin-independent proliferation of malignant T cells appears to be in part a result of deficient expression and/or function of negative regulators such as Suppressors Of Cytokine Signaling-3 (SOCS3) and the protein tyrosine phosphatases (e.g. SHP1) [16-18]. Importantly, the Jak3/STAT pathway (i) promotes the expression of IL-5, IL-10, IL-17A, IL-17F cytokines, (ii) regulates the production of angiogenetic factors, and (iii) confers resistance to treatment with HDAC inhibitors in malignant T cells [19, 7, 15, 20-24]. Yet, relatively little is known about STAT target genes in CTCL and how STAT3, STAT5, and their co-activators and/or co-repressors regulate of expression of disease-associated genes.
Recently, a new class of post-transcriptional regulators, non-coding microRNAs (miRNAs), has received considerable attention in relation to CTCL. Skin lesions from CTCL patients displayed a distinct miRNA expression signature. Furthermore, a minimal miRNA classifier consisting of only three miRNAs (miR-155, miR-203, and miR-205) was able to distinguish between malignant and benign dermatoses with high accuracy suggesting that miRNAs profiling is a powerful diagnostic tool in CTCL [25-27]. Importantly, expression of several miRNAs including miR-155 and miR-21 were associated with progressive disease and a poor prognosis [27-31]. miR-21 confers resistance to apoptosis, while miR-155 promotes malignant proliferation [14, 29]. Conversely, the expression of other miRNAs is often lost in CTCL skin lesions when compared to benign dermatoses and normal skin [25, 28, 30]. Some of these miRNAs are now recognized to act as tumor suppressors as documented by their ability to inhibit tumor growth and/or metastasis [32, 33]. mir-22 expression is down-regulated in a number of cancers and has been assigned a role of tumor suppressor miRNA in colon and breast cancers [33-37]. In a recent study of miRNA expression in Sézary syndrome, a leukemic variant of CTCL, Ballabio et al. described a loss of miR-22 expression in circulating CD4+ T cells [38].
It is largely unknown what promotes the expression of onco-miRNAs and repression of tumor-suppressor miRNAs in CTCL. In the current work, we demonstrate that malignant T cells display down-regulation of miR-22 expression and provide mechanistic evidence that Jak3/STAT signaling inhibits the expression of this gene. This, in turn, contributes to elevated expression of MYCBP and MAX (co-activators of the c-Myc oncogene), and NCoA1, a transcriptional regulator previously associated with neoplastic transformation events [34]. Our work provides evidence that aberrant Jak3/STAT activation represses a novel tumor suppressor miRNA in CTCL.
Results
Loss of miR-22 expression in malignant T cells
Decreased expression of miR-22 was recently reported in several solid cancers and in circulating CD4+ T cells in Sézary syndrome [35-39]. As shown in Figure 1A, miR-22 expression was significantly downregulated in malignant CTCL T cell lines (MyLa2000, MyLa2059, SeAx, and PB2B) when compared to non-malignant (i.e. reactive) T cell lines obtained from CTCL skin lesions (MyLa1850 and MySi) and psoriatic skin (PSOR). Notably, the expression of pri-miR-22 was also decreased in the malignant T cell line MyLa2059 when compared to non-malignant T- cell line (Figure 1B). Similarly in Sézary syndrome peripheral mononuclear cells miR-22 expression was decreased when compared to the cells from healthy volunteers (Figure 1C).
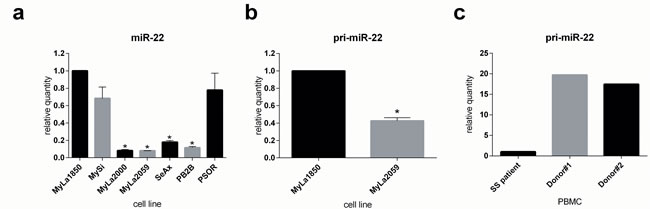
Figure 1: Expression of mature and primary miR-22 in CTCL as determined by qPCR. a. miR-22 expression in non-malignant (MyLa1850, MySi) and malignant (MyLa2000, MyLa2059, SeAx, PB2B) CTCL T cell lines as well as one T cell line derived from psoriasis vulgaris patient (PSOR), reference U6, n = 3. b. primary miR-22 (pri-miR-22) expression in non-malignant (MyLa1850) and malignant (MyLa2059) CTCL T-cell line. Reference GAPDH, n = 3 c. pri-miR-22 expression in primary Peripheral Blood Mononuclear Cells (PBMCs) derived from two healthy donors relative to one patient diagnosed with Sézary Syndrome, reference GAPDH.
Jak3/STAT signaling represses miR-22 expression
Il-2Rg-signaling cytokines regulate expression of multiple miRNAs through the Jak/STAT pathway. As shown in Figure 2, IL-2 induced a significant decrease in miR-22 expression in non-malignant T cell lines MyLa1850 (Figure 2, left panel) and MySi (Figure 2, right panel). Conversely, inhibition of IL-2R signaling by curcumin (a broad-range Janus kinase inhibitor) triggered in IL-2 treated non-malignant T cells an increased miR-22 expression when compared to the vehicle control (Figure 3A, left). Likewise, in malignant T cells that are known display a constitutive, aberrant Jak3 activation [40], curcumin produced an up-regulation of miR-22 (Figure 3A, right).. Notably, curcumin also enhanced pri-miR-22 expression in malignant MyLa2059 and SeAx T cells (Figure 3B, right and central panels) and in IL-2-treated non-malignant T cells (Figure 3B, left panel). Since curcumin inhibits other kinases in addition to Jak3 in malignant T cells, we tested the effect of a more selective Jak inhibitor, Jak3- inhibitor II, on miR-22 expression in malignant T cells. As shown in Figure 4, Jak3- inhibitor II triggered an increase in miR-22 expression comparable to the effect of curcumin in an earler experiment (Figure 3). Overall, these findings indicate that Jak3 activation repress miR-22 expression in malignant T cells. Since the active Jak3 mediates tyrosine phosphorylation and subsequent activation of STAT3 and STAT5 [1-3, 40], we examined whether Jak3-mediated repression of miR-22 was regulated via these transcription factors. Figure 5A shows expression changes in miR-22 (Figure 5A) and STAT3, STAT5A, and STAT5b (Figure 5B) following siRNA-mediated depletion of these STATs in malignant T cells. Inhibition of STAT3, STAT5A, and STAT5B induced a significant increase in the expression of miR22 (Figure 5A) Indicating that Jak3 regulates the expression of miR-22 via both STAT3 and STAT5.
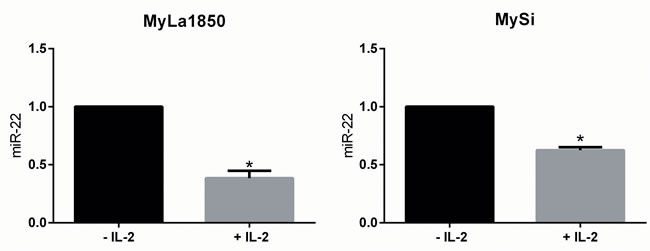
Figure 2: Effect of the T cell growth factor, IL-2, on miR-22 expression. Expression of miR-22 in IL-2 sensitive, non-malignant, CTCL T cells (MyLa1850 and MySi). Cells were depleted of IL-2 for 48 hours (– IL-2) or depleted of IL-2 for 24 hours, followed by 24 hours of IL-2 supplementation (+ IL-2). miR-22 expression was determined by qPCR using U6 as a reference n = 3.
