
Introduction
Despite initially successful multimodal therapy that includes resection, chemotherapy and for some cases radiation therapy, tumor recurrence remains a major etiology of the morbidity and mortality in cancer patients. A systematic review of acquired relapse in cancer patients suggested that tumor cells undergo dynamic clonal evolution under the strong selective pressure of chemotherapy, radiation therapy and any other therapeutic intervention [1, 2]. These treatment-resistant clones of neoplastic cells show somatic mutations and phenotypic variations not present in their state of origin. Over the past decade, these subpopulations have been isolated using novel surface markers of CSCs to dissect the causes of inter- and intra- tumors heterogeneity [3]. The source of this subset of treatment-resistant, relapse-initiating and the dynamic evolution of these clones must be understood.
The location of a tumor recurrence relative to the primary tumor (local, regional or distant) is influenced predominantly by microenvironmental factors that provide an adaptive landscape for relapsed tumor cells. The adhesion of tumor cells to the extracellular matrix (ECM) drives the activation of certain signature genes that promote cancer progression or tissue-dependent dormancy [4, 5]. Thus, identifying these genetic alterations could reveal new avenues for preventing or treating tumor relapse and could improve the long-term survival of patients. As shown in Table 1, survival rate is associated with tumor recurrence in various types of cancer.
A major cause of tumor relapse is an increasing number of CTCs and their downstream transformation into CSCs which initiate recurrence [6-9]. Notably, cases demonstrating chemo- or radio-resistance have high numbers of EMT transformed CTCs [10, 11]. Evidence from clinical studies suggests that poor survival of cancer patients has been linked with EMT phenotypes in malignant cancer cells [12-14].
Accumulating evidence shows that a subset of CTCs and CSCs have an EMT phenotype [15-19]. Another notable finding demonstrates that a subclone of CTCs can be induced to express phenotypes of CSCs [18, 20-23]. These discoveries suggest that EMT links CTCs and CSCs, enabling these cells to survive in the peripheral circulation and actively causing relapse. A better understanding of the etiology of the reprogramming switches that determine the progression through EMT, CTCs, dormancy and CSCs could pave the way towards clinically relevant drug targets.
In this review, we revisit the concept of relapse to introduce the notion of EMT transformed CTCs and CSCs. We highlight the most recent studies demonstrating the potential contributions of EMT positive CTCs and CSCs to recurrence and recommend a redesign of the therapeutic research on CSCs. Improving our understanding of these cells may help to categorize potential targets for novel therapies to preclude relapse.
Table 1: Tissue-specific tumor recurrence rates and 5-year survival rates in patients with cancer.
Tumor sites |
Recurrence rate |
5 years survival rate |
References |
Bladder |
~40-70% |
~15-98% |
|
Bone |
~50% |
. ~60-80% |
[88] |
Breast |
~15-20% |
~90-20% |
|
Brain |
~85% |
~10% |
[91] |
Colon |
~18-32% |
~6-74% |
|
Head and Neck |
~24-33% |
~50-66% |
|
Kidney |
~20-40% |
~8-81% |
[97] |
Liver |
~70% |
~25-50% |
|
Lung |
~10-24% |
~40-60% |
|
Ovary |
~20-50% |
~18-89% |
|
Pancreatic |
~80% |
~5-14% |
[105] |
Prostate |
~30-44% |
~99-100% |
[106] |
Testis |
~4-14% |
~74-99% |
|
Thyroid |
~5-10% |
~51-100% |
|
Uterus |
~14-25% |
~17-95% |
Tumor dormancy and relapse
Tumor dormancy is a state of clinical remission in which cancer cells remain occult, i.e., indiscernible and asymptomatic for an extended period of time. Primary tumors often employ this strategy as a means of resisting the first line of treatment. The phenomenon of dormancy is associated with numerous epithelial tumors, including melanoma and breast, prostate, liver, and thyroid cancer with variable latency periods [24-29]. Dormancy is frequently observed in patients with cancer who have undergone frequent chemo- or radiation therapy [30, 31]. Thus, it is critical to delineate gene signatures associated with the sequential stages of dormancy including mitotic-arrest in dividing tumor cells, the angiogenic switch, and the escape from immune surveillance and transformation into relapse initiating cells (Figure 1).
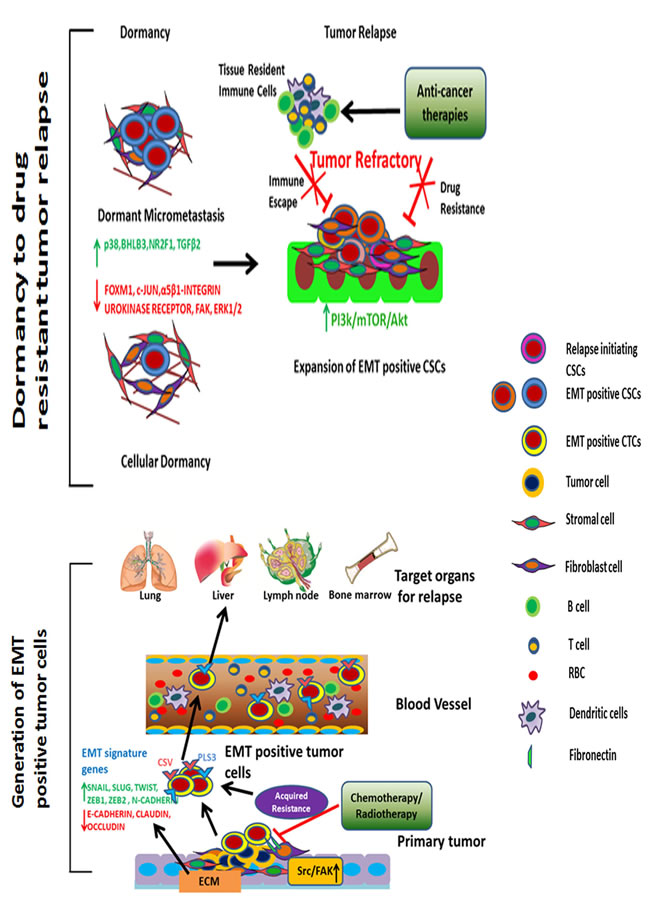
Figure 1: Tumor relapse driven by EMT-positive CTCs and CSCs. Upon anti-cancer therapy of primary tumors, EMT-positive CTCs are detected in large numbers in the peripheral blood. These CTCs migrate through organs such as the liver, lungs, lymph nodes, and bone marrow. Once the tumor cells arrive at their site of relapse, they remain dormant for an extended period and transform into CSCs. ECM remodeling; p38α, NR2F1, and TGFβ2 signaling; and inhibition of ERK1/2, FAK, FOXM1, and c-JUN pathways facilitate dormancy. Furthermore, tumor-associated tissue environments provide an embedded niche to protect these cells from anti-cancer therapies or any other lethal damage. Under ambient conditions, with ECM remodeling and activation of proliferative, angiogenic signaling pathways, EMT-positive CSCs undergo proliferation to initiate recurrence. These cells are highly resistant to anti-cancer drugs and are capable of evading immune surveillance.
