
Introduction
Germline mutations in both alleles of a Fanconi Anemia (FA) gene lead to FA, a rare human genetic disease, which is also referred to a chromosomal abnormality syndrome [1-5]. Homozygous germline mutations in each individual FA gene account for a corresponding FA complementation group. Common features shared among all complementation groups indicate that encoded proteins function in a similar or common signaling transduction pathway, named the FA pathway or the FA-BRCA pathway, in regard to the direct or indirect involvement of breast cancer susceptibility genes, BRCA1 and BRCA2 [6-8]. We recently reported that the functional heterozygosity occurring in the FA signaling pathway during the course of cancer development plays a crucial role in promoting the development of human cancer in patients without FA [9, 10]. These studies, for the first time, demonstrated the tumor suppressor role of the FA signaling pathway predicated in 1971 by Dr. Swift [11]. To date, there are seventeen FA genes (FANC/A, B, C, D1, D2, E, F, G, I, G, L, M, N O, P, Q and S) that have been identified [6, 7] and the functions of these genes have emerged as an intense area of investigation in cancer research [6, 9, 10, 12, 13]. It is not too hard to recognize how important this signaling pathway is in enhancing our understanding of human tumorigenesis. However, the relevant knowledge of how the genetically mutated FA pathway is involved in the development of non-FA human cancers remains to be limited. Here we report common occurrences of the mutated FA pathway and its strong association with the development of human bladder cancer that was only further analyzed. This study, for the first time, demonstrated the importance of the FA tumor suppressor pathway at the genetic level among the general population.
Results
The recent technological advances offer us research tools that are much more articulated than what we could imagine before. Whole metabolome, transcriptome or genome analysis is of growing importance in advancing our views on the cancer development and treatment [14-16]. However, the annotation of the relationship between the mutated FA pathway and human cancer in a genome-wide manner is lacking. Here we analyzed a total of 68 publicly available DNA sequence datasets for mutations occurring in the 17 FA genes and compounded a sum rate for the mutated FA pathway via c-BioPortal [17, 18]. These sum rates are scattered from 1 to 55% with a frequency in a range of 15-35% (Table 1 and Supplementary Table 1). This is the first report to comprehensively show the scale of detectable mutations in the FA pathway in non-FA human cancers, firmly supporting our prior report that the FA tumor suppressor pathway plays a crucial role in suppressing cancer development in the patients without FA [9, 10]. Again, for the first time at the genetic level, the frequently mutated FA pathway as indicated (Table 1 and Supplementary Table 1) conveys its essential nature in suppressing the development of human cancers in the general population. Mutations occurring in any of the FA genes certainly contribute to the genetic hetero- or homo-zygosity of a given individual FA gene, and hereafter compromise the FA tumor suppressor signaling and promote the development of non-FA human cancer.
Table 1: The rates of the mutated FA pathway in non-FA human cancers |
||
Tumors (sequence datasets from TCGA) |
Percent of Cases Carrying Mutated Pathway |
No. of Cases Sequenced |
Acute myeloid Leukemia |
4.30% |
188 |
Adrenocortical Carcinoma |
26.10% |
88 |
Bladder Urothelial Carcinoma |
52.80% |
127 |
Brain Lower Grade Glioma |
11.90% |
286 |
Breast Invasive Carcinoma |
36.10% |
962 |
Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma |