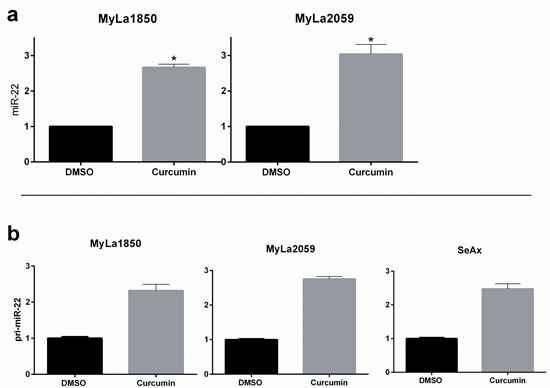
Figure 3: Curcumin treatment increases expression of mature and primary miR-22. miR-22 a. and pri-miR-22 b. expression measured by qPCR in non-malignant (MyLa1850) and malignant (MyLa2059, SeAx) CTCL T cells subjected to 24h treatment with 20µM curcumin or DMSO (control).a. Reference U6, n = 2. b. Reference GAPDH, error bars reflect variation in technical triplicates.
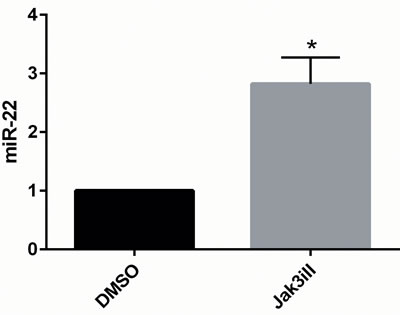
Figure 4: Inhibition of JAK3 increases expression of mature miR-22 in malignant CTCL cell line MyLa2059. miR-22 expression in MyLa2059 following 24 hours treatment with Jak3iII (40ug/mL) or DMSO control. Measured by qPCR, reference U6, n = 3.
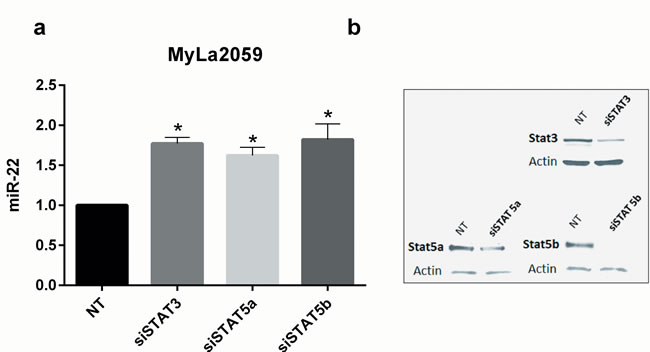
Figure 5: Transient knockdown of STAT3 and STAT5 genes increases expression of mature miR-22 in malignant CTCL cell line, Myla2059. a. miR-22 expression in MyLa2059 48h following transient transfection with siSTAT3, siSTAT5a, siSTAT5b or non-target (NT) control. Reference U6, n = 3, error bars reflect variation in technical triplicates. b. Representative Western Blot showing knockdown efficiency of siRNA transfections, 48h.
STAT binding to the miR-22 host gene promoter
In addition to their role as transcriptional activators, STAT3 can also function as repressors of transcription. Thus, in some cancer models STAT3 represses the expression of multiple genes including key regulators of stress responses and neoplastic transformation such as p53 [41]. Interestingly, STAT3 has been implicated in both transcriptional activation and repression of the oncomiR-21 [29, 42, 43], which probably reflects a cell-context dependent recruitment of co-activators and co-repressors such as BLIMP-1 [43]. Likewise, STAT5 can function as a transcriptional activator or a transcriptional repressor [44-47]. Accordingly, we examined whether siRNA- mediated depletion of STAT3 and STAT5 influenced the expression of the primary transcript of the miR-22 gene (miR-22HG/C17Orf91), pri-miR-22. As shown in Figure 6A, siRNA-mediated inhibition of STAT3, STAT5A, and STAT5B triggered increased expression of pri-miR-22 suggesting that they - directly or indirectly - repress transcription of miR-22HG.
To elucidate whether STAT3 and STAT5 directly bind to the miR-22HG, we performed oligo-nucleotide-based binding assay using a synthetic oligonucleotide corresponding to a STAT binding sequence in the miR-22HG. As shown in Figure 6C, pY-STAT5 and to a lesser extend pY-STAT3 co-precipitated with the miR-22HG oligonucleotide. Notably, the phosphorylation of STAT3 and STAT5 was almost completely blocked by pre-treatment of malignant T cells with a Jak3 inhibitor, CP-690550, (Figure 6C, left panel). Since this inhibitor blocks STAT3 and STAT5 tyrosine phosphorylation (Figure 6C, left) and transcriptional activity[48], these findings suggest that the activated form of the STAT proteins bind to miR-22HG.
To address further whether STAT proteins directly bind the miR-22HG promoter in malignant T cells, we performed Chromatin Immune-Precipitation followed by DNA sequencing (ChIP-seq) to identify transcriptional targets of STAT and other putative relevant transcription factors in a more physiological setting. ChIP-seq analysis of STAT5-precipitated chromatin from malignant MyLa2059 cells yielded an enrichment of reads comprising a promotor region of the miR-22HG (C17Orf91, Figure 6D). In contrast, only few reads for the miR-22HG promoter were detected in chromatin precipitated with STAT3 antibody (Figure 6D). Since curcumin also inhibits NFκB signaling [49], we also performed chromatin immunoprecipitation with RelA and RelB antibodies. As shown in Figure 6D, precipitation with RelA and RelB antibodies did not yield any reads for the miR-22HG promoter region.
Based on the information obtained from ChIP-seq, we designed primers flanking the STAT-binding site within the promoter (Supplementary Figure S1). These were used in an independent ChIP experiment to detect enrichment by PCR amplification of this sequence in chromatin from malignant MyLa2059 cells precipitated with STAT3 and STAT5 antibodies relative to a negative control antibody (rabbit IgG). Consistent with our earlier experiemental findings, ChIP-seq, PCR amplification (Figure 6E) showed a marked enrichment of the sequence representing the miR-22HG promoter in samples precipitated with STAT5 antibody, whereas STAT3-precipitated samples produced a less pronounced band. In a parallel experiment, we observed enrichment of the sequence representing the STAT5 binding site in the BIC promoter in samples precipitated with STAT5 antibody whereas STAT3-precipitated samples were negative (Figure 6E, lower panel) showing the specificity of the ChIP assay and confirming previous data on a selective STAT5 binding to the BIC promoter [14]. Taken together these data indicate that STAT5, and seemingly to a lesser degree, STAT3 bind to the miR-22HG promoter.