In the era of translational studies and extensive genomic sequencing, numerous genes have been linked to dormancy in different types of cancer [32-35]. For example breast and prostate cancers, NR2F1, SHARP1, BMP7high and COCOlow signatures induce quiescence and delayed metastasis [36]. Many investigations recently interpreted that EMT-transformed cells are linked with decreased proliferation or quiescence [37-39]. Notably, the EMT program drives tumor cells to become quiescent CTCs. Identifying the molecular characteristics of EMT positive CTCs and CSCs during the latency period are thus instrumental to determine whether these cells relapse or remain dormant.
According to experimental and clinical studies, the microenvironments of certain organs such as the bone marrow, lung, liver, and brain promote dormancy [40]. The host microenvironments in these tissues maintain reciprocal signaling with CTCs and CSCs and thus induce the expression of pro-dormancy genes. Furthermore these cancer cells are embedded in a niche that provides a shield from immune surveillance, extending the period of dormancy.
Survival signals rather than proliferative ones can be identified in dormant tumor cells and used to prevent recurrence. In multiple myeloma, bortezomib treatment causes tumor cells to enter a quiescent phase owing to activation of the unfolded protein response pathway [41]. Inhibiting eIF2α dephosphorylation in this type of cancer using the GADD34-PP1c inhibitor decreased the number of dormant tumor cells and reduced recurrences in this type of cancer [41]. In breast cancer, low expression of extracellular-signal-regulated kinase (i.e., ERK) and high levels of p38α were detected in quiescent cancer cells [42]. Activation of p38α induced at least three transcription factors- p53 (R213Q), BHLHB3 and NR2F1 and inhibited the expression of FOXM1 and c-JUN, which are associated with G1-S transition [42, 43]. Some of these dormancy signature genes were found to delay recurrence in breast and prostate cancers by suppressing malignant behavior [44]. Furthermore knockdown or systemic inhibition of p38α in vivo showed that dormant cells were capable of regaining tumorigenicity [45]. A recent study using a head and neck cancer model identified that transforming growth factor-β 2 (TGFβ2) was increased in dormant cells [45]. TGFβ2 created a unique signal through TGFβRIII to induce canonical pathway SMAD1/2/5 to upregulate p27 and induce non-canonical activation of p38 for dormancy [45].
In addition to p38α and TGFβ2, the composition of the ECM has the potential to determine proliferation and dormancy. Adhesion to the ECM initiates intracellular signaling pathways that can increase cell cycle progression, motility, survival, and other metastatic phenotypes of tumor cells. For example, downregulation of the urokinase receptor in squamous cell carcinoma (HEp3) inactivates α5β1-integrins [46]. Subsequently, focal adhesion kinase (FAK) signaling is inhibited owing to the cells inability to bind to fibronectin. This results in dormancy of cancer cells. Conversely, dormancy to proliferative response in a fibrotic environment requires collagen-I mediated integrin β1 signaling, which requires activation of Src and FAK to phosphorylate myosin light chain kinase in an ERK dependent manner [47]. Clearly cytoskeletal rearrangement and ECM composition are critical in determining whether tumor cells will remain dormant or metastasize. Thus, inhibiting the growth promoting changes in an ECM-associated microenvironment may help prevent relapse.
EMT and cancer advancement
The EMT program is now known to facilitate the metastatic spread and progression of cancer cells from the site of the primary tumor to the surrounding tissues and distant organ(s). The identification and biological characterization of the EMT inducing transcription factors Snail, Slug and Twist showed the cascade of the tissue remodeling process of epithelial tumors [48, 49]. Overexpression of these EMT signatures changes the polarity of epithelial cells such that they acquire the morphological and biochemical traits of mesenchymal cells. Numerous genes linked with EMT, evidence that this program is essential for tumor cells to circumvent apoptosis, anoikis, oncogene addiction, and cellular senescence and to escape immune surveillance [50].
Understanding and targeting the adaptive growth of EMT driven cancer cells could lengthen progression-free survival [51]. A prospective study of 46 patients with liver cancer showed the EMT markers twist and Vimentin in 84.8% and 80.4% of those patients’ CTC samples respectively [52]; tumor progression was closely correlated with the presence of EMT positive CTCs in those patients. In patients with non–small cell lung cancer (NSCLC), resistance to EGFR inhibitors was associated with EMT induction [53, 54]; in this subset, EMT may have been promoted through Zeb1 and Src activation upon overexpression of the growth factor CRIPTO1 [53]. A seminal study by Shao et al highlighted that a loss of K-Ras signaling was compensated by the transcriptional coactivator YAP1 to maintain the EMT program during relapse in a murine lung cancer model [55]. Similarly, the functional study with High Mobility Group A1 (HMGA1) protein has emphasized its role as a key regulator of the mesenchymal transition and linked with stem-like phenotypes in breast cancer [56]. Apart from dynamics of cellular proteins, metabolic reprogramming is an essential step to maintain EMT state for CSCs [57]. Under nutrient starvation condition, EMT positive CSCs utilizes glycolytic and ketogenic end products to catabolize exogenous mitochondrial fuel [57, 58]
These findings suggest that the differentiation state of tumor cells contributes significantly to acquired drug resistance. The mechanisms by which tumor cells sustain the EMT phenotype in the relapse state are highly diverse between different types of cancer. The enhancement of mesenchymal-like features may epigenetically reprogram epithelial cancer cells to adapt well to new microenvironments and thus may contribute to distant recurrence.
CTCs and risk of relapse
CTCs have gained huge importance in the design of therapeutic regimens and monitoring cancer progression in the era of personalized medicine. Owing to advancements in single-cell molecular analysis, CTCs are considered a precursor for metastatic transformation and a predictive factor of tumor relapse. Compared with the traditional single-biopsy approach, the analysis of CTCs captures a broad range of genomic variations. With the use of next-generation sequencing, CTC profiles are a powerful clinical indicator for the transition from chemotherapeutic susceptibility to chemoresistance. Also, the genomic landscape obtained from these sequencing data greatly facilitates the identification of druggable therapeutic targets.
CTCs are heterogeneous and can be broadly classified into three categories- epithelial, transitioning from epithelial to mesenchymal and mesenchymal. We will focus on epithelial and EMT CTCs and their association with metastatic potential and acquired drug resistance in adult cancers. Epithelial-origin CTCs are detected in the peripheral circulation and are believed to shed periodically from primary or metastatic tumor sites (Figures 1, 2). Extensive, seminal studies in the past decade have implicated EMT and CSCs in metastasis and relapse [15, 18, 59].
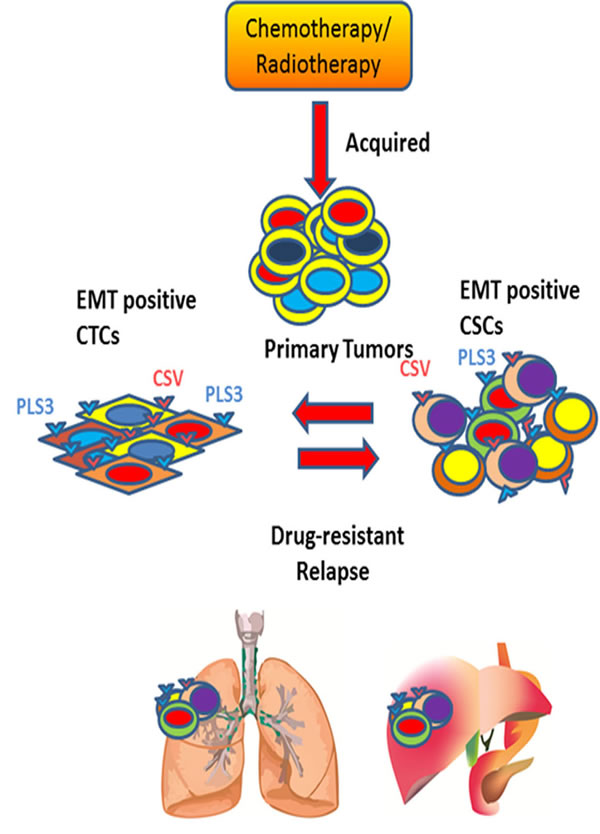
Figure 2: Understanding the dynamic equilibrium between EMT positive CTCs and CSCs to define tumor relapse. Periodic chemo- or radiotherapy on primary tumor induces EMT positive tumor cells. These EMT positive tumor cells are transformed into quiescent CTCs upon entering into the bloodstream. These EMT positive CTCs express Plastin 3 (PLS3) and cell surface Vimentin (CSV) on its surface. During relapse phase, EMT positive CTCs reprogram into drug-resistant EMT positive CSCs under ambient condition to cause poor survival rate for cancer patients.