23.60% |
191 |
Colorectal Adenocarcinoma |
24.10% |
220 |
Esophageal Carcinoma |
26.60% |
184 |
Glioblastoma Multiforme |
12.80% |
273 |
Head and Neck Squamous Cell Carcinoma |
34.40% |
302 |
Kidney Chromophobe |
10.60% |
66 |
Kidney Renal Clear Cell Carcinoma |
20.70% |
415 |
Kidney Renal Papillary Cell Carcinoma |
16.10% |
161 |
Liver Hepatocellular Carcinoma |
29% |
193 |
Lung Adenocarcinoma |
39% |
172 |
Lung Squamous Cell Carcinoma |
50.60% |
178 |
Lymphoid Neoplasm Diffuse Large B-cell Lymphoma |
27.10% |
48 |
Ovarian Serous Cystadenocarcinoma |
46.60% |
311 |
Pancreatic Adenocarcinoma |
35.60% |
90 |
Pheochromocytoma and Paraganglioma |
7.50% |
161 |
Prostate Adenocarcinoma |
25.20% |
258 |
Sarcoma |
29.60% |
257 |
Skin Cutaneous Melanoma |
44.20% |
278 |
Stomach Adenocarcinoma |
39.10% |
220 |
Thyroid Carcinoma |
5% |
399 |
Uterine Carcinosarcoma |
28.60% |
56 |
Uterine Corpus Endometrial Carcinoma |
36.40% |
242 |
To further understand the importance, at the genetic level, of those detectable mutations in the FA pathway in contributing to human tumorigenesis among the general population, we asked how significant the rate of the mutated FA pathway is during the development of human bladder cancer by cross-referencing the available clinical information, provided by the Cancer Genome Atlas (TCGA). We defined the mutated FA pathway, resulting from mutations found in one or 2 to 17 FA genes known so far; and the corresponding wild type FA pathway, meaning that no mutations are found in any of the 17 FA genes. We manually extracted the patient information of bladder tumor stages from the clinic notes available on TCGA. As shown in Figure 1, the mutated FA pathway, in contrast to the non-mutated one, is significantly correlated with the tumor promotion with a p value = 0.044 (more tumor cases at the high (late) stage when the FA pathway is mutated), indicating the tumor suppressor role played by the non-mutated FA pathway during the development of human bladder cancer. Next, we suspected this correlation might be heavily attributed to the mutated pathway resulting from multiple mutated FA genes, not the sole influence of the mutated FA pathway, noting that each FA gene has multiple functions in addition to its role in the FA pathway [19-22]. We regrouped cases by counting the mutated FA pathway resulting from a single mutated FA gene, and found that this redefined mutated FA pathway remains to be significantly correlated with tumor promotion (p = 0.032) (Figure 2). This further indicates the importance of the FA tumor suppressor pathway during the development of human bladder cancer. Our studies on human bladder cancer once again genetically validate that the FA tumor-suppressor signaling is not restricted to the FA cells.
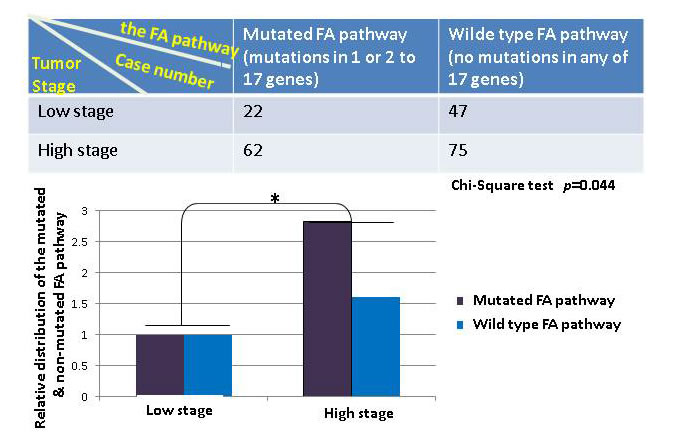
Figure 1: The mutated FA pathway is associated with the development of human bladder cancer. On the basis of the clinic notes available for a set of human bladder cancer samples on TCGA, we divided this set of cancer samples into two groups with or without a mutated FA pathway. We further divided each group into two subgroups upon tumor stages. We combined cases at the clinic stage 0, I, and II as the low stage, and those at the clinic stage III and IV as the high stage considering the limited sample sizes. The top table shows the distribution of bladder cancer cases in the groups we defined. The relative distribution of the mutated or non-mutated FA pathway in bladder cancer was plotted in bars at the bottom to suggest the role of a mutated FA pathway is statistically significant in promoting tumor growth from the low stage to the high stage in comparison with the wild type FA pathway. Chi-square test was performed with a p value = 0.044.