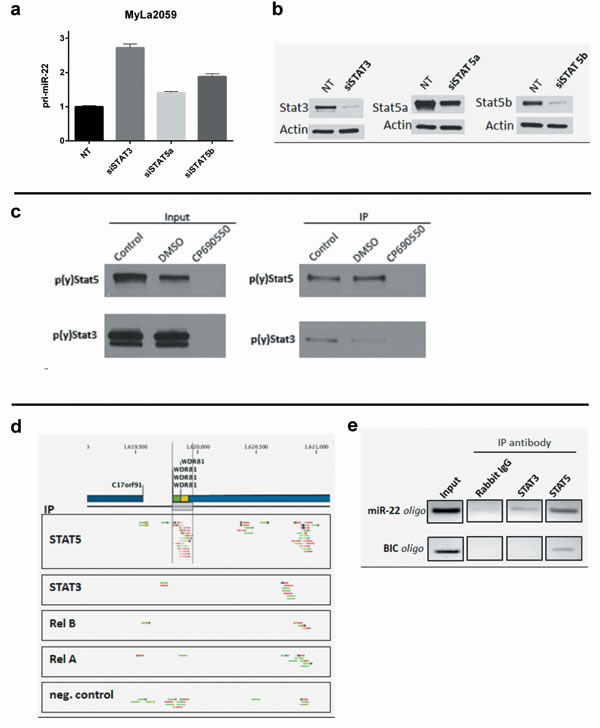
Figure 6: Binding of STAT transcription factors to the the miR-22HG C17orf91 and upstream promoter regions. a. primary miR-22 expression in MyLa2059 24h following transient transfection with siSTAT3, siSTAT5a, siSTAT5b or non-target (NT) control. Reference GAPDH, error bars reflect variation in technical triplicates. b. Knockdown efficiency is evaluated by Western Blot. c. Binding of p(y)STAT3 and p(y)STAT5 to an oligonucleotide sequence designed to mimic a putative STAT binding side inside the miR-22HG, C17orf91. Malignant CTCL cell line, Myla2059, was treated for 24h with JAK inhibitor, CP690550 (50µM), DMSO or untreated (control). Protein extracts were subjected to oligonucleotide pulldown and investigated for p(y)STAT3 and p(y)STAT5 presence by Western Blotting before (input) and after (IP) pulldown. d. ChIP-seq reads from the C17orf91 promoter region in malignant MyLa2059 cells. Reads obtained from immunoprecipitation with STAT5, STAT3, RelB, RelA and a negative control (Rabbit IgG, bottom). The chromosomal position of C17orf91 and upstream WDR81 refer to hg19. Forward reads are indicated in green and reverse reads are shown in red. e. PCR analysis of ChIP samples using the primer set indicated in Supplementary Figure 1. For the C17orf91 promoter the 120bp amplicon was detected in the STAT5- and to a lesser degree the STAT3-precipitated samples. For the BIC (miR-155HG) promoter 190bp amplicon was detected only in the STAT5-precipitated sample.
HDAC inhibitors induce expression of miR-22 in malignant T cells
In addition to a direct transcriptional repression of target genes, STAT3 and STAT5 may also repress transcription through the recruitment of transcriptional co-repressors such as EZH2 and HDACs [44, 46]. Hence, we treated malignant T cells with corresponding inhibitors to address whether repression of pri-miR-22 expression was mediated through these co- repressors. While the inhibitor of EZH2 (DZ-Nep) had no effect on pri-miR-22 expression at concentrations up to 10 µΜ uM (Supplementary Figure S2), a broad HDAC inhibitor, SAHA, used clinically for the treatment of CTCL, triggered a profound, time- and concentration- dependent up-regulation of pri-miR-22 expression (Figure 7). These findings indicate that HDACs play a key role in pri-miR-22 repression, whereas EZH2 most likely do not.
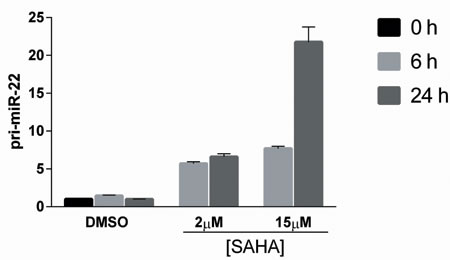
Figure 7: HDAC inhibitor, SAHA, induces time- and concentration-dependent increase in primary miR-22 expression. Malignant CTCL cell line, MyLa2059, was treated for 0, 6 or 24 hours with 2µM or 15 µM SAHA or DMSO control. Relative expression of pri-miR-22 was determined by qPCR, reference GAPDH. Error bars reflect variation in technical triplicates.
Elucidating miR-22 targets in malignant T cells
miR-22 has been assigned a role of a tumor suppressor in various solid tumors (reviewed in [38]) as well as some hematological malignancies (acute lymphoblastic leukemia, multiple myeloma and anaplastic large cell lymphoma) [50-53]. While the molecular mechanisms of this miRNA have not been fully elucidated, several miR-22 targets have been identified and validated. Several these targets including HDACs and transcriptional co-activators/regulators of c-Myc [37,39,54,55] could be involved in the pathogenesis of CTCL. To determine whether these proteins were susceptible to inhibition by miR-22 in malignant T cells, we used electroporation to transfect these cells with a synthetic miR-22 (miR-22 mimic). As shown in Figure 8, transfection with a miR-22 mimic inhibited to various degree the expression of all seven verified miR-22 targets examined when compared to transfection with scrambled miR-22 serving as a negative control (Figure 8A). Accordingly, a marked inhibition by 30-60% of MAX, MYCBP, NCoA1, and PTEN was observed in both the malignant T cell lines analyzed: MyLa2059 and SeAx (Figure 8A). Other verified miR-22 targets (CDK6, HDAC4 and HDAC6 [39, 56, 57]) were also inhibited by miR-22 but the effect was much weaker (Figure 8A).
The profound inhibition of NCoA1 by miR-22 is of particular interest as NCoA1 is a transcriptional co-activator regulating multiple oncogenic pathways associated with disease progression and metastasis formation in breast and prostate cancer [34]. When correlating miR-22 and NCoA1 expression in malignant and non-malignant T cells by RT-qPCR (Figure 8B), a relatively high miR-22 expression was associated with a low NCoA1 expression in non-malignant T cells and conversely a relatively low miR-22 expression in malignant T cells was associated with a high expression of NCoA1 mRNA (Figure 8B). These NCoA1 expression levels were also confirmed on a protein level (Figure 8C).
To further substantiate and extend these findings, we transfected malignant T cell lines with a miR-22 mimic and a scrambled control miRNA and measured the miR-22 effect on NCoA1 protein expression. As shown in Figure 8D, miR-22 triggered a significant decrease in NCoA1 protein expression therefore, supporting the notion that miR-22 directly down-regulates the expression of NCoA1 in malignant T cells.