Acquisition of the EMT phenotype in CTCs can indicate the risk of relapse and survival (Figure 1). Compared with epithelial CTCs detected by the conventional markers EpCAM and cytokeratin, the high rates of EMT-positive CTCs were associated with prognosis in patients with hepatocellular carcinoma [52]. In breast, prostate, liver, colorectal, head and neck, pancreatic, endometrial, and lung cancers, the number of CTCs exhibiting EMT markers increased from early- to late-stage cases [4, 16, 52, 60, 61]. In a comparative study between early and metastatic breast cancer cases, CTCs expressing the EMT markers vimentin and twist increased from ~73-77% to ~100% of CTCs [16]. In another study, 14 of 52 primary breast cancers between stages I and III received neoadjuvant therapy [10]. Interestingly, EMT-inducing transcription factors were overexpressed in neoadjuvant therapy-treated patients compared with those not treated with neoadjuvant therapy. Similarly, in patients with colorectal cancer, the novel marker Plastin3 identified the most aggressive CTCs undergoing EMT in one-third of 711 patients with colorectal cancers [11]; these Plastin3-positive CTCs showed inducible staining of the EMT marker vimentin.
Another novel marker, cell surface Vimentin (CSV), has recently been shown to detect EMT-positive CTCs in breast and colorectal cancer patients [62, 63]. In a pilot study of 58 patients with metastatic breast cancer, CSV antibodies demonstrated superior sensitivity (85% vs 48%) and specificity (94% vs 83%) compared with EpCAM-based detection for progressive disease upon treatment. This difference could be attributed to a shifting of the CTC population toward drug-resistant, dormant, or both phenotypes. Also, the low detection of CTCs by the EpCAM antibody was likely due to its nuclear localization upon disease progression. These studies clearly concluded the possibility of shortcomings in U.S. Food and Drug Administration–approved EpCAM-based CTC capture techniques [16, 64, 65].
More than 300 clinical trials are using CTC counts as an indicator for disease progression and overall survival (Table 2) [66, 67]. These clinical trials reflect the translational significance of CTC for monitoring therapeutic responses to adjuvant therapies. In general, higher numbers of CTCs (≥5 per 7.5 ml of blood) were associated with shorter median survival and higher tumor burden (Table 2). Upon chemotherapeutic treatment, a decrease in CTC count was associated with an improvement in median overall survival. Hence, CTC kinetics has the potential to indicate whether to maintain current medication or switch therapy. However, most of these clinical trials used EpCAM as a CTC marker, which may mean that the assessments of the pharmacodynamics of the drugs under trial are not optimally accurate. It is imperative to include the markers Plastin3 and CSV to detect EMT-positive CTCs for comprehensive, more precise characterization of CTCs.
Characterizing the EMT phenotype in CTCs is not sufficient to explain their transformation to a proliferative state at distant organs; it is critical to shed light on the variable duration of dormancy and how these cells are breaking quiescence and are modified into relapse-initiating cells at a secondary site. Recent studies validated the existence of stem cell–like CTCs, which have the ability to self-renew, clonally expand, and initiate tumors, like CSCs can [20, 59, 68, 69]. In 2013, Sun et al reported the CSC biomarkers CD133 and ABCG2 in EpCAM-positive CTCs in 82 patients with hepatocellular carcinoma [70], and they identified the nuclear localization of β-catenin in 10 of 17 of these patients with EpCAM-positive CTCs. The authors concluded that EpCAM-positive CTCs with stem cell–like phenotypes might represent a subset of CTCs with a more aggressive phenotype, earlier recurrence, and worse survival. Further studies are required to explore these stem cell–like CTCs to predict the recurrence timeframe and determine the therapeutic window of treatment for better survival.
Table 2: Prognostic significance of CTC counts in phase II and III clinical trials in cancers of various tissues.
Tumor Tissue |
CTC detection rate |
Phase of Trial |
Prognostic relevance |
References |
Breast |
91% (n = 41 of 45) |
II |
42.9% of patients had 12 months of survival with CTCs (≥1). |
[114] |
Breast |
11.2%(n = 51 of 455) |
III |
Not applicable. |
[115] |
Breast |
39% (n = 148 of 378) |
II |
75% of trastuzumab-treated group showed CK19-negative CTCs. |
[116] |
Colon |
37.5% (n = 180 of 480) |
III |
41% of CTC-positive (≥5) patients had 24 months survival. |
[117] |
Lung |
44.4% (n = 8 of 18) |
II |
76.5% patients showed favorable CTC counts. |
[118] |
Lung |
78% (n = 32 of 41) |
II |
18% of patients with ≥5 CTCs converted to favorable CTCs (<5). |
[119] |
Ovary |
32.1% (n = 216 of 672) |
III |
CTC count was not correlated with survival. |
[120] |
Prostate |
66% (n = 263 of 400) |
III |
Median progression-free survival times, 25.1 months (<3 CTCs) and 16.2 months (≥3 CTCs). |
[121] |
Prostate |
71.5% (n = 88 of 123) |
I/II |
47% of patients with ≥2 positive and 28% of patients with <2 CTC biomarkers showed distant relapse. |
[122] |
Prostate |
35.4% (n = 11 of 31) |
II |
Overall survival rate for 36 months was positive for 55% of patients with 1 positive and 42% of patients with 2-3 positive CTC biomarkers. |
[123] |
Pancreas |
37.5% (n = 19 of 51) |
II |
Median overall survival times, 17.4 to 25.3 months (<2 CTCs) and 12.4 months (≥2 CTCs). |
[124] |
Skin |
~13-17.5% (n = 44-56 of 320) |
III |
Unable to correlate with disease characteristics owing to low CTC counts. |
[125] |
Skin |
86% (n = 214 of 269) |
III |
Increased progression-free survival with decreased CTC counts. |
[126] |
CSCs and relapse
CSCs are a rare subset of tumor cells that bear properties of stem cells, and they show the greatest diversity in cancer progression. Recently, substantial progress has been made in understanding CSCs by characterizing genetic and epigenetic changes occur in their dormant and relapsed stages (Figures 1, 2); however, the surface markers may not unequivocally enrich all CSCs. To date, researchers have identified a few surface markers that enrich various CSCs from the primary tumor for the majority of cancer types. Tumor dormancy and therapeutic refractoriness in different types of cancer are due largely to CSCs and their clonal evolution [71-74]. However, because of the repeated refinement of the CSCs on the basis of new markers, it is difficult to categorize the exact or overlapping populations responsible for promoting the processes of dissemination, intravasation, dormancy, and relapse. Also, self-replicative and non-differentiating cancer stemloids are a topic of considerable interest to pursue effective anti-cancer therapy [74, 75]. These stem-like cells play a seminal role in therapy-resistant relapse due to diverse oncogenic mutations in their clonal populations [75]. Thus, selectively targeting cancer stemloids might provide better therapeutic response for cancer patients.