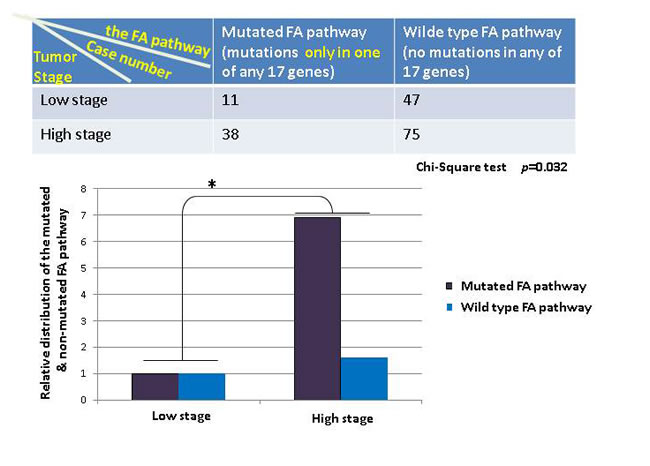
Figure 2: The mutated FA pathway resulting only from one mutated FA gene appears to potently promotes the development of human balder cancer. Bladder cancer cases were regrouped according to the mutated FA pathway harboring only one of the mutated FA genes, accompanying the same criterion used previously (Figure 1) for the subgroups. The top table shows the distribution of bladder cancer cases in each group we redefined. The relative distribution of the mutated or non-mutated FA pathway in bladder cancer was plotted in bars at the bottom to suggest the role of a single mutated FA gene-containing pathway is statistically significant in promoting tumor growth from the low stage to the high stage in contract to the wild type status of the FA pathway. Chi-square test was conducted with a p value = 0.032.
Discussion
We here demonstrate a previously unrecognized fact that the mutated FA pathway frequently occurs in the non-FA human tumors (Table 1 and Supplementary Table 1). We analyze the frequency of mutations to FA genes in the FA pathway, by including point mutations, deletions, as well as gene amplification events present in FANC/A, B, C, D1/2, E, F, G, I, G, L, M, N O, P, Q or S [6, 7]. The latter type of mutation might be subject to a point relevant to an over-activating FA pathway, rather than the loss of function. However the reported studies suggested that the over-activated FA signaling is tumorigenic [13, 23] and, thus, generally leads to the loss of tumor suppressive effect as a point mutation or deletion would. As a matter of fact, such scale rates of the mutated FA pathway occurring in human cancers ascertain extreme importance of the FA tumor suppressor pathway; once mutated, it promotes the development of human cancer as indicated in Figures 1 and 2. Our studies and many others [9, 19, 24-26] showed that somatic inactivation of the FA pathway could be at the level of any FA proteins or others functionally relevant. The compromised FA signaling pathway, if including epigenetic modifications of the FA genes [27] and other factors directly or indirectly on FA proteins [9, 28], would have a much higher rate in non-FA cancer patients than those accordingly shown in Table 1 and Supplementary Table 1 only on the basis of mutations in the 17 FA genes.
The irreparable, accumulated DNA damage contributes to the development of human cancer; on the other hand, those accumulated DNA damage resulting from cancer therapeutic agents [29-33], would be beneficial to the outcome of cancer treatment. FA cells carrying a defective FA pathway are sensitive to DNA crosslinking agents and die over time [1-5]. Therefore, tumor cells, harboring a mutated FA pathway, and eventually carrying accumulated DNA damage, will die after the exposure to therapeutic drugs [12, 34]. Consequently, the genetic profiling of FA genes may be a strategy to predict the sensitivity of cancer treatment. This would be very helpful for avoiding the drawbacks of general therapies in clinic. For instance, Bacillus Calmette-Guerin (BCG) [35-37] is given to patients with non-muscle-invasive bladder cancer mainly as immunotherapy without prior knowledge of clear functioning mechanisms. Ideally, BCG would be administered only to those patients who do not carry the mutated FA signaling and to give platinum-related drugs to patients who would have functional hetero- or homo-zygosity of the FA pathway (FA gene mutations, oncogenic factors compromising FA signaling, etc.). Looking at the explicit rates of the mutated FA pathway in all types of human cancers (Table 1 and Supplementary Table 1), there is a very low rate (<5%), for example, in medulloblastoma. This could be a rational explanation for the unclear benefit reported for medulloblastoma chemotherapy in using a combination including cisplatin or carboplatin [38]. Because of the low rate of the mutated FA pathway in medulloblastoma (1.8 or 4.3%, calculated from two separate datasets) (Supplementary Table 1), most tumor cells carrying a fully functioning FA pathway genetically commit to repair the damaged DNA caused by cisplatin or carboplatin and presumably continue to proliferate.