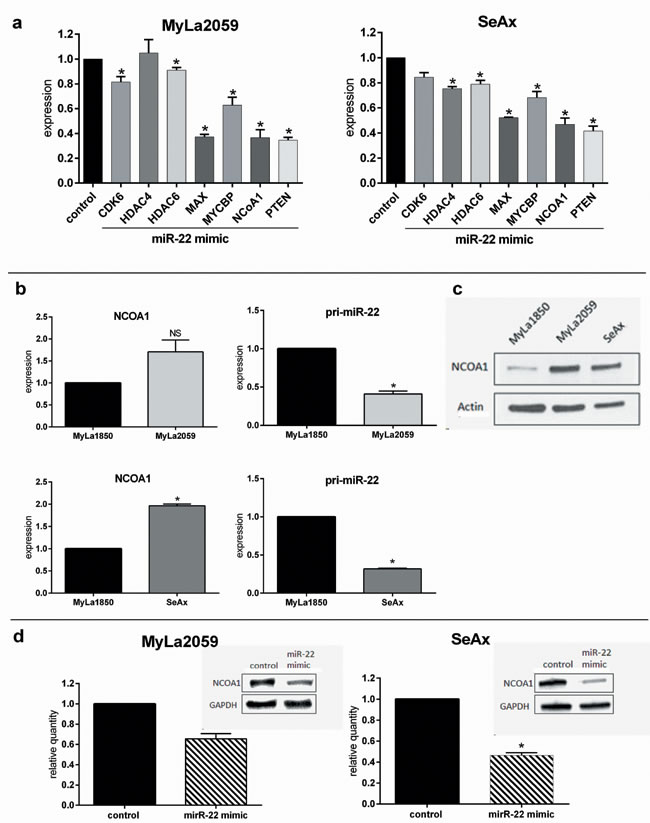
Figure 8: Expression of previously verified miR-22 targets are decreased by miR-22 mimic in malignant CTCL cell lines. a. Expression of CDK6, HDAC4, HDAC6, MYX, MYCBP, NCoA1 and PTEN following transient transfection with miR-22 mimic relative to a scrambled control in malignant CTCL cell lines (MyLa2059, SeAx) as determined by qPCR. Reference GAPDH, n = 7. b. qPCR comparison of relative NCoA1-(left panel) and pri-miR-22-(right panel) mRNA expression levels in malignant versus non-malignant cell lines (upper panel: MyLa2059 vs. MyLa1850, lower panel: SeAx vs. MyLa1850). Reference GAPDH, n = 3 c. Western Blot showing NCoA1 protein expression levels in non-malignant (MyLa1850) and malignant (MyLa2059, SeAx) CTCL cell lines. d. Relative expression of NCoA1 protein in malignant CTCL cells (left: MyLa2059, right: SeAx) following transfection with miR-22 mimic or scrambled control. Upper inset is a representative of the Western Blots results for NCoA1 and GAPDH quantification, n = 3.
Discussion
miR-22 has recently been assigned the role of a tumor suppressor miRNA and low expression has been reported in advanced disease and metastasis in several solid cancers (reviewed in [38]). In this work, we provide mechanistic evidence of deficient expression and putative tumor suppressive function of miR-22 in malignant T cells. Our data confirm and extend the recent findings by Ballabio et al. on down-regulation of miR-22 expression in peripheral blood malignant (Sézary) CD4+ T cells [58].
Our experimental molecular studies indicate that the observed low expression of miR-22 in CTCL was due to constitutive repression of miR-22 transcription by aberrant Jak3/STAT signaling in malignant T cells. We document that STAT5 and to a lesser extend STAT3 bind to a STAT-binding motif in the miR-22HG promoter and down-regulate this gene. Thus, inhibition of the Jak3, STAT5A, and STAT5B and STAT3 led to up-regulation of pri-miR-22 and miR-22. We further document that HDAC inhibitors play an important role in regulating miR-22 expression. Most importantly, our results demonstrate that forced expression of miR-22 in malignant cell lines inhibits expression of a number of critical oncogenes including MAX, MYCBP, NCoA1, and CDK6.
It is now well established that in addition to their function as transcription factors, STAT5 and STAT3 can function as transcriptional repressors. For instance STAT5 represses the expression of Cyclin D2 and BCL6 [47, 59], whereas STAT3 represses Glucose-6-Phosphatase and p53 expression [41, 60]. However, relatively little is known about the mechanisms involved in STAT3 and STAT5- mediated repression of gene transcription. Steric hindrance by STAT proteins and recruitment of other co-repressors might account for their function as transcriptional repressors (reviewed in [61]). Thus, STAT5 inhibits expression of the Ig kappa light chain locus by recruiting EZH2, which in turn triggers histone-trimethylation and chromatin conformational changes [42, 44, 62].
Likewise, STAT3 and STAT5 recruit HDACs which, depending on the cellular context, repress or activate transcription through de-acetylation of STAT proteins and other molecules in the transcriptional complexes. An inhibitor of EZHs had no effect on the expression of pri-miR-22, whereas inhibitors of HDACs induced a profound up-regulation in miR-22 expression suggesting that HDACs, but not EZHs, might be involved in the repression of pri-miR-22 transcription.
To assess a putative regulatory function of miR-22 in malignant T cells, we evaluated expression changes of validated miR-22 targets. Specifically, we investigated the effect of miR-22 on the expression of NCoA1, MAX, MYCBP, HDAC4, HDAC6, CDK6, and PTEN, all of which have independently been validated as miR-22 targets [37, 39, 54-57, 63, 64]. Constitutive expression of miR-22 inhibited all of these targets. Four targets (NCoA1, MAX, MYCBP, and PTEN) demonstrated a very high sensitivity to miR-22- mediated inhibition, whereas the other targets were only weakly inhibited by miR-22. The inhibition of MAX and MYCBP was of particular relevance because they have previously been proposed to play a pathogenic role in CTCL due to their effect on the C-Myc oncogene [19, 64, 65].
We, therefore, propose a model where miR-22 expression is low in malignant T cells and is down regulated by IL-2 in normal T cells, while MAX and MYCBP are constitutively active in malignant T cells and are activated by IL-2Rgc cytokines in non-malignant T cells [64]. Therefore, we believe that the repression of miR-22 by IL-2Rgc cytokines and Jak3/STAT activation is not only relevant in relation to CTCL, but might have implications for T cell biology in general.
NCoA1, also known as the steroid receptor co-activator (SRC-1), is a transcriptional co-activator and repressor not previously described in lymphoma but linked to oncogenesis in malignancies such as carcinoma of breast, ovarian, and prostate (reviewed by Walsh [34]). Indeed, global gene repression by NCoA1 promotes oncogenesis and metastatic spread in breast cancer [66,67]. Interestingly, the major clinical importance of an increased expression of NCoA1 in patients with advanced cancer relates to the development of drug resistance [68-71]. For instance, NCoA1 protects cancer cells from tamoxifen induced apoptosis [66,72]. In malignant T cells, as demonstrated in our study, constitutive expression of miR-22 inhibited NCoA1 expression on mRNA and protein levels, confirming its role in regulating NCoA1 expression and function [73]. As NCoA1 regulates retinoid signaling and several other nuclear receptors in addition to the steroid receptor, studies are in progress to address whether NCoA1 plays a role in drug resistance in CTCL and whether co-transfection of miR-22 potentiates the efficacy of anti-cancer therapies and drugs such as bexarotene and tazarotene and other treatment modalities. Indeed, in a study on the efficacy of extracorporeal photochemotherapy (ECP) in CTCL, Berger et al. [74] reported on a dramatic effect on leukocyte gene expression. The study did not include information of miR-22 expression but analysis of the raw microarray data-set obtained from NCBI (GSE23604) (Supplementary Figure 3S (A-C)) indicates that a subset of miR-22 target genes (74 genes) were significantly altered following ECP ( > 2 fold change, p < 0.001) (Suppl. Figure S3D). These included validated miR-22 targets (NCoA1 and PTEN) examined in the current study, which were both significantly inhibited post-ECP (Suppl. Figure S3E). These data indicate that NCoA1 and other miR-22 target- genes are inhibited following ECP suggesting a potential role of miR-22 target genes, and by inference restoration of miR-22, in mediating the effect of ECP [74].
Recent data in mice demonstrated that locked nucleotide acid (LNA)-modified oligonucleotides have promising therapeutic potentials and are now tested in a phase one clinical trial [75]. Since LNA-modified oligonucleotides can be designed to mimic both miR-agonists and antagonists, it may be speculated that LNA-based miR-22 mimics might also have a therapeutic potential in cancer treatment.