Our increasing understanding of the molecular biology and aberrantly activated cellular pathways of CSCs has revealed a number of novel targets for targeted therapeutic regimens that have successfully reduced CSCs both in vitro and in preclinical models (Table 3). An example is that upregulation of anti-apoptotic pathway has been detected to maintain mesenchymal state and chemoresistance in breast cancer cells [76]. Using this molecular concept in preclinical study showed that BH3-mimetics were capable to remove both epithelial and mesenchymal HMLE (Human Mammary with Large T and TERT) cells [76]. Therefore, drugs suppressing CSCs hold great promise for redefining cancer therapy in advanced-stage cases. However, CSCs undergo dynamic clonal modification during the metastatic cascade, chemotherapeutic treatments, dormancy, and relapse. Because of their highly heterogeneous nature, relapse-initiating CSCs must be captured and characterized, as most conventional anti-cancer therapies have limited success in eradicating them in patients. A recent study on prostate cancer cells showed that EMT-positive CSCs exhibit resistance to radiation therapy via the PI3K/Akt/mTOR pathway [19]. The biologic link between EMT phenotypes and CSCs has recently been evidenced by epigenetic programming in many types of cancer [77, 78]. In breast cancer cells, Snail interacts with methyltransferase G9a to recruit DNA methyltransferase at the E-cadherin promoter region to silence its expression under low-glucose conditions [78]. In malignant pediatric brain tumor ATRT (Atypical teratoid/rhabdoid tumor), activated STAT3 regulates EMT phenotypes in association with Snail in cisplatin resistant cells [79]. A recent work found that nuclear localized PKCθ acts as a chromatin-anchored switch for EMT to induce expression of mesenchymal genes [78]. Furthermore, the long noncoding RNA Hotair is overexpressed upon TGFβ pathway activation in many cases of cancer [80]; Hotair interacts with polycomb repressive complex 2 to promote methylation at the promoter regions of epithelial genes and involved EMT progression [80, 81].
Table 3: Novel therapeutic compounds targeting CSCs in various tissue-specific cancers. ATRA, all-trans retinoic acid; AML, acute myelogenous leukemia; CML, chronic myelognous leukemia; MM, multiple myeloma.
Tissue stem cells |
Drug |
In vivo study |
In vitro inhibition |
References |
Breast |
ATRA |
No |
Mammosphere |
[127] |
Breast |
IMD-0354+ Doxorubicin |
Yes |
Sphere |
[128] |
Breast |
Salinomycin |
Yes |
Mammosphere |
[129] |
Brain |
Disulfiram |
No |
Ubiquitin-proteasomal pathway |
[130] |
Brain |
γ-secretase inhibitor |
Yes |
Notch pathway |
[131] |
Blood (AML) |
ABT-263 |
Yes |
Oxidative phosphorylation |
[132] |
Blood (CML) |
FTY720 |
Yes |
PP2A agonist |
[133] |
Blood (MM) |
Palcitaxel-Fe3O4 |
Yes |
Not tested |
[134] |
Colon |
Metformin + FuOx |
No |
Colonosphere |
[135] |
Colon |
CC188 |
No |
Carbohydrate epitope on the surface |
[136] |
Colon |
α-DLL4 |
Yes |
Not tested |
[137] |
Gallbladder |
Emodin |
Yes |
ABCG2 pump |
[138] |
Liver |
Lupeol |
Yes |
Hepatosphere |
[139] |
Lung, Breast and Ovary |
VS-5584 |
No |
PI3K-mTOR |
[140] |
Pancreas |
GDC-0449 |
No |
Hedgehog pathway |
[141] |
Pancreas |
GNT-61 |
Yes |
Hedgehog pathway |
[142] |
It has been demonstrated that a subset of isolated CSCs expresses EMT phenotypes in numerous cancers [82-84]. Unfortunately, no suitable marker can enrich EMT-transformed CSCs that are phenotypically different from primary tumor–derived CSCs. To avoid relapse, these cells must be detected in a dormant phase, which may last from a few months to many years.
Some studies validated stem cell–like properties in CTCs [20, 59], suggesting that CSCs transformed into these CTCs and then became dormant. During relapse, they may then become EMT-positive CSCs that proliferate as relapse-initiating cells forming aggressive tumors. Considering the immense clinical significance of these CSCs, it is important to develop strategies to enrich these cells for molecular understanding of relapse. Application of conventional surface markers of CSCs has proven difficult owing to the dynamic clonal evolution of these cells in response to chemotherapy, dormancy, and new tumor microenvironments.
One potential alternative strategy for enriching CSCs is using CTC markers, which can enrich EMT-positive populations, as EMT-positive cancer cells are indicators of aggressive relapse in cancer patients [85]. Thus, isolating EMT-transformed CSCs using CTC surface markers from the relapse site and from the primary tumor could provide a comprehensive picture of the etiologies of relapse. Substantial and systematic research focusing on drug-resistant, relapse-initiating CSCs could promote the development of effective treatment for aggressive cancer, and the identification and culture of these CSCs could be a powerful tool in the investigation of cancer relapse. Also, the identification of aberrant pathways in relapse-initiating CSCs could facilitate the development of therapies for patients for whom traditional chemotherapies and radiation treatments have poor clinical outcomes.
Summary
The etiology of tumor recurrence with a variable time frame remains elusive. Major obstacles include the heterogeneity of tumors in patients and the difficulty of capturing residual drug-resistant tumor cells. Thus, recent research has aimed to identify suitable clinical models that can accurately catalog the steps of cancer recurrence. Clinical studies indicate that harnessing EMT-transformed CTCs and CSCs could shed light on the transition from dormancy to relapse in cancer patients. Future therapeutic studies of CSCs should focus on EMT positive CSCs or relapse-initiating tumor cells rather than just CSCs enriched from primary tumors. The molecular and cellular plasticity of EMT-positive cells needs to be characterized to categorize aberrant molecular pathways and heterotypic interactions with tumor microenvironments. Furthermore, ECM remodeling that supports the EMT program in tumor cells to initiate drug resistance and relapse is required to allow more in-depth tracing.
Currently, neoadjuvant therapy is recommended for patients who are at risk of recurrence after resection of the primary tumor. However, because of the dynamic interaction between tumor microenvironments and cancer cells, EMT-positive CSCs frequently undergo genetic drift and clonal evolution, so novel pharmacologic agents that demonstrate better therapeutic efficacy than current neoadjuvant therapies need to be generated. As translational research is streamlined toward more personalized therapy, suppressing EMT-transformed CTCs and CSCs should prove useful for preventing relapse and extending the lifespans of patients with recurrent cancer.
Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
CTCs, Circulating tumor cells; CSCs, Cancer stem cells; EMT, Epithelial mesenchymal transition; NAT, Neo adjuvant therapy; NR2F1, Nuclear receptor subfamily group 2; TGFβ2, Transforming growth factor β 2; FAK, Focal adhesion kinase; ERK, Extra-cellular-signal related kinase ;MLCK, Myosin light chain kinase; BHLHB3, Basic helix-loop-helix domain containing, class B, 3; PLS3, Plastin 3.
References
1. Aktipis CA, Kwan VS, Johnson KA, Neuberg SL and Maley CC. Overlooking evolution: a systematic analysis of cancer relapse and therapeutic resistance research. PLoS One. 2011; 6:e26100.
2. Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006; 20:385-391.
3. Marusyk A, Almendro V and Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012; 12:323-334.
4. Tsai JH and Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013; 27:2192-2206.
5. Sosa MS, Bragado P and Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014; 14:611-622.
6. Giuliano M, Giordano A, Jackson S, Hess KR, De Giorgi U, Mego M, Handy BC, Ueno NT, Alvarez RH, De Laurentiis M, De Placido S, Valero V, Hortobagyi GN, Reuben JM and Cristofanilli M. Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment. Breast Cancer Res. 2011; 13:R67.
7. Pierga JY, Hajage D, Bachelot T, Delaloge S, Brain E, Campone M, Dieras V, Rolland E, Mignot L, Mathiot C and Bidard FC. High independent prognostic and predictive value of circulating tumor cells compared with serum tumor markers in a large prospective trial in first-line chemotherapy for metastatic breast cancer patients. Ann Oncol. 2012; 23:618-624.
8. Dahan P, Martinez Gala J, Delmas C, Monferran S, Malric L, Zentkowski D, Lubrano V, Toulas C, Cohen-Jonathan Moyal E and Lemarie A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis. 2014; 5:e1543.
9. Zhang S, Cui B, Lai H, Liu G, Ghia EM, Widhopf GF, 2nd, Zhang Z, Wu CC, Chen L, Wu R, Schwab R, Carson DA and Kipps TJ. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A. 2014.
10. Mego M, Mani SA, Lee BN, Li C, Evans KW, Cohen EN, Gao H, Jackson SA, Giordano A, Hortobagyi GN, Cristofanilli M, Lucci A and Reuben JM. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int J Cancer. 2012; 130:808-816.
11. Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H, Iwatsuki M, Ota D, Ohkuma M, Iwaya T, Nishida N, Kogo R, Sudo T, Tanaka F, Shibata K, Toh H, et al. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer research. 2013; 73:2059-2069.
12. Wik E, Raeder MB, Krakstad C, Trovik J, Birkeland E, Hoivik EA, Mjos S, Werner HM, Mannelqvist M, Stefansson IM, Oyan AM, Kalland KH, Akslen LA and Salvesen HB. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013; 19:1094-1105.
13. Oishi N, Kumar MR, Roessler S, Ji J, Forgues M, Budhu A, Zhao X, Andersen JB, Ye QH, Jia HL, Qin LX, Yamashita T, Woo HG, Kim YJ, Kaneko S, Tang ZY, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology. 2012; 56:1792-1803.
14. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA and Mani SA. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010; 107:15449-15454.
15. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013; 339:580-584.
16. Kallergi G, Papadaki MA, Politaki E, Mavroudis D, Georgoulias V and Agelaki S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011; 13:R59.
17. Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F, Sessa G, Carriero MV, Normanno N, Rocco G and Pirozzi G. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell death & disease. 2013; 4:e620.
18. Giordano A, Gao H, Anfossi S, Cohen E, Mego M, Lee BN, Tin S, De Laurentiis M, Parker CA, Alvarez RH, Valero V, Ueno NT, De Placido S, Mani SA, Esteva FJ, Cristofanilli M, et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol Cancer Ther. 2012; 11:2526-2534.
19. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH and Li Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013; 4:e875.
20. Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R and Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009; 11:R46.
21. Ksiazkiewicz M, Markiewicz A and Zaczek AJ. Epithelial-mesenchymal transition: a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012; 79:195-208.
22. Pirozzi G, Tirino V, Camerlingo R, La Rocca A, Martucci N, Scognamiglio G, Franco R, Cantile M, Normanno N and Rocco G. Prognostic value of cancer stem cells, epithelial-mesenchymal transition and circulating tumor cells in lung cancer. Oncol Rep. 2013; 29:1763-1768.
23. Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC and Farace F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer. 2011; 105:1338-1341.
24. Uhr JW and Pantel K. Controversies in clinical cancer dormancy. Proc Natl Acad Sci U S A. 2011; 108(30):12396-12400.
25. Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, Krebsbach PH, Pienta KJ, Shiozawa Y and Taichman RS. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013; 15:1064-1074.
26. Marshall JC, Collins JW, Nakayama J, Horak CE, Liewehr DJ, Steinberg SM, Albaugh M, Vidal-Vanaclocha F, Palmieri D, Barbier M, Murone M and Steeg PS. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J Natl Cancer Inst. 2012; 104:1306-1319.
27. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, Benten D, Forbes SJ, Wells RG and Iredale JP. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011; 53:1192-1205.
28. Kucerova L, Feketeova L, Matuskova M, Kozovska Z, Janega P, Babal P and Poturnajova M. Local bystander effect induces dormancy in human medullary thyroid carcinoma model in vivo. Cancer Lett. 2013; 335:299-305.
29. Osella-Abate S, Ribero S, Sanlorenzo M, Maule MM, Richiardi L, Merletti F, Tomasini C, Marra E, Macripo G, Fierro MT and Quaglino P. Risk factors related to late metastases in 1,372 melanoma patients disease free more than 10 years. Int J Cancer. 2014.
30. Moore N, Houghton J and Lyle S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012; 21:1822-1830.
31. Liang H, Deng L, Burnette B, Weichselbaum RR and Fu YX. Radiation-induced tumor dormancy reflects an equilibrium between the proliferation and T lymphocyte-mediated death of malignant cells. Oncoimmunology. 2013; 2:e25668.
32. Straume O, Shimamura T, Lampa MJ, Carretero J, Oyan AM, Jia D, Borgman CL, Soucheray M, Downing SR, Short SM, Kang SY, Wang S, Chen L, Collett K, Bachmann I, Wong KK, et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci U S A. 2012; 109:8699-8704.
33. Gao H, Chakraborty G, Lee-Lim AP, Mavrakis KJ, Wendel HG and Giancotti FG. Forward genetic screens in mice uncover mediators and suppressors of metastatic reactivation. Proc Natl Acad Sci U S A. 2014; 111:16532-16537.
34. Chen H, Ko JM, Wong VC, Hyytiainen M, Keski-Oja J, Chua D, Nicholls JM, Cheung FM, Lee AW, Kwong DL, Chiu PM, Zabarovsky ER, Tsao SW, Tao Q, Kan R, Chan SH, et al. LTBP-2 confers pleiotropic suppression and promotes dormancy in a growth factor permissive microenvironment in nasopharyngeal carcinoma. Cancer Lett. 2012; 325:89-98.
35. Indraccolo S, Minuzzo S, Masiero M, Pusceddu I, Persano L, Moserle L, Reboldi A, Favaro E, Mecarozzi M, Di Mario G, Screpanti I, Ponzoni M, Doglioni C and Amadori A. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer research. 2009; 69:1314-1323.