The field of FA cancer research progresses rapidly, and there are FA cases yet to be defined [39]. This may raise an issue of how to accurately define the wild type status of the FA pathway. We could have counted the cases harboring mutations in any of unidentified FA genes for the wild type group (Figures 1 and 2). However, the wild type status of the FA pathway named upon no mutation in any of the 17 FA genes would be the closest condition we can possibly provide as far (Main functions of the 17 genes outlined in Supplementary Table 2). Most importantly, there is a statistically significant correlation between the mutated FA pathway and the tumor promotion of human bladder cancer in the settings we studied (Figures 1 and 2), supporting the genetic evidence that the FA pathway has a role as a tumor suppressor in non-FA individuals and further concurring with our studies in this exciting field [9, 10, 12, 13, 21, 40-43]. While we had an in-depth look at the mutated FA pathway causing from only one mutated FA gene (Figure 2), we wanted to see how the mutated FA pathway resulting from two or more mutated FA genes is related to the tumor promotion. Unfortunately, it did not show statistical significance in terms of tumor promotion potentiated by the mutated FA pathway, but remains to hold a higher tendency of promoting tumor growth as compared to the wild type pathway (2.4 folds versus 1.6 folds for tumors at the high/late stage carrying the mutated or non-mutated status of the pathway, respectively). This may result from a limited sample size or/and the state of tumors that would have entered into the phase of the highest malignancy at the genetic level. In addition, owing to a tremendous amount of time needed in manually extracting clinical information, we did not go on performing similar analyses for other types of cancers in this study. We believe the finding would be similar to what shown in Figures 1 and 2, indicating the importance, at the prospective of cancer genetics [44, 45], of the tumor suppressor role played by the FA pathway during the development of non-FA human cancers. Of note, we realized that c-BioPortal does not give the clear information whether the output mutations affect both alleles or/and the all functions of any given gene. Possibly, the given gene may have a certain level of normal functions though mutated. Nonetheless, either heterozygous or homozygous mutations occurring in FA genes will compromise the tumor suppressor function of the FA signaling pathway and promote the development of human cancers. In fact, this has been evidenced by numerous studies reporting an increased incidence of breast, ovarian, pancreatic, or prostate cancer tightly associated with the heterozygosity of several FA genes (FANCD1, FANCJ, FANCN, FANCO or/and FANCS) [22, 46-57].
materials and Methods
Publicly available DNA sequence datasets
A total of 68 DNA sequence datasets were used and analyzed upon the known 17 FA genes as far via c-BioPortal [17, 18] to determine a mutation rate of the FA pathway in a variety of human cancers. The sources of those sequence datasets are TCGA, MSKCC, BGI, BCCRC, Nature, Nature Genetics, Cell, Science, AMC, Cancer Cell, and PNAS. The rates of the mutated pathway were calculated upon 27 sequence datasets of TCGA (Table 1), and others on the basis of 41 sequence datasets available in the rest of resources herein stated (Supplementary Table 1).
Source of clinic information of human bladder cancer samples
The clinic information for a total of 206 human bladder cancer cases was downloaded from TCGA database (updated on Jan 17, 2015). The related information for tumor stage according to The American Joint Committee On Cancer (AJCC) was collected manually and plotted as described in the text.
Statistical analysis
The correlation between the mutated FA pathway and the cancer stage was analyzed using Chi-square test. A p-value <0.05 was considered as statistical significance. All analyses were processed by SPSS 20 software.