In conclusion, we demonstrate for the first time, that miR-22 expression is down-regulated in CTCL malignant T cells and propose a molecular mechanism, where aberrant Jak3/STAT signaling leads to STAT3/STAT5 activation and binding to its cognate sequence on the miR-22HG promoter. This binding leads to a direct transcriptional repression of the gene. We further demonstrate that normal function of this tumor suppressor microRNA is to down-regulate a number of putative oncogenes including validated miR-22 targets MAX, MYCBP, HDAC4, HDAC6, CDK6, and NCoA1. Taken together, these findings suggest that Jak3/STAT-mediated repression of miR-22 plays a key role in the pathogenesis and progression of CTCL. Combined with other reports, our results indicate that the Jak3-STAT3/STAT5 pathway may serve as therapeutic targets in CTCL.
Materials and methods
Cell lines and culture
Malignant T cell lines, MyLa2059, MyLa2000, PB2B and SeAx were established from patients diagnosed with CTCL [75, 76]. MyLa1850 and MySi are non-malignant T cell lines that were also established from CTCL patients [77]. PSOR2 is a non-malignant cell line established from a patient with psoriasis vulgaris [78].
All cell lines were cultured in RPMI1640 medium (#R2405, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 5% penicillin/streptomycin (Sigma-Aldrich). Furthermore the medium used for MyLa2059, MyLa2000 and PB2B was supplemented with 10% fetal bovine serum (Life Technologies, Roskilde, Denmark). The medium used for SeAx, MyLa1850, MySi and PSOR2 was supplemented with 10% human serum (Blood Bank, State University Hospital, Copenhagen, Denmark) and 103U/mL IL-2 (Proleukin) (Chiron, Emeryville, CA, USA) and 25ng/mL IL-4 (Leinco, St. Louis, MO, USA) (the latter only for PSOR2). Peripheral blood mononuclear cells (PBMCs) were isolated from a patient diagnosed with CTCL as well as from two healthy donors using a Ficoll Gradient, Lymphoprep (Axis-Shield PoC AS, Oslo, Norway) and were described elsewhere [79].
Inhibitors
JAK inhibitor II (Calbiochem, San Diego, CA, USA); JAK3 inhibitor, CP690550 (Pfizer, Ballerup, Denmark); Curcumin (Alexis, Laufelfigen, Switzerland); SAHA (Vorinostat) (Cayman Chemical, Ann Arbor, MI, USA); Trichostatin A, MS-275 (Entinostat) and Sodium Butyrate (Enzo Life Sciences, Plymouth Meeting, PA, USA); DzNep hydrochloride (Sigma-Aldrich).
Antibodies
Antibodies used in this work are commercially available and were obtained as follows: NCOA1 antibody from LifeSpan Bioscience (Seattle, WA, USA) (LS-B1702). pySTAT3 antibody from Nanotools (Denzlingen, Germany) (#0036-100/STAT3-9E12). STAT5a and STAT5b antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA) (L-20, SC-1081; C-17, sc-835-G). py-STAT5, STAT5, STAT3, RelA, RelB, Histone H3 and rabbit IgG antibodies from Cell Signaling Technologies (Beverly, MA, USA) (#9351, #9358, #4904, #8242, #4922, #4620, #2729). GAPDH antibody from Abcam (Cambridge, UK) (Ab9485) ; and β- Actin antibody from Sigma-Aldrich (A4700).
Oligonucleotide pulldown assay
Biotinylated oligonucleotides were designed complementary to the predicted STAT binding site (according to the transcription factor search database TFSEARCH version 1.3 http://www.cbrc.jp/papia/howtouse/howtouse_tfsearch.html inside C17orf91/miR-22HG (forward: 5’BIO- TCACTCTTCAGGGAAAAGTGATA-3’; reverse: 5’BIO-TATCACTTTTCCCTGAAGAGTGA- 3’) and were synthesized by Eurofins MWG GmbH (Martinsried, Germany). Lyophilized Ooligonucleotides were resuspended in nuclease-free water and annealed at 95°C for 5 min. Samples were lysed, and the supernatant was pre-cleared with streptavidin agarose beads (Invitrogen, Paisley, UK) for 3h rotating at 4°C. The supernatant was transferred to a new tube and agarose beads as well as 250 pmol oligos per sample were added. Samples were incubated overnight rotating at 4°C. After being washed 5 times with lysis buffer, sample loading buffer was added; the samples were denatured at 100°C for 10 min and subsequently subjected to SDS-PAGE and blotted for pY-STAT3 and pY-STAT5.
Chromatin Immunoprecipitation (ChIP) was performed using SimpleChIP® Enzymatic Chromatin IP kit (Agarose Beads) from Cell Signaling Technologies (Beverly, MA, USA) as previously described [14]. Cross-linked chromatin fragments were captured with antibodies against STAT3, STAT5, RELA, RELB, Histone H3 (positive control) or rabbit IgG (negative control). PCR was performed on immunoprecipitated and control DNA samples using primers for regions upstream of the promoters C17orf91 (miR-22HG) (forward sequence: 5`TCC TAA AGA GCA GGC GAA AG3`; reverse sequence: 5`AAA AAT GCC AAC TCA CAG AGC3` ) and BIC (miR-155HG) (forward sequence: 5` GAA AGG GAA AGG GGA AAA CA3`; reverse sequence: 5` CGA ACG TGC GAC CCT TTT AT3`) respectively. Both sets of primers were designed to encompass putative STAT binding sites. PCR annealing temperature was 56ᵒC and amplicon sizes were 120bp and 190bp for miR-22HG- and miR-155HG genes, respectively. The PCR products were separated on a 3% agarose gel using ethidium bromide for visualization.
ChIPseq library construction and sequencing was described in detail in Kopp et al. [14].
Transient transfections
2*106 cells per sample were transfected with small interfering RNA (siRNA) against JAK3, STAT3, STAT5a, STAT5b or non-targeting control #1 (ON-TARGETplus SMARTpool, Thermo Scientific, Lafayette, CO, USA). Cell pellets were resuspended in 100uL transfection solution (Ingenio Electroporation solution, Mirus Bio, Madison, WI, USA) with 0.5 nmol of the respective siRNAs and transfected with an Amaxa Nucleofector (Amaxa GmbH, Cologne, Germany).
For transient transfection with microRNA mimic (agomiR), 7*106 cells/sample were transfected with 0.25 nmol miR-hsa-miR-22-3p mimic or mimic control (both miRVana® miRNA mimics, Ambion (Sigma-Aldrich, Saint Louis, MO, USA). For SeAx cells a single transfection was performed whereas MyLa2059 cells were subjected to transfections on two consecutive days.
Protein extraction and western blotting
Protein was extracted from 1*106 cells persample and subjected to SDS-PAGE and Western blotting as described previously [79], [80].
RNA extraction and qRT-PCR
Total RNA was purified with miR-Neasy Mini Kit (Qiagen, Valencia, CA, USA), cDNA was transcribed from 10ng RNA using TaqMan® miRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed using TaqMan® miRNA assays (Applied Biosystems, Foster City, CA, USA) for miR-22 (#000398) according to the manufacturer’s instructions. Expression of U6 (#001973) was used as a reference. For quantification of expression of pri-miR-22 or various mRNAs, RNA extraction was performed with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA was subsequently transcribed using the High Capacity cDNA Reverse Transcription Kit followed by PCR analysis using TaqMan® Gene Expression Assays: pri-miR-22 (#Hs03302632_pri), NCOA1 (#Hs00186661), CDK6 (Hs 01026371_m1), HDAC4 (Hs 01041638_m1), HDAC6 (Hs 00195869_m1), MAX (Hs 00811069_g1), MYCBP (Hs 00429315_g1), PTEN (Hs 02621230_s1) (all from Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. GAPDH (#Hs02758991_g1) was included as a reference. The amplification was performed on an Mx3005P qPCR System (Agilent Technologies, Santa Clara, CA, USA) real-time cycler on standard settings.