36. Aguirre-Ghiso JA, Bragado P and Sosa MS. Metastasis awakening: targeting dormant cancer. Nat Med. 2013; 19:276-277.
37. Brabletz T. To differentiate or not—routes towards metastasis. Nat Rev Cancer. 2012; 12:425-436.
38. Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, Klein-Szanto AJ, Herlyn M, Diehl JA, Rustgi AK and Nakagawa H. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer research. 2010; 70:4174-4184.
39. Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D, Gimotty PA, Klein-Szanto AJ, Diehl JA, Herlyn M and Nakagawa H. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer research. 2011; 71:6836-6847.
40. Kang Y and Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell. 2013; 23:573-581.
41. Schewe DM and Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer research. 2009; 69:1545-1552.
42. Sosa MS, Avivar-Valderas A, Bragado P, Wen HC and Aguirre-Ghiso JA. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17:5850-5857.
43. Adam AP, George A, Schewe D, Bragado P, Iglesias BV, Ranganathan AC, Kourtidis A, Conklin DS and Aguirre-Ghiso JA. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer research. 2009; 69:5664-5672.
44. Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, Basso G, Leo G, Rosato A, Bicciato S, Cordenonsi M and Piccolo S. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012; 487:380-384.
45. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, Schewe DM and Aguirre-Ghiso JA. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013; 15:1351-1361.
46. Aguirre Ghiso JA, Kovalski K and Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999; 147:89-104.
47. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, Webster JD, Hoover S, Simpson RM, Gauldie J and Green JE. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer research. 2010; 70:5706-5716.
48. Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010; 21 Suppl 7:vii89-92.
49. Velpula KK, Dasari VR, Tsung AJ, Dinh DH and Rao JS. Cord blood stem cells revert glioma stem cell EMT by down regulating transcriptional activation of Sox2 and Twist1. Oncotarget. 2011; 2:1028-1042.
50. Tiwari N, Gheldof A, Tatari M and Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012; 22:194-207.
51. Canadas I, Rojo F, Taus A, Arpi O, Arumi-Uria M, Pijuan L, Menendez S, Zazo S, Domine M, Salido M, Mojal S, Garcia de Herreros A, Rovira A, Albanell J and Arriola E. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014; 20:938-950.
52. Li YM, Xu SC, Li J, Han KQ, Pi HF, Zheng L, Zuo GH, Huang XB, Li HY, Zhao HZ, Yu ZP, Zhou Z and Liang P. Epithelial-mesenchymal transition markers expressed in circulating tumor cells in hepatocellular carcinoma patients with different stages of disease. Cell Death Dis. 2013; 4:e831.
53. Park KS, Raffeld M, Moon YW, Xi L, Bianco C, Pham T, Lee LC, Mitsudomi T, Yatabe Y, Okamoto I, Subramaniam D, Mok T, Rosell R, Luo J, Salomon DS, Wang Y, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest. 2014; 124:3003-3015.
54. Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan JP, Kirkpatrick DS and Settleman J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014; 5:7328-7341.
55. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T and Hahn WC. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014; 158:171-184.
56. Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A, Sgarra R, Del Sal G and Manfioletti G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget. 2013; 4:1293-1308.
57. Cuyas E, Corominas-Faja B and Menendez JA. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget. 2014; 5:3970-3982.
58. Cheong H. Integrating autophagy and metabolism in cancer. Archives of pharmacal research. 2015; 38(3):358-371.
59. Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D and Georgoulias V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010; 288:99-106.
60. Alonso-Alconada L, Muinelo-Romay L, Madissoo K, Diaz-Lopez A, Krakstad C, Trovik J, Wik E, Hapangama D, Coenegrachts L, Cano A, Gil-Moreno A, Chiva L, Cueva J, Vieito M, Ortega E, Mariscal J, et al. Molecular profiling of circulating tumor cells links plasticity to the metastatic process in endometrial cancer. Mol Cancer. 2014; 13:223.
61. Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ and Garcia-Blanco MA. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011; 9:997-1007.
62. Satelli A, Brownlee Z, Mitra A, Meng QH and Li S. Circulating Tumor Cell Enumeration with a Combination of Epithelial Cell Adhesion Molecule- and Cell-Surface Vimentin-Based Methods for Monitoring Breast Cancer Therapeutic Response. Clin Chem. 2014.
63. Satelli A, Mitra A, Brownlee Z, Xia X, Bellister S, Overman MJ, Kopetz S, Ellis LM, Meng QH and Li S. Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015; 21:899-906.
64. Gorges TM, Tinhofer I, Drosch M, Rose L, Zollner TM, Krahn T and von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012; 12:178.
65. Grover PK, Cummins AG, Price TJ, Roberts-Thomson IC and Hardingham JE. Circulating tumour cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Ann Oncol. 2014.
66. Alix-Panabieres C and Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014; 14(9):623-631.
67. Ghazani AA, Castro CM, Gorbatov R, Lee H and Weissleder R. Sensitive and direct detection of circulating tumor cells by multimarker micro-nuclear magnetic resonance. Neoplasia. 2012; 14:388-395.
68. Raimondi C, Gradilone A, Naso G, Vincenzi B, Petracca A, Nicolazzo C, Palazzo A, Saltarelli R, Spremberg F, Cortesi E and Gazzaniga P. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res Treat. 2011; 130:449-455.
69. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, Scharpff M, Marme F, Sinn HP, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013; 31:539-544.
70. Sun YF, Xu Y, Yang XR, Guo W, Zhang X, Qiu SJ, Shi RY, Hu B, Zhou J and Fan J. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013; 57:1458-1468.
71. Paez D, Labonte MJ, Bohanes P, Zhang W, Benhanim L, Ning Y, Wakatsuki T, Loupakis F and Lenz HJ. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18:645-653.
72. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C and Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5:219-234.
73. Cojoc M, Mabert K, Muders MH and Dubrovska A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin Cancer Biol. 2014.
74. Blagosklonny MV. Why therapeutic response may not prolong the life of a cancer patient: selection for oncogenic resistance. Cell cycle. 2005; 4:1693-1698.
75. Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer biology & therapy. 2007; 6:1684-1690.
76. Keitel U, Scheel C and Dobbelstein M. Overcoming EMT-driven therapeutic resistance by BH3 mimetics. Oncoscience. 2014; 1:706-708.
77. Tam WL and Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013; 19:1438-1449.
78. Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM and Zhou BP. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012; 122:1469-1486.
79. Liu WH, Chen MT, Wang ML, Lee YY, Chiou GY, Chien CS, Huang PI, Chen YW, Huang MC, Chiou SH, Shih YH and Ma HI. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget. 2015; 6:1750-1768.
80. Padua Alves C, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, Valente V, Zago MA and Silva WA, Jr. Brief report: The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells. 2013; 31:2827-2832.
81. Wu L, Murat P, Matak-Vinkovic D, Murrell A and Balasubramanian S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry. 2013; 52:9519-9527.
82. Baccelli I and Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012; 198:281-293.
83. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G, Gulotta G, Dieli F, De Maria R and Stassi G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014; 14:342-356.
84. Singh A and Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29:4741-4751.
85. Mitra A, Satelli A, Xia X, Cutrera J, Mishra L and Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin CD133 novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer. 2014.
86. Aldousari S and Kassouf W. Update on the management of non-muscle invasive bladder cancer. Can Urol Assoc J. 2010; 4:56-64.