Acknowledgments
The work is supported partly by R01CA188251 & R01CA136532 to PF and University of Hawaii Cancer Center. We thank Dr. Bing Han and Dr. Manoj Nepal for helping collect a part of clinic data from TCGA. We also thank all public resources we used, which were listed in Table 1 and Supplementary Table 1.
Conflicts of Interest
There is no conflict of interest.
References
1. Bagby GC, Jr. Genetic basis of Fanconi anemia. Curr Opin Hematol. 2003; 10: 68-76.
2. Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006; 107: 4223-4233.
3. Huang TT, D’Andrea AD. Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol. 2006; 7: 323-334.
4. Fei P, Yin J, Wang W. New advances in the DNA damage response network of Fanconi anemia and BRCA proteins. FAAP95 replaces BRCA2 as the true FANCB protein. Cell Cycle. 2005; 4: 80-86.
5. Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D’Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006; 8: 339-347.
6. Wang AT, Smogorzewska A. SnapShot: Fanconi Anemia and Associated Proteins. Cell. 2015; 160: 354-354 e351.
7. Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, Moilanen JS, Greenberg RA, University of Washington Centre for Mendelian G. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 2015; 5: 135-142.
8. Pickering A, Zhang J, Panneerselvam J, Fei P. Advances in the understanding of the Fanconi anemia tumor suppressor pathway. Cancer Biol Ther. 2013; 14: 1089-1091.
9. Zhang J, Zhao D, Park HK, Wang H, Dyer RB, Liu W, Klee GG, McNiven MA, Tindall DJ, Molina JR, Fei P. FAVL elevation in human tumors disrupts Fanconi anemia pathway signaling and promotes genomic instability and tumor growth. J Clin Invest. 2010; 120: 1524-1534.
10. Panneerselvam J, Park HK, Zhang J, Dudimah FD, Zhang P, Wang H, Fei P. FAVL impairment of the Fanconi anemia pathway promotes the development of human bladder cancer. Cell Cycle. 2012; 11: 2947-2955.
11. Swift M. Fanconi’s anaemia in the genetics of neoplasia. Nature. 1971; 230: 370-373.
12. Zhang J, Wang X, Lin CJ, Couch FJ, Fei P. Altered expression of FANCL confers mitomycin C sensitivity in Calu-6 lung cancer cells. Cancer Biol Ther. 2006; 5: 1632-1636.
13. Zhang J, Zhao D, Wang H, Lin CJ, Fei P. FANCD2 monoubiquitination provides a link between the HHR6 and FA-BRCA pathways. Cell Cycle. 2008; 7: 407-413.
14. Li XL, Lu X, Parvathaneni S, Bilke S, Zhang H, Thangavel S, Vindigni A, Hara T, Zhu Y, Meltzer PS, Lal A, Sharma S. Identification of RECQ1-regulated transcriptome uncovers a role of RECQ1 in regulation of cancer cell migration and invasion. Cell Cycle. 2014; 13: 2431-2445.
15. Garrobo I, Marion RM, Dominguez O, Pisano DG, Blasco MA. Genome-wide analysis of in vivo TRF1 binding to chromatin restricts its location exclusively to telomeric repeats. Cell Cycle. 2014; 13: 3742-3749.
16. Bisio A, Zamborszky J, Zaccara S, Lion M, Tebaldi T, Sharma V, Raimondi I, Alessandrini F, Ciribilli Y, Inga A. Cooperative interactions between p53 and NFkappaB enhance cell plasticity. Oncotarget. 2014; 5: 12111-12125.
17. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6: pl1.
18. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2: 401-404.
19. Raghunandan M, Chaudhury I, Kelich SL, Hanenberg H, Sobeck A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle. 2015; 14: 342-353.
20. Kehrli KR, Sidorova JM. Mitomycin C reduces abundance of replication forks but not rates of fork progression in primary and transformed human cells. Oncoscience. 2014; 1: 540-555.
21. Panneerselvam J, Pickering A, Han B, Li L, Zheng J, Zhang J, Zhang Y, Fei P. Basal level of FANCD2 monoubiquitination is required for the maintenance of a sufficient number of licensed-replication origins to fire at a normal rate. Oncotarget. 2014; 5: 1326-1337.