Statistics
For statistical analysis a two-tailed Student’s t-test with a significance level of p = 0.05 was used. A significant difference (p < 0.05) between a sample and control is indicated with an asterisk.
Acknowledgments
This project was funded by The Calsberg Foundation, The Novo Nordic Research Foundation, The Lundbeck Foundation, The Danish Research Council (FSS under Det Frie Forskningsråd), The Danish Cancer Society (Kræftens Bekæmpelse) and Dansk Kræftforsknings Fond.
We thank K. Kaltoft for the gift of the MyLa cell lines.
Conflicts of interest
The authors declare no conflict of interest.
References
1. Zhang Q, Nowak I, Vonderheid EC, Rook a H, Kadin ME, Nowell PC, Shaw LM, Wasik MA. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc. Natl. Acad. Sci. U. S. A. 1996; 93:9148-53.
2. Nielsen M, Kaltoft K, Nordahl M, Röpke C, Geisler C, Mustelin T, Dobson P, Svejgaard A, Odum N. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc. Natl. Acad. Sci. U. S. A. 1997; 94:6764-9.
3. Kopp KLM, Kauczok CS, Lauenborg B, Krejsgaard T, Eriksen KW, Zhang Q, Wasik MA, Geisler C, Ralfkiaer E, Becker JC, Ødum N, Woetmann A. COX-2-dependent PGE(2) acts as a growth factor in mycosis fungoides (MF). Leukemia 2010; 24:1179-85.
4. Krejsgaard T, Vetter-Kauczok CS, Woetmann A, Kneitz H, Eriksen KW, Lovato P, Zhang Q, Wasik MA, Geisler C, Ralfkiaer E, Becker JC, Ødum N. Ectopic expression of B-lymphoid kinase in cutaneous T-cell lymphoma. Blood 2009; 113:5896-5904.
5. Zhang Y, Wang Y, Yu R, Huang Y, Su M, Xiao C, Martinka M, Dutz JP, Zhang X, Zheng Z, Zhou Y. Molecular Markers of Early-Stage Mycosis Fungoides. J. Invest. Dermatol. 2012; 132:1698-1706.
6. Huang Y, Su M-W, Jiang X, Zhou Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T cell lymphoma. Blood 2015; 125:1435-43.
7. Krejsgaard T, Gjerdrum LM, Ralfkiaer E, Lauenborg B, Eriksen KW, Mathiesen A-M, Bovin LF, Gniadecki R, Geisler C, Ryder LP, Zhang Q, Wasik MA, Odum N et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sézary syndrome. Leukemia 2008; 22:2230-2239.
8. Chang T-P, Vancurova I. NFκB function and regulation in cutaneous T-cell lymphoma. Am. J. Cancer Res. 2013; 3:433-45.
9. Geskin LJ, Viragova S, Stolz DB, Fuschiotti P. Interleukin-13 is over-expressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015. doi:http://dx.doi.org/10.1182/blood-2014-07-590398.
10. Dulmage BO, Geskin LJ. Lessons learned from gene expression profiling of cutaneous T- cell lymphoma. Br. J. Dermatol. 2013; 169:1188-97.
11. Litvinov I V, Netchiporouk E, Cordeiro B, Zargham H, Pehr K, Gilbert M, Zhou Y, Moreau L, Woetmann A, Ødum N, Kupper TS, Sasseville D. Ectopic expression of embryonic stem cell and other developmental genes in Cutaneous T-Cell Lymphoma. Oncoimmunology 2014; 3:e970025, DOI:10.4161/21624011.2014.970025.
12. Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, Nissen MH, Röpke C, Wasik MA, Odum N. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 2001; 15:787-93.
13. Sommer VH, Clemmensen OJ, Nielsen O, Wasik M, Lovato P, Brender C, Eriksen KW, Woetmann A, Kaestel CG, Nissen MH, Ropke C, Skov S, Ødum N. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia 2004; 18:1288-95.
14. Kopp KL, Ralfkiaer U, Gjerdrum LMR, Helvad R, Pedersen IH, Litman T, Jønson L, Hagedorn PH, Krejsgaard T, Gniadecki R, Bonefeld CM, Skov L, Geisler C et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle 2013; 12:1939-47.
15. Abraham RM, Zhang Q, Odum N, Wasik MA. The role of cytokine signaling in the pathogenesis of cutaneous T-cell lymphoma. Cancer Biol. Ther. 2011; 12:1019-1022.
16. Brender C, Lovato P, Sommer VH, Woetmann a, Mathiesen a-M, Geisler C, Wasik M, Ødum N. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNalpha. Leukemia 2005; 19:209-13.
17. Brender C, Nielsen M, Kaltoft K, Mikkelsen G, Zhang Q, Wasik M, Billestrup N, Odum N. STAT3-mediated constitutive expression of SOCS-3 in cutaneous T-cell lymphoma. Blood 2001; 97:1056-1062.
18. Zhang Q, Raghunath PN, Vonderheid E, Odum N, Wasik MA. Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promoter. Am. J. Pathol. 2000; 157:1137-46.
19. Qin JZ, Dummer R, Burg G, Döbbeling U. Constitutive and interleukin-7/interleukin-15 stimulated DNA binding of Myc, Jun, and novel Myc-like proteins in cutaneous T-cell lymphoma cells. Blood 1999; 93:260-7.
20. Nielsen M, Nissen MH, Gerwien J, Zocca MB, Rasmussen HM, Nakajima K, Röpke C, Geisler C, Kaltoft K, Ødum N. Spontaneous interleukin-5 production in cutaneous T-cell lymphoma lines is mediated by constitutively activated Stat3. Blood 2002; 99:973-977.
21. Marzec M, Halasa K, Kasprzycka M, Wysocka M, Liu X, Tobias JW, Baldwin D, Zhang Q, Odum N, Rook AH, Wasik MA. Differential effects of interleukin-2 and interleukin-15 versus interleukin-21 on CD4+ cutaneous T-cell lymphoma cells. Cancer Res. 2008; 68:1083-91.
22. Krejsgaard T, Ralfkiaer U, Clasen-linde E, Eriksen KW, Kopp KL, Bonefeld CM, Geisler C, Dabelsteen S, Wasik MA, Ralfkiaer E, Woetmann A, Odum N. Malignant Cutaneous T-Cell Lymphoma Cells Express IL-17 Utilizing the Jak3 / Stat3 Signaling Pathway. J. Invest. Dermatol. 2011:1331-1338.
23. Krejsgaard T, Litvinov I V, Wang Y, Xia L, Willerslev-olsen A, Koralov SB, Kopp KL, Bonefeld CM, Wasik MA, Geisler C, Woetmann A, Zhou Y, Sasseville D et al. Elucidating the role of interleukin-17F in cutaneous T-cell lymphoma. Blood: 2015; 122:943-951.
24. Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth J a, Li L, Gooden F, Korenchuk S, Hou XS, Harrington E a, Randolph S, Reilly JF et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008; 68:3785-94.
25. Ralfkiaer U, Hagedorn PH, Bangsgaard N, Løvendorf MB, Ahler CB, Svensson L, Kopp KL, Vennegaard MT, Lauenborg B, Zibert JR, Krejsgaard T, Bonefeld CM, Søkilde R et al. Diagnostic microRNA profiling in cutaneous T-cell lymphoma (CTCL). Blood 2011; 118:5891-900.