87. Kobayashi H, Kikuchi E, Mikami S, Maeda T, Tanaka N, Miyajima A, Nakagawa K and Oya M. Long term follow-up in patients with initially diagnosed low grade Ta non-muscle invasive bladder tumors: tumor recurrence and worsening progression. BMC Urol. 2014; 14:5.
88. Gelderblom H, Jinks RC, Sydes M, Bramwell VH, van Glabbeke M, Grimer RJ, Hogendoorn PC, McTiernan A, Lewis IJ, Nooij MA, Taminiau AH, Whelan J and European Osteosarcoma I. Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. European journal of cancer. 2011; 47:895-902.
89. Wang Y, Yin Q, Yu Q, Zhang J, Liu Z, Wang S, Lv S and Niu Y. A retrospective study of breast cancer subtypes: the risk of relapse and the relations with treatments. Breast cancer research and treatment. 2011; 130:489-498.
90. Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO and Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010; 28:1684-1691.
91. Greenspoon JN, Sharieff W, Hirte H, Overholt A, Devillers R, Gunnarsson T and Whitton A. Fractionated stereotactic radiosurgery with concurrent temozolomide chemotherapy for locally recurrent glioblastoma multiforme: a prospective cohort study. Onco Targets Ther. 2014; 7:485-490.
92. O’Connell MJ, Campbell ME, Goldberg RM, Grothey A, Seitz JF, Benedetti JK, Andre T, Haller DG and Sargent DJ. Survival following recurrence in stage II and III colon cancer: findings from the ACCENT data set. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008; 26:2336-2341.
93. Ding P, Liska D, Tang P, Shia J, Saltz L, Goodman K, Downey RJ, Nash GM, Temple LK, Paty PB, Guillem JG, Wong WD and Weiser MR. Pulmonary recurrence predominates after combined modality therapy for rectal cancer: an original retrospective study. Annals of surgery. 2012; 256:111-116.
94. Begg AC. Predicting recurrence after radiotherapy in head and neck cancer. Seminars in radiation oncology. 2012; 22:108-118.
95. Baxi SS, Pinheiro LC, Patil SM, Pfister DG, Oeffinger KC and Elkin EB. Causes of death in long-term survivors of head and neck cancer. Cancer. 2014; 120:1507-1513.
96. Mell LK, Dignam JJ, Salama JK, Cohen EE, Polite BN, Dandekar V, Bhate AD, Witt ME, Haraf DJ, Mittal BB, Vokes EE and Weichselbaum RR. Predictors of competing mortality in advanced head and neck cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010; 28:15-20.
97. Chin AI, Lam JS, Figlin RA and Belldegrun AS. Surveillance strategies for renal cell carcinoma patients following nephrectomy. Rev Urol. 2006; 8:1-7.
98. Yamamoto S, Tomita Y, Nakamori S, Hoshida Y, Nagano H, Dono K, Umeshita K, Sakon M, Monden M and Aozasa K. Elevated expression of valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003; 21:447-452.
99. Forner A, Llovet JM and Bruix J. Hepatocellular carcinoma. Lancet. 2012; 379:1245-1255.
100. Goodgame B, Viswanathan A, Zoole J, Gao F, Miller CR, Subramanian J, Meyers BF, Patterson AG and Govindan R. Risk of recurrence of resected stage I non-small cell lung cancer in elderly patients as compared with younger patients. J Thorac Oncol. 2009; 4:1370-1374.
101. Maeda R, Yoshida J, Hishida T, Aokage K, Nishimura M, Nishiwaki Y and Nagai K. Late recurrence of non-small cell lung cancer more than 5 years after complete resection: incidence and clinical implications in patient follow-up. Chest. 2010; 138:145-150.
102. Zeliadt SB, Sekaran NK, Hu EY, Slatore CC, Au DH, Backhus L, Wu DY, Crawford J, Lyman GH and Dale DC. Comparison of demographic characteristics, surgical resection patterns, and survival outcomes for veterans and nonveterans with non-small cell lung cancer in the Pacific Northwest. J Thorac Oncol. 2011; 6:1726-1732.
103. Kim ML, Kim JM, Seong SJ, Lee SY, Han M and Cho YJ. Recurrence of ovarian endometrioma after second-line, conservative, laparoscopic cyst enucleation. Am J Obstet Gynecol. 2014; 210:216 e211-216.
104. Brown AP, Jhingran A, Klopp AH, Schmeler KM, Ramirez PT and Eifel PJ. Involved-field radiation therapy for locoregionally recurrent ovarian cancer. Gynecol Oncol. 2013; 130:300-305.
105. Chang DK, Johns AL, Merrett ND, Gill AJ, Colvin EK, Scarlett CJ, Nguyen NQ, Leong RW, Cosman PH, Kelly MI, Sutherland RL, Henshall SM, Kench JG and Biankin AV. Margin clearance and outcome in resected pancreatic cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009; 27:2855-2862.
106. Botrel TE, Clark O, dos Reis RB, Pompeo AC, Ferreira U, Sadi MV and Bretas FF. Intermittent versus continuous androgen deprivation for locally advanced, recurrent or metastatic prostate cancer: a systematic review and meta-analysis. BMC Urol. 2014; 14:9.
107. Tandstad T, Smaaland R, Solberg A, Bremnes RM, Langberg CW, Laurell A, Stierner UK, Stahl O, Cavallin-Stahl EK, Klepp OH, Dahl O and Cohn-Cedermark G. Management of seminomatous testicular cancer: a binational prospective population-based study from the Swedish norwegian testicular cancer study group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011; 29:719-725.
108. Travis LB, Beard C, Allan JM, Dahl AA, Feldman DR, Oldenburg J, Daugaard G, Kelly JL, Dolan ME, Hannigan R, Constine LS, Oeffinger KC, Okunieff P, Armstrong G, Wiljer D, Miller RC, et al. Testicular cancer survivorship: research strategies and recommendations. J Natl Cancer Inst. 2010; 102:1114-1130.
109. Kruijff S, Petersen JF, Chen P, Aniss AM, Clifton-Bligh RJ, Sidhu SB, Delbridge LW, Gill AJ, Learoyd D and Sywak MS. Patterns of structural recurrence in papillary thyroid cancer. World J Surg. 2014; 38:653-659.
110. Amin A, Badwey A and El-Fatah S. Differentiated thyroid carcinoma: an analysis of 249 patients undergoing therapy and aftercare at a single institution. Clin Nucl Med. 2014; 39:142-146.
111. Sugiyama T, Fujiwara K, Ohashi Y, Yokota H, Hatae M, Ohno T, Nagai Y, Mitsuhashi N, Ochiai K and Noda K. Phase III placebo-controlled double-blind randomized trial of radiotherapy for stage IIB-IVA cervical cancer with or without immunomodulator Z-100: a JGOG study. Ann Oncol. 2014; 25:1011-1017.
112. Hoogendam JP, Zaal A, Rutten EG, Heijnen CJ, Kenter GG, Veldhuis WB, Verheijen RH and Zweemer RP. Detection of cervical cancer recurrence during follow-up: a multivariable comparison of 9 frequently investigated serum biomarkers. Gynecol Oncol. 2013; 131:655-660.
113. Desai NB, Kiess AP, Kollmeier MA, Abu-Rustum NR, Makker V, Barakat RR and Alektiar KM. Patterns of relapse in stage I-II uterine papillary serous carcinoma treated with adjuvant intravaginal radiation (IVRT) with or without chemotherapy. Gynecol Oncol. 2013; 131:604-608.