22. Park JY, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, Meetei AR, Andreassen PR. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. 2014; 33: 4803-4812.
23. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D’Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009; 16: 314-320.
24. Boisvert RA, Howlett NG. The Fanconi anemia ID2 complex: dueling saxes at the crossroads. Cell Cycle. 2014; 13: 2999-3015.
25. Cantor SB, Brosh RM, Jr. What is wrong with Fanconi anemia cells? Cell Cycle. 2014; 13: 3823-3827.
26. Karanja KK, Lee EH, Hendrickson EA, Campbell JL. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle. 2014; 13: 1540-1550.
27. Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003; 9: 568-574.
28. Kalvala A, Gao L, Aguila B, Reese T, Otterson GA, Villalona-Calero MA, Duan W. Overexpression of Rad51C splice variants in colorectal tumors. Oncotarget. 2014.
29. Su WP, Hsu SH, Wu CK, Chang SB, Lin YJ, Yang WB, Hung JJ, Chiu WT, Tzeng SF, Tseng YL, Chang JY, Su WC, Liaw H. Chronic treatment with cisplatin induces replication-dependent sister chromatid recombination to confer cisplatin-resistant phenotype in nasopharyngeal carcinoma. Oncotarget. 2014; 5: 6323-6337.
30. Choi YE, Battelli C, Watson J, Liu J, Curtis J, Morse AN, Matulonis UA, Chowdhury D, Konstantinopoulos PA. Sublethal concentrations of 17-AAG suppress homologous recombination DNA repair and enhance sensitivity to carboplatin and olaparib in HR proficient ovarian cancer cells. Oncotarget. 2014; 5: 2678-2687.
31. Friboulet L, Postel-Vinay S, Sourisseau T, Adam J, Stoclin A, Ponsonnailles F, Dorvault N, Commo F, Saulnier P, Salome-Desmoulez S, Pottier G, Andre F, Kroemer G, Soria JC, Olaussen KA. ERCC1 function in nuclear excision and interstrand crosslink repair pathways is mediated exclusively by the ERCC1-202 isoform. Cell Cycle. 2013; 12: 3298-3306.
32. Aggarwal M, Banerjee T, Sommers JA, Brosh RM, Jr. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle. 2013; 12: 3329-3335.
33. Singh S, Shemesh K, Liefshitz B, Kupiec M. Genetic and physical interactions between the yeast ELG1 gene and orthologs of the Fanconi anemia pathway. Cell Cycle. 2013; 12: 1625-1636.
34. Bagby GC, Olson SB. Cisplatin and the sensitive cell. Nat Med. 2003; 9: 513-514.
35. Hsu JW, Yin PN, Wood R, Messing J, Messing E, Lee YF. 1 alpha, 25-dihydroxylvitamin D3 promotes Bacillus Calmette-Guerin immunotherapy of bladder cancer. Oncotarget. 2013; 4: 2397-2406.
36. Spaliviero M, Dalbagni G, Bochner BH, Poon BY, Huang H, Al-Ahmadie HA, Donahue TF, Taylor JM, Meeks JJ, Sjoberg DD, Donat SM, Reuter VE, Herr HW. Clinical outcome of patients with T1 micropapillary urothelial carcinoma of the bladder. J Urol. 2014; 192: 702-707.
37. Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer—a current perspective. Nat Rev Urol. 2014; 11: 153-162.
38. Michiels EM, Schouten-Van Meeteren AY, Doz F, Janssens GO, van Dalen EC. Chemotherapy for children with medulloblastoma. Cochrane Database Syst Rev. 2015; 1: CD006678.
39. Pagano G, d’Ischia M, Pallardo FV. Fanconi anemia (FA) and crosslinker sensitivity: Re-appraising the origins of FA definition. Pediatr Blood Cancer. 2015.