26. Marstrand T, Ahler CB, Ralfkiaer U, Clemmensen A, Kopp KL, Sibbesen N a, Krejsgaard T, Litman T, Wasik MA, Bonefeld CM, Grønbæk K, Gjerdum LMR, Gniadecki R et al. Validation of a diagnostic microRNA classifier in cutaneous T-cell lymphomas. Leuk. Lymphoma 2013; 55:957-958.
27. Sandoval J, Díaz-Lagares A, Salgado R, Servitje O, Climent F, Ortiz-Romero PL, Pérez-Ferriols A, Garcia-Muret MP, Estrach T, Garcia M, Nonell L, Esteller M, Pujol RM et al. MicroRNA Expression Profiling and DNA Methylation Signature for Deregulated MicroRNA in Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2015; 135:1128-37.
28. Ralfkiaer U, Lindal L, Litman T, Gjerdrum L-M, Busch Ahler C, Gniadecki R, Marstrand T, Fredholm S, Iveren L, Wasik M, Bonefeld CM, Geisler C, Krejsgaard T et al. MicroRNA Expression in Early Mycosis Fungoides Is Distinctly Different from Atopic Dermatitis and Advanced Cutaneous T-Cell Lymphoma. Anticancer Res 2014; 34:7207-7218.
29. Van der Fits L, van Kester MS, Qin Y, Out-Luiting JJ, Smit F, Zoutman WH, Willemze R, Tensen CP, Vermeer MH. MicroRNA-21 expression in CD4+ T cells is regulated by STAT3 and is pathologically involved in Sézary syndrome. J. Invest. Dermatol. 2011; 131:762-8.
30. Van Kester MS, Ballabio E, Benner MF, Chen XH, Saunders NJ, van der Fits L, van Doorn R, Vermeer MH, Willemze R, Tensen CP, Lawrie CH. miRNA expression profiling of mycosis fungoides. Mol. Oncol. 2011; 5:273-80.
31. Moyal L, Barzilai A, Gorovitz B, Hirshberg A, Amariglio N, Jacob-Hirsch J, Maron L, Feinmesser M, Hodak E. miR-155 is involved in tumor progression of mycosis fungoides. Exp. Dermatol. 2013; 22(Lcm):431-433.
32. Di Leva G, Garofalo M, Croce CM. MicroRNAs in Cancer. Annu. Rev. Pathol. Mech. Dis. 2014; 9(1):287-314.
33. Baranwal S, Alahari SK. miRNA control of tumor cell invasion and metastasis. Int. J. Cancer 2011; 126:1283-1290.
34. Walsh C a, Qin L, Tien JC-Y, Young LS, Xu J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 2012; 8:470-85.
35. Li B, Song Y, Liu T-J, Cui Y-B, Jiang Y, Xie Z-S, Xie S-L. miRNA-22 suppresses colon cancer cell migration and invasion by inhibiting the expression of T-cell lymphoma invasion and metastasis 1 and matrix metalloproteinases 2 and 9. Oncol. Rep. 2013; 29:1932-8.
36. Alvarez-Díaz S, Valle N, Ferrer-Mayorga G, Lombardía L, Herrera M, Domínguez O, Segura MF, Bonilla F, Hernando E, Muñoz A. MicroRNA-22 is induced by vitamin D and contributes to its antiproliferative, antimigratory and gene regulatory effects in colon cancer cells. Hum. Mol. Genet. 2012:1-9.
37. Ting Y, Medina DJ, Strair RK, Schaar DG. Differentiation-associated miR-22 represses Max expression and inhibits cell cycle progression. Biochem. Biophys. Res. Commun. 2010; 394:606-11.
38. Xiong J. Emerging roles of microRNA-22 in human disease and normal physiology. Curr. Mol. Med. 2012; 12:247-58.
39. Zhang J, Yang Y, Yang T, Liu Y, Li A, Fu S, Wu M, Pan Z, Zhou W. microRNA-22 , downregulated in hepatocellular carcinoma and correlated with prognosis , suppresses cell proliferation and tumourigenicity. Br. J. Cancer 2010; 103:1215-1220.
40. Krejsgaard T, Vetter-Kauczok CS, Woetmann A, Lovato P, Labuda T, Eriksen KW, Zhang Q, Becker JC, Ødum N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 2006; 20:1759-66.
41. Niu G, Wright KL, Ma Y, Gabriela M, Huang M, Irby R, Briggs J, Karras J, Cress WD, Pardoll D, Jove R, Wright GM, Chen J et al. Role of Stat3 in Regulating p53 Expression and Function Role of Stat3 in Regulating p53 Expression and Function. Mol. Cell. Biol. 2005; 25:7432-7440.
42. Zhang Q, Wang HY, Liu X, Bhutani G, Kantekure K, Wasik M. IL-2R common gamma-chain is epigenetically silenced by nucleophosphin-anaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc. Natl. Acad. Sci. U. S. A. 2011; 108:11977-82.
43. Barnes NA, Stephenson S, Cocco M, Tooze RM, Doody GM. BLIMP-1 and STAT3 counterregulate microRNA-21 during plasma cell differentiation. J. Immunol. 2012; 189:253-60.
44. Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR, Clark MR. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011; 12:1212-20.
45. Mitchell T, Whittaker S, John S. Dysregulated expression of COOH-terminally truncated Stat5 and loss of IL2-inducible Stat5-dependent gene expression in Sezary Syndrome. Cancer Res. 2003; 63:9048-9054.
46. Heltemes-Harris LM, Willette MJL, Vang KB, Farrar MA. The role of STAT5 in the development, function, and transformation of B and T lymphocytes. Ann. N. Y. Acad. Sci. 2011; 1217:18-31.
47. Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene 2007; 26:224-233.
48. Miklossy G, Hillard TS, Turkson J. Therapeutic modulators of STAT signalling for human disease. Nat. Rev. Drug Discov. 2013; 12:611-629.
49. Hour T-C, Chen J, Huang C-Y, Guan J-Y, Lu S-H, Pu Y-S. Curcumin enhances cytotoxicity of chemotherapeutic agents in prostate cancer cells by inducing p21(WAF1/CIP1) and C/EBPbeta expressions and suppressing NF-kappaB activation. Prostate 2002; 51:211-218.
50. Ninomiya S, Tyybäkinoja A, Borze I, Räty R, Saarinen-Pihkala UM, Usvasalo A, Elonen E, Knuutila S. Integrated analysis of gene copy number, copy neutral LOH, and microRNA profiles in adult acute lymphoblastic leukemia. Cytogenet. Genome Res. 2012; 136:246-55.
51. Lionetti M, Musto P, Martino M Di. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin. cancer Res. 2013; 19:3130-42.
52. Lionetti M, Agnelli L, Mosca L, Fabris S, Andronache A, Todoerti K, Ronchetti D, Lambertenghi D. G, Neri A. Integrative High-resolution Microarray Analysis of Human Myeloma Cell Lines Reveals Deregulated miRNA Expression Associated with Allelic Imbalances and Gene Expression Profiles. Genes. Chromosomes Cancer 2009; 48:521-531.
53. Liu C, Iqbal J, Teruya-Feldstein J, Shen Y, Dabrowska MJ, Dybkaer K, Lim MS, Piva R, Barreca A, Pellegrino E, Spaccarotella E, Lachel CM, Kucuk C et al. MicroRNA expression profiling identifies molecular signatures associated with anaplastic large cell lymphoma. Blood 2013; 122:2083-92.