114. Pierga JY, Bidard FC, Cropet C, Tresca P, Dalenc F, Romieu G, Campone M, Mahier Ait-Oukhatar C, Le Rhun E, Goncalves A, Leheurteur M, Domont J, Gutierrez M, Cure H, Ferrero JM, Labbe-Devilliers C, et al. Circulating tumor cells and brain metastasis outcome in patients with HER2-positive breast cancer: the LANDSCAPE trial. Ann Oncol. 2013; 24:2999-3004.
115. Azim HA, Jr., Rothe F, Aura CM, Bavington M, Maetens M, Rouas G, Gebhart G, Gamez C, Eidtmann H, Baselga J, Piccart-Gebhart M, Ellis C, Vuylsteke P, Cure H, Domont J, Ferro A, et al. Circulating tumor cells and response to neoadjuvant paclitaxel and HER2-targeted therapy: a sub-study from the NeoALTTO phase III trial. Breast. 2013; 22:1060-1065.
116. Georgoulias V, Bozionelou V, Agelaki S, Perraki M, Apostolaki S, Kallergi G, Kalbakis K, Xyrafas A and Mavroudis D. Trastuzumab decreases the incidence of clinical relapses in patients with early breast cancer presenting chemotherapy-resistant CK-19mRNA-positive circulating tumor cells: results of a randomized phase II study. Ann Oncol. 2012; 23:1744-1750.
117. Sastre J, Maestro ML, Gomez-Espana A, Rivera F, Valladares M, Massuti B, Benavides M, Gallen M, Marcuello E, Abad A, Arrivi A, Fernandez-Martos C, Gonzalez E, Tabernero JM, Vidaurreta M, Aranda E, et al. Circulating tumor cell count is a prognostic factor in metastatic colorectal cancer patients receiving first-line chemotherapy plus bevacizumab: a Spanish Cooperative Group for the Treatment of Digestive Tumors study. Oncologist. 2012; 17:947-955.
118. Besse B, Planchard D, Veillard AS, Taillade L, Khayat D, Ducourtieux M, Pignon JP, Lumbroso J, Lafontaine C, Mathiot C and Soria JC. Phase 2 study of frontline bortezomib in patients with advanced non-small cell lung cancer. Lung Cancer. 2012; 76:78-83.
119. Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BG, Hicks RJ, Hampton GM, Amler LC, Pirzkall A and Lackner MR. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18:2391-2401.
120. Poveda A, Kaye SB, McCormack R, Wang S, Parekh T, Ricci D, Lebedinsky CA, Tercero JC, Zintl P and Monk BJ. Circulating tumor cells predict progression free survival and overall survival in patients with relapsed/recurrent advanced ovarian cancer. Gynecol Oncol. 2011; 122:567-572.
121. Goldkorn A, Ely B, Quinn DI, Tangen CM, Fink LM, Xu T, Twardowski P, Van Veldhuizen PJ, Agarwal N, Carducci MA, Monk JP, 3rd, Datar RH, Garzotto M, Mack PC, Lara P, Jr., Higano CS, et al. Circulating tumor cell counts are prognostic of overall survival in SWOG S0421: a phase III trial of docetaxel with or without atrasentan for metastatic castration-resistant prostate cancer. J Clin Oncol. 2014; 32:1136-1142.
122. Dreicer R, MacLean D, Suri A, Stadler WM, Shevrin D, Hart L, MacVicar GR, Hamid O, Hainsworth J, Gross ME, Shi Y, Webb IJ and Agus DB. Phase I/II trial of orteronel (TAK-700)—an investigational 17,20-lyase inhibitor—in patients with metastatic castration-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014; 20:1335-1344.
123. Antonarakis ES, Heath EI, Posadas EM, Yu EY, Harrison MR, Bruce JY, Cho SY, Wilding GE, Fetterly GJ, Hangauer DG, Kwan MF, Dyster LM and Carducci MA. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013; 71:883-892.
124. Chee CE, Krishnamurthi S, Nock CJ, Meropol NJ, Gibbons J, Fu P, Bokar J, Teston L, O’Brien T, Gudena V, Reese A, Bergman M, Saltzman J, Wright JJ, Dowlati A and Brell J. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist. 2013; 18:1091-1092.
125. Hoshimoto S, Shingai T, Morton DL, Kuo C, Faries MB, Chong K, Elashoff D, Wang HJ, Elashoff RM and Hoon DS. Association between circulating tumor cells and prognosis in patients with stage III melanoma with sentinel lymph node metastasis in a phase III international multicenter trial. J Clin Oncol. 2012; 30:3819-3826.
126. Hoshimoto S, Faries MB, Morton DL, Shingai T, Kuo C, Wang HJ, Elashoff R, Mozzillo N, Kelley MC, Thompson JF, Lee JE and Hoon DS. Assessment of prognostic circulating tumor cells in a phase III trial of adjuvant immunotherapy after complete resection of stage IV melanoma. Ann Surg. 2012; 255:357-362.
127. Bhat-Nakshatri P, Goswami CP, Badve S, Sledge GW, Jr. and Nakshatri H. Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Sci Rep. 2013; 3:2530.
128. Gomez-Cabrero A, Wrasidlo W and Reisfeld RA. IMD-0354 targets breast cancer stem cells: a novel approach for an adjuvant to chemotherapy to prevent multidrug resistance in a murine model. PLoS One. 2013; 8:e73607.
129. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA and Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009; 138:645-659.
130. Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon JG, Reynolds B and Foltz G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget. 2012; 3:1124-1136.
131. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL and Eberhart CG. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010; 28:5-16.
132. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW and Jordan CT. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013; 12:329-341.
133. Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, Ferenchak G, Dorrance AM, Paisie CA, Eiring AM, Ma Y, Mao HC, Zhang B, Wunderlich M, May PC, Sun C, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013; 123:4144-4157.
134. Yang C, Wang J, Chen D, Chen J, Xiong F, Zhang H, Zhang Y, Gu N and Dou J. Paclitaxel-Fe3O4 nanoparticles inhibit growth of CD138(-) CD34(-) tumor stem-like cells in multiple myeloma-bearing mice. Int J Nanomedicine. 2013; 8:1439-1449.
135. Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E and Majumdar AP. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014; 9:e84369.
136. Xu M, Yuan Y, Xia Y and Achilefu S. Monoclonal antibody CC188 binds a carbohydrate epitope expressed on the surface of both colorectal cancer stem cells and their differentiated progeny. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14:7461-7469.
137. Fischer M, Yen WC, Kapoun AM, Wang M, O’Young G, Lewicki J, Gurney A and Hoey T. Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer research. 2011; 71:1520-1525.
138. Li XX, Dong Y, Wang W, Wang HL, Chen YY, Shi GY, Yi J and Wang J. Emodin as an effective agent in targeting cancer stem-like side population cells of gallbladder carcinoma. Stem Cells Dev. 2013; 22:554-566.
139. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S and Ng IO. Lupeol targets liver tumor-initiating cells through phosphatase and tensin homolog modulation. Hepatology. 2011; 53:160-170.
140. Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, Weaver DT, Padval MV, Pachter JA and Xu Q. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer research. 2014.
141. Singh BN, Fu J, Srivastava RK and Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One. 2011; 6:e27306.
142. Fu J, Rodova M, Roy SK, Sharma J, Singh KP, Srivastava RK and Shankar S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013; 330:22-32.