40. Fu D, Dudimah FD, Zhang J, Pickering A, Paneerselvam J, Palrasu M, Wang H, Fei P. Recruitment of DNA polymerase eta by FANCD2 in the early response to DNA damage. Cell Cycle. 2013; 12: 803-809.
41. Pickering A, Panneerselvam J, Zhang J, Zheng J, Zhang Y, Fei P. In vitro FANCD2 monoubiquitination by HHR6 and hRad18. Cell Cycle. 2013; 12: 3448-3449.
42. Panneerselvam J, Pickering A, Zhang J, Wang H, Tian H, Zheng J, Fei P. A hidden role of the inactivated FANCD2: upregulating DeltaNp63. Oncotarget. 2013; 4: 1416-1426.
43. Park HK, Wang H, Zhang J, Datta S, Fei P. Convergence of Rad6/Rad18 and Fanconi anemia tumor suppressor pathways upon DNA damage. PLoS One. 2010; 5: e13313.
44. Song M, Giovannucci EL. Cancer risk: many factors contribute. Science. 2015; 347: 728-729.
45. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347: 78-81.
46. Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, Besenbacher S, Lundin P, Stacey SN, Gudmundsson J, Magnusson OT, le Roux L, Orlygsdottir G, Helgadottir HT, Johannsdottir H, Gylfason A, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011; 43: 1104-1107.
47. Ahlborn LB, Steffensen AY, Jonson L, Djursby M, Nielsen FC, Gerdes AM, Hansen TV. Identification of a breast cancer family double heterozygote for RAD51C and BRCA2 gene mutations. Fam Cancer. 2015; 14: 129-133.
48. Iwama E, Takayama K, Baba E, Nakanishi Y. [Personalized medicine in non-small-cell carcinoma]. Fukuoka Igaku Zasshi. 2014; 105: 57-66.
49. Kohlhase S, Bogdanova NV, Schurmann P, Bermisheva M, Khusnutdinova E, Antonenkova N, Park-Simon TW, Hillemanns P, Meyer A, Christiansen H, Schindler D, Dork T. Mutation analysis of the ERCC4/FANCQ gene in hereditary breast cancer. PLoS One. 2014; 9: e85334.
50. Pickholtz I, Saadyan S, Keshet GI, Wang VS, Cohen R, Bouwman P, Jonkers J, Byers SW, Papa MZ, Yarden RI. Cooperation between BRCA1 and vitamin D is critical for histone acetylation of the p21waf1 promoter and growth inhibition of breast cancer cells and cancer stem-like cells. Oncotarget. 2014; 5: 11827-11846.
51. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121: 269-275.
52. Guaoua S, Ratbi I, Lyahyai J, El Alaoui SC, Laarabi FZ, Sefiani A. Novel nonsense mutation of BRCA2 gene in a Moroccan man with familial breast cancer. Afr Health Sci. 2014; 14: 468-471.
53. Li D, Bi FF, Chen NN, Cao JM, Sun WP, Zhou YM, Li CY, Yang Q. A novel crosstalk between BRCA1 and poly (ADP-ribose) polymerase 1 in breast cancer. Cell Cycle. 2014; 13: 3442-3449.
54. Ow GS, Ivshina AV, Fuentes G, Kuznetsov VA. Identification of two poorly prognosed ovarian carcinoma subtypes associated with CHEK2 germ-line mutation and non-CHEK2 somatic mutation gene signatures. Cell Cycle. 2014; 13: 2262-2280.
55. Fridlich R, Annamalai D, Roy R, Bernheim G, Powell SN. BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair (Amst). 2015; 30: 11-20.
56. Downs B, Kim YC, Xiao F, Snyder C, Chen P, Fleissner EA, Becirovic D, Wen H, Sherman S, Cowan KH, Lynch HT, Wang SM. Two PALB2 germline mutations found in both BRCA1+ and BRCAx familial breast cancer. Breast Cancer Res Treat. 2015.
57. Erturk E, Cecener G, Tezcan G, Egeli U, Tunca B, Gokgoz S, Tolunay S, Tasdelen I. BRCA mutations cause reduction in miR-200c expression in triple negative breast cancer. Gene. 2015; 556: 163-169.