54. Takata A, Otsuka M, Kojima K, Yoshikawa T, Kishikawa T, Yoshida H, Koike K. MicroRNA-22 and microRNA-140 suppress NF-κB activity by regulating the expression of NF-κB coactivators. Biochem. Biophys. Res. Commun. 2011; 411:826-31.
55. Xiong J, Du Q, Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene 2010; 22:4980-4988.
56. Xu D, Takeshita F, Hino Y, Fukunaga S, Kudo Y, Tamaki A, Matsunaga J, Takahashi R-U, Takata T, Shimamoto A, Ochiya T, Tahara H. miR-22 represses cancer progression by inducing cellular senescence. J. Cell Biol. 2011; 193:409-24.
57. Huang S, Wang S, Bian C, Yang Z, Zhou H, Zeng Y, Li H, Han Q, Zhao RC. Upregulation of miR-22 Promotes Osteogenic Differentiation and Inhibits Adipogenic Differentiation of Human Adipose Tissue-Derived Mesenchymal Stem Cells by Repressing HDAC6 Protein Expression. Stem Cells Dev. 2012. doi:10.1089/scd.2012.0014.
58. Ballabio E, Mitchell T, van Kester MS, Taylor S, Dunlop HM, Chi J, Tosi I, Vermeer MH, Tramonti D, Saunders NJ, Boultwood J, Wainscoat JS, Pezzella F et al. MicroRNA expression in Sezary syndrome: identification, function, and diagnostic potential. Blood 2010; 116:1105-13.
59. De Mattos SF, Essafi A, Soeiro I, Pietersen AM, Birkenkamp KU, Edwards CS, Martino A, Nelson BH, Francis JM, Jones MC, Brosens JJ, Coffer PJ, Lam EW-F. FoxO3a and BCR-ABL Regulate cyclin D2 Transcription through a STAT5/BCL6-Dependent Mechanism. Mol. Cell. Biol. 2004; 24:10058-10071.
60. Ramadoss P, Unger-Smith NE, Lam FS, Hollenberg AN. STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol. Endocrinol. 2009; 23(June 2009):827-837.
61. Heltemes-Harris LM, Farrar MA. The Role of STAT5 in Lymphocte Decelopment and Transformation. Curr. Opin. Immunol. 2012; 24:146-152.
62. Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3-and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. PNAS 2005; 102:1-6.
63. Tan G, Shi Y, Wu Z-H. MicroRNA-22 promotes cell survival upon UV radiation by repressing PTEN. Biochem. Biophys. Res. Commun. 2012; 417:546-51.
64. Bissonnette RP, McGahon A, Mahboubi A, Green DR. Functional Myc-Max heterodimer is required for activation-induced apoptosis in T cell hybridomas. J. Exp. Med. 1994; 180:2413-2418.
65. Van Doorn R. Aberrant Expression of the Tyrosine Kinase Receptor EphA4 and the Transcription Factor Twist in Sezary Syndrome Identified by Gene Expression Analysis. Cancer Res. 2004; 64:5578-5586.
66. Walsh C a, Bolger JC, Byrne C, Cocchiglia S, Hao Y, Fagan A, Qin L, Cahalin A, McCartan D, McIlroy M, O’Gaora P, Xu J, Hill AD et al. Global gene repression by the steroid receptor coactivator SRC-1 promotes oncogenesis. Cancer Res. 2014. doi:10.1158/0008-5472.CAN-13-2133.
67. Wang S, Yuan Y, Liao L, Kuang S-Q, Tien JC-Y, O’Malley BW, Xu J. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc. Natl. Acad. Sci. U. S. A. 2009; 106:151-156.
68. McCartan D, Bolger JC, Fagan A, Byrne C, Hao Y, Qin L, McIlroy M, Xu J, Hill AD, Ó Gaora P, Young LS. Global characterization of the SRC-1 transcriptome identifies ADAM22 as an ER-independent mediator of endocrine-resistant breast cancer. Cancer Res. 2012; 72:220-229.
69. McBryan J, Theissen SM, Byrne C, Hughes E, Cocchiglia S, Sande S, O’Hara J, Tibbitts P, Hill ADK, Young LS. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 2012; 72:548-59.
70. Redmond AM, Bane FT, Stafford AT, Mcllroy M, Dillon MF, Crotty TB, Hill AD, S.Young L. Coassociation of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clin. Cancer Res. 2009; 15:2098-2106.
71. Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill ADK, Young LS. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. Pathol. 2004; 57:1069-1074.
72. Graham JD, Bain DL, Richer JK, Jackson TA, Tung L, Horwitz KB. Thoughts on tamoxifen resistant breast cancer. Are coregulators the answer or just a red herring? J. Steroid Biochem. Mol. Biol. 2000; 74:255-259.
73. Yu H, Wu M, Zhao P, Huang Y, Wang W, Yin W. Neuroprotective effects of viral overexpression of microRNA-22 in rat and cell models of cerebral ischemia-reperfusion injury. J Cell Biochem 2014; 241:233-241.
74. Berger C, Hoffmann K, Vasquez JG, Mane S, Lewis J, Filler R, Zhao H, Durazzo T, Baird A, Lin W, Foss F, Christensen I, Girardi M et al. contribution to the clinical efficacy of extracorporeal photochemotherapy Rapid generation of maturationally synchronized human dendritic cells : contribution to the clinical efficacy of extracorporeal photochemotherapy. Blood 2011; 116:4838-4847.
75. Lanford RE, Hildebrandt-eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198-201.
76. Kaltoft K, Hansen BH, Pedersen CB, Pedersen S, Thestrup-Pedersen K. Common clonal chromosome aberrations in cytokine-dependent continuous human T-lymphocyte cell lines. Cancer Genet. Cytogenet. 1995; 85:68-71.
77. Woetmann A, Lovato P, Eriksen KW, Krejsgaard T, Labuda T, Zhang Q, Mathiesen A-M, Geisler C, Svejgaard A, Wasik MA, Odum N. Nonmalignant T cells stimulate growth of T-cell lymphoma cells in the presence of bacterial toxins. Blood 2007; 109:3325-3332.
78. Eriksen KW, Woetmann A, Skov L, Krejsgaard T, Bovin LF, Hansen ML, Grønbaek K, Billestrup N, Nissen MH, Geisler C, Wasik M a, Ødum N. Deficient SOCS3 and SHP-1 expression in psoriatic T cells. J. Invest. Dermatol. 2010; 130:1590-1597.
79. Krejsgaard T, Willerslev-Olsen A, Lindahl LM, Bonefeld CM, Koralov SB, Geisler C, Wasik MA, Gniadecki R, Kilian M, Iversen L, Woetmann A, Odum N. Staphylococcal enterotoxins stimulate lymphoma-associated immune dysregulation. Blood 2014; 124:761-770.
80. Geisler C, Dietrich J, Nielsen BL, Kastrup J, Lauritsen JPH, Ødum N, Christensen MD. Leucine-based receptor sorting motifs are dependent on the spacing relative to the plasma membrane. J. Biol. Chem. 1998; 273:21316-21323.
81. Hofmann B, Odum N, Platz P, Ryder LP, Svejgaard A, Neilsen JO. Immunological studies in acquired immunodeficiency syndrome. Functional studies of lymphocyte subpopulations. Scand. J. Immunol. 1985; 21:235-